
Abstract
In their active hypophosphorylated state, members of the retinoblastoma family of pocket proteins negatively regulate cell cycle progression at least in part by repressing expression of E2F-dependent genes. Mitogen-dependent activation of G1 and G1/S Cyclin Dependent Kinases (CDKs) results in coordinated hyperphosphorylation and inactivation of these proteins, which no longer bind and repress E2Fs. S and G2/M CDKs maintain pocket protein hyperphosphorylated through the end of mitosis. The inactivating action of inducible CDKs is opposed by the Ser/Thr protein phosphatases PP2A and PP1. Various trimeric PP2A holoenzymes have been implicated in dephosphorylation of pocket proteins in response to specific cellular signals and stresses or as part of an equilibrium with CDKs throughout the cell cycle. PP1 has specifically been implicated in dephosphorylation of pRB in late mitosis and early G1. This review is particularly focused on the emerging role of PP2A as a major hub for integration of growth suppressor signals that require rapid inactivation of pocket proteins. Of note, activation of particular PP2A holoenzymes triggers differential activation of pocket proteins in the presence of active CDKs.
Introduction
This review is focused on how the phosphorylation state of pocket proteins is maintained throughout the cell cycle and perturbed in response to signals that promote growth arrest via rapid dephosphorylation of pocket proteins. Because reactivation of pocket proteins by upregulation of CDK inhibitors has been extensively studied and been the epicenter of numerous excellent reviews, (reviewed in refs.Citation1-4) this manuscript is centered on the role played by serine/threonine protein phosphatases and in particular PP2A.
Holoenzymes, Substrates and Other Factors in the Equilibrium that Determines the Phosphorylation State of Pocket Proteins
Pocket protein structure and function
The cell cycle is negatively regulated by the retinoblastoma family of proteins, commonly designated “pocket proteins,” which mediate transcriptional repression of many cell cycle genes. (reviewed in refs.Citation5-7) The pocket protein family includes the pRB tumor suppressor protein and the 2 closely related paralog proteins p107 and p130 (also designated RBL1 and RBL2, respectively). The RB gene is named for the disease in which it was first found to be mutated.Citation8
Pocket proteins consist of 5 major domains: the N- and C- terminal domains, 2 “pocket” domains designated “A” and “B,” and a spacer region that links the 2 pocket domains. (reviewed in refs.Citation9-12) The highest degree of homology among all 3 pocket proteins lies in the pocket domains, whereas the spacer and N-terminal domains are more divergent. p107 and p130 share ∼54% homology with each other, while pRB is only 25% homologous to p107 and p130 (reviewed in ref.Citation5). Adenoviral E1A was the first viral oncoprotein found to bind pRB,Citation13 and 2 conserved regions containing amino acids ∼40-70 and ∼121-139 were found to be required for pRB binding.Citation13-15 Sequence alignments revealed that SV40 T-antigen and HPV E7 also contained these conserved motifs.Citation16 These 3 viral proteins all share one common motif- LXCXE- required to bind pocket proteins, which is also present in some Cyclins and other cellular proteins. Deletion of this motif rendered HPV E7 unable to bind pRB.Citation17 The pocket region of the pRB family is comprised of 2 domains, “A” and “B,” that resemble cyclin folds (reviewed in refs.Citation5,18). These folds form a structure that serves as a binding site for a majority of the proteins known to interact with pocket proteins, including Cyclins, E2Fs, histone deacetylases, and c-Myc, among others (reviewed in refs.Citation9,10,12,19). p107 and p130 contain a Cyclin/CDK binding site in their spacer in addition to CDK inhibitory regions in their N-termini that inhibit Cyclin/CDK2 complexes.Citation20,21 In contrast, pRB does not contain a kinase inhibitory domain, and its Cyclin/CDK binding site is located at the C-terminus.Citation22,23
The expression of pocket proteins is differentially regulated throughout the cell cycle. p130 is expressed highly in quiescent cells and is present at promoters of genes required for cell cycle exit. During G1, p130 is hyperphosphorylated to form 3, the slowest migrating hyperphosphorylated form of p130, which is quickly downregulated.Citation24-26 This is due to targeted degradation by the ubiquitin ligase SCFSkp2.Citation27,28 p107 is undetectable or expressed at low levels in quiescent cells, as its E2F dependent promoter is repressed in G0.Citation29 p107 is expressed starting in mid-G1 following mitogenic stimulation.Citation30 pRB levels increase slightly from G1 as it is also an E2F responsive gene, however, changes in pRB expression are not as dramatic as they are for p107 or p130 (reviewed in refs.Citation12,31).
Pocket proteins are recruited to promoters when complexed with members of the E2F family. There are 2 main varieties of E2Fs that associate with pocket proteins: the “activator” E2Fs and the “repressor” E2Fs. The activator E2Fs- E2F1, E2F2, and E2F3a- are reported to primarily associate with pRB in G1Citation32,33 and are responsible for transcription of the E2F gene program from mid to late G1 through S phase when released from pocket proteins.Citation34 Phosphorylation by the coordinated action of Cyclin D/CDK4/6 and Cyclin E/CDK2 complexes in mid to late G1 dissociates pocket proteins from E2Fs allowing binding of positive transcription cofactors such as p300, CBP, P/CAF and other histone acetylases (reviewed in ref.Citation35,36). Association of “activator” E2Fs (aE2Fs) with p107 and p130 has also been reported.Citation37,38 While these complexes are less abundant than pRB/aE2Fs complexes,Citation38,39 changes in the expression of both E2Fs and the hypophosphorylated forms of pocket proteins are likely to regulate their relative abundance. However, whether their function is different than that of pRB/aE2F complexes is not known.
p107 and p130 are preferential partners of E2F4 and E2F5, referred to as “repressor” E2Fs. These E2Fs are not found at gene promoters unless they are bound by pocket proteins.Citation33 Following Cyclin/CDK inactivation of p107 or p130 in mid to late-G1, E2F4 is exported to the cytoplasm,Citation40 where it remains until it binds hypophosphorylated p107 or p130 again, as cells exit mitosis and progress through G1 or exit the cell cycle into G0. At this point the newly formed E2F4/pocket protein complexes translocate to the nucleus to repress transcription of E2F-dependent genes.Citation34 Although not so exhaustively studied, E2F5 is believed to behave similarly in cells where it is expressed (reviewed in ref.Citation36,41).
Thus, pocket proteins are negative regulators of the cell cycle. In their active- or hypophosphorylated- state, which normally occurs in quiescent cells or the G1 phase of the cell cycle, they are associated with E2F and DP transcription factor heterodimers and bound to the promoters of E2F dependent genes, which result in the active repression of cell cycle genes. Upon mitogenic stimulation, G1 and G1/S Cyclin/CDK complexes phosphorylate pocket proteins, resulting in their release from E2F/DP complexes. In the absence of pocket protein binding, activator E2Fs 1-3a become positive transcription factors or transactivators, allowing p300, CBP, P/CAF and other histone acetylases to bind, thus promoting cell cycle progression through the restriction point (reviewed in refs.Citation12,35).
Pocket protein activity is determined by Cyclin/CDK complexes and Ser/Thr Protein Phosphatases throughout the cell cycle
pRB and p130 are inactivated by the coordinated action of Cyclin D/CDK4/6 and Cyclin E/CDK2 complexes. p107 appears to be mostly inactivated by Cyclin D/CDK4/6, but phosphorylation of certain residues by Cyclin E/CDK2 has been observed (reviewed in refs.Citation11,42). Once phosphorylated, pRB, p107 and p130 remain in a hyperphosphorylated (inactive) state throughout the remainder of the cell cycle until late mitosis, where they are abruptly dephosphorylated coinciding with Cyclin/CDK inactivation in preparation for the next round of the cell cycle. More precisely, pRB was found to be rather rapidly dephosphorylated in anaphase following nocodazole block and release in various cell types, suggesting that a phosphatase was responsible. Treatment of late mitotic cell extracts with metal ion chelators revealed the dephosphorylation of pRB was not mediated by PP2B and/or PP2C, as they require calcium and magnesium for their catalytic activity, respectively. pRB dephosphorylation in the same extracts was sensitive to >10 nM okadaic acid, which suggested PP1 was involved.Citation43 Around the same time, another laboratory identified the PP1α2 isoform as a pRB binding partner in a yeast 2-hybrid screen, and showed that the timing of the interaction of PP1α2 and pRB lasts from M phase to mid-G1, when cell cycle CDKs are inactive. Interestingly, it also was shown that PP1α2 cannot bind a pRB C-terminal deletion mutant.Citation44 From this evidence, a model emerged suggesting that from mid-G1 to mitosis, CDKs are “switched on,” resulting in the inactivation of pocket proteins, until late mitosis, where the CDKs are “switched off.” Conversely, PP1 is “switched on” in anaphase, leading to the reactivation of pocket proteins until early/mid-G1, when PP1 would be “switched off.”Citation45,46 Although PP1 had been implicated in targeting pRB, it was not shown to interact with p107 or p130 at the time, and a later study showed that p107 only weakly bound to PP1α while p130 failed to bind.Citation47
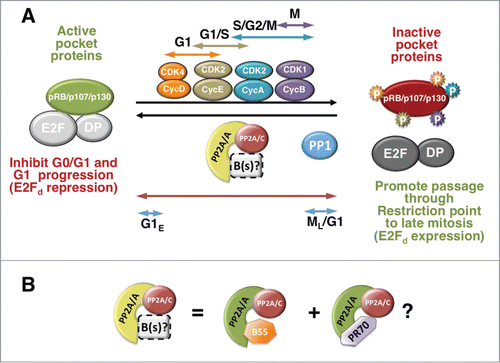
PP2A holoenzymes have been shown to interact with all 3 pocket proteins upon cellular stresses or extracellular signaling.Citation48-52 Subsequently, PP2A holoenzymes were hypothesized to mediate an equilibrium with CDKs that determines the phosphorylation state of all 3 pocket proteins during the cell cycle.Citation52 This idea arose from the observation that pocket proteins were rapidly dephosphorylated in asynchronous cells following treatments with cycloheximide or the pan CDK inhibitor flavopiridol, suggesting rapid dephosphorylation following CDK inhibition independently of cell cycle position. Treatment of cells with the phosphatase inhibitors calyculin A and okadaic acid at concentrations that preferentially inhibit PP2A rather than PP1 in vivo suggested the phosphatase responsible was PP2A. Consistently, expression of SV40 small t antigen, known to displace B subunits from the PP2A core enzyme, inhibited the dephosphorylation of p107 triggered by CDK inhibition. Finally, the catalytic subunit of PP2A was found to immunoprecipitate with p107 and p130 in lysates from serum starved T98G cells at different points upon re-stimulation with FBS, showing that PP2A targets pocket proteins throughout the cell cycle.Citation52 These observations started to shape a “dynamic equilibrium” model described in (), where PP2A holoenzymes modulate pocket protein activity throughout the cell cycle in coordination with inducible CDKs, while PP1 resets pRB specifically in mitosis (reviewed in ref.Citation42,52). However, PP2A consists of a collection of trimeric holoenzymes that come in many “flavors” resulting from combination of its 3 subunits: the “C” catalytic subunit, the “A” scaffolding subunit and a “B” regulatory subunit. Hence, the nature of the PP2A holoenzyme targeting pocket proteins during the cell cycle was unknown because the B subunit(s) implicated had yet to be identified.
Figure 1. A dynamic equilibrium between inducible CDKs and PP2A modulate the phosphorylation state of pocket proteins through the cell cycle. (A) Pocket proteins are active in their hypophosphorylated state. Pocket proteins are hypophosphorylated in early to mid G1 or in G0, when they are found associated with E2F/DP complexes and other proteins. Hypophosphorylated pocket proteins are also thought to bind transcription factors involved in differentiation (not shown). Pocket proteins are inactivated by CDK-dependent hyperphosphorylation. G1 Cyclin D1/CDKs start the process, which is coordinated with G1/S Cyclin E/CDK2 and maintained through S phase and mitosis by Cyclin A/CDK2 and Cyclin B/CDK1. PP2A and PP1 oppose the effects of CDKs. PP2A is active toward the 3 pocket proteins through the cell cycle and in quiescent cells, and this activity may be mediated by the cooperation of various trimeric PP2A holoenzymes (B(s)/PP2A) most prominently PP2A/B55α and PP2A/B55δ and perhaps PP2A/PR70 (see details in the text and 1B). This activity is regulated by a variety of signals and likely down-modulated in mitosis (see text for details). The hyperphosphorylation mediated by inducible CDKs, inactivates pocket proteins by disrupting/preventing their association with E2Fs, which mediates passage through the restriction point by triggering the expression of E2F-dependent (E2Fd) genes needed for DNA synthesis and, later, mitosis. When CDKs are inactivated late in mitosis (ML), B55/PP2A and in the case of pRB, PP1 abruptly dephosphorylate pocket proteins, resetting them to their active state (G1E, designates ealy G1). Arrows show the cell cycle activity span of the kinase or phosphatase with the same color. (B) B55α/PP2A appears to be one of the trimeric holoenzymes in equilibrium with CDKs through the cell cycle. This holoenzyme primarily targets p107 and p130 to a lesser extent. Since an interaction of this holoenzyme with pRB in chondrocytes has been detected, it is possible that it modulates all 3 pocket proteins, although likely with differential affinity. PR70/PP2A interacts with pRB in cells and with pRB and p130 in vitro. It remains to be determined if PR70/PP2A holoenzymes contribute to maintain the equilibrium with CDKs during the cell cycle or only target pRB (and perhaps the other pocket proteins) in response to certain stimuli (i.e., oxidative stress).
PP2A holoenzyme subunit composition and structure
PP2A is an abundant serine/threonine phosphatase that is responsible for the dephosphorylation of a myriad of substrates: The A/C dimer is referred to as the “core enzyme;” when the core dimer is bound to a regulatory subunit it is referred to as a holoenzyme (). PP2A is involved in a vast number of cellular functions and some of its major targets include the pocket protein family, p53, the MAPK and AKT pathways and many mitotic substrates often phosphorylated by CDKs (reviewed in refs.Citation42,53,54).
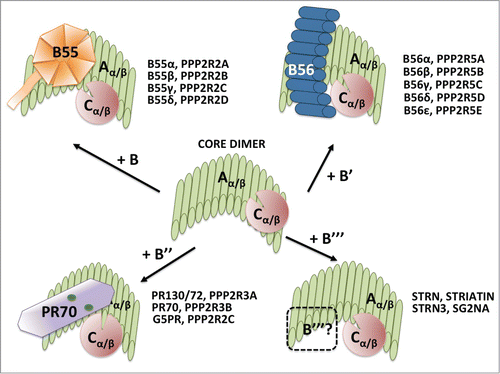
Figure 2. Trimeric PP2A holoenzyme and core dimer composition. The cartoon depicts the 4 types of trimeric holoenzymes and the core dimer. All the B regulatory subunit genes in each family are listed. The crystal structure of the core dimer and the trimeric holoenzymes containing B (B55α), B’ (B56γ) or B" (PR70) have been solved (see text for details). The bending of the scaffold changes with each B subunit, and the bending is maximum with PR70. It is thought that the particular B/C interfaces create B-family specific pockets for substrates. The structure of the B"’/PP2A holoenzymes has not ben solved (represented by a dashed B"’ subunit). B55α is a β-propeller with 7 blades and make less contacts with the catalytic subunit than B56γ. B56γ contains HEAT repeats like the scaffold. The PR70 structure is elongated and contains calcium binding sites, represented by green spheres. See text for additional details.
PP2A/C- the catalytic subunit
There are 2 isoforms of PP2A/C, termed PP2A/Cα and PP2A/Cβ, with the former being more abundant. They are 98% identical, with most of the divergence in the N-terminus.Citation55 The knockout of the α isoform of the catalytic subunit is lethal around embryonic day 6, shortly after implantation.Citation56 The catalytic subunit contacts the scaffold subunit on its C-terminal HEAT repeats (see below). In contrast, the contacts that PP2A/C makes with B subunits are unique for each B-family.
The C-terminal tail of the catalytic subunit is highly conserved, and is required for the binding of B family subunits. Mutation and deletion of the 304-TPDYFL-309 tail prevents the regulatory B subunit (B55) from binding to the core enzyme.Citation57 The T304, Y307, and L309 residues can be modified by phosphorylationCitation58,59 or methylationCitation60 respectively. Phosphorylation of Y307 by tyrosine kinases including v-SRC and EGFRCitation58 and T304 inhibits phosphatase activity. Phosphorylation of these residues is increased in the presence of okadaic acid, suggesting PP2A autodephosphorylates.Citation58,61 Methylation of L309 is modulated by the methyltransferase LCMT-1,Citation60 the methylesterase PME-1Citation62,63 and PTPA.Citation64-67 Methylation of the catalytic subunit is believed to play a role in specific B subunit recruitment to the core enzyme.Citation59,68-70 For example, methylation of L309 was found to be necessary for the binding of the B55α subunit in cells, but not members of the B′, B″ and B″′ family.Citation59,70 Because it appears to be necessary for binding in cells but not in vitro,Citation71 these results may suggest that L309 methylation is not required for B subunit binding but it may enhance affinity, or alternatively, it may serve as a recruitment signal to a specific cellular location or for other cofactors needed for holoenzyme assembly (reviewed in ref.Citation72).
PP2A/A- the scaffold subunit
The “A” subunit of the PP2A holoenzyme, PP2A/A, also called PR65, serves as the scaffold for the regulatory and catalytic subunits. There are 2 isoforms, PP2A/Aα and PP2A/Aβ, which share 87% sequence identity.Citation73 The α isoform is expressed in most tissues, while the β isoform is enriched in the testis.Citation74 The scaffold subunit is structurally composed of a series of antiparallel α-helices known as HEAT (Huntington, elongation factor 3, PR65/A, TOR) repeats,Citation75 which give these proteins a horseshoe-like structure. The catalytic subunit makes contacts with the C-terminal HEAT repeats 11-15,Citation76 whereas the B subunit contacts the N-terminal repeats. Crystal structures of the B, B’ and B" families have revealed that although very structurally divergent, the B and B" subunits contact HEAT repeats 1–7Citation77,78 and the B’ subunits contact HEAT repeats 2–8.Citation76,79 The scaffolding subunit also demonstrates flexibility, as the HEAT repeats twist and shift to varying degrees upon binding of the regulatory and catalytic subunits (reviewed in ref.Citation72).
PP2A/B- the regulatory subunit
The B subunit is the major determinant in substrate specificity and subcellular localization. To date, 15 separate B subunits in 4 major families have been identified. Aside from the 4 known families of subunits, other cellular interactors and viral oncogenes such as SV40 small t and Adenovirus E4ORF4 can also associate with the core enzyme, resulting in over 200 biochemically distinct combinations (reviewed in ref.Citation80)().
The B family, also called PR55, B55, or PPP2R2, has 4 major isoforms, most commonly referred to as B55α, B55β, B55γ, and B55δ. The members of the B family are highly conserved; most of their structural differences lie in the N-terminus. B55α and B55δ are expressed in most tissues and are much more structurally similar to each other than B55β and B55γ, which are reported to be enriched in brain tissue (reviewed in ref.Citation53). B family subunits have a β-propeller structure comprised of 7 blades, each formed by WD-40 repeats. The crystal structure of the PP2A holoenzyme containing the B55α subunitCitation77 demonstrates that B-family subunits make multiple contacts with the first 7 HEAT repeats in the A scaffolding subunit but very little contact with the catalytic subunit, which is bound to the C-terminal HEAT repeats of the scaffold.Citation81 Given the high similarities within the members of this family, it is likely the other isoforms contact the core enzyme in a similar fashion. On the exposed top face of the B55α subunit is an acidic “groove” that is believed to serve as the substrate binding site, and mutation of specific residues in this area prevent the dephosphorylation of Tau, a known substrate of B55α, in vitro.Citation77 Additionally, one of these mutants, B55α-D197K, is also unable to bind p107, another substrate of B55α,Citation82 and is also noticeably deficient in binding the core enzyme. Of note, other sites that are important for B55α to mediate dephosphorylation of Tau have little effect on B55α/p107 binding, suggesting that different combinations of residues within the charged top of B55α determine specific binding to a variety of unrelated substrates.Citation82
The B’, also called the PR61, B56 or PPP2R5 family is encoded by 5 genes, some with multiple isoforms, for a total of 10 known products.Citation83 The crystal structure of B56γ revealed that B56 family members are composed of HEAT repeats, a structure similar to that of the scaffold subunit. B56γ interacts with the N-terminal HEAT repeats of the scaffolding subunit and lie almost perpendicularly across the N-terminus. However, unlike the B55α subunit, B56γ makes substantial contacts with the catalytic subunit.Citation76,79 It is believed that the substrate binding site of B56 family members also lies on the top side of the subunit, as the B56γ subunit also has a highly acidic concave groove.Citation76,79 At least one residue, E153, which is conserved in all B56 family members, when mutated in B56β, remained bound to the core enzyme but failed to dephosphorylate tyrosine hydroxylase, a known substrate.Citation84
The B″, or PR72/PPP2R3 family of subunits, has 3 known members- PR130/PR72, PR70/PR48 and G5PR. This particular family is unique in that they possesses Ca2+-binding EF-hand motifs, as their binding to the core enzyme is calcium dependent.Citation85 The PR72 and PR130 subunits are differentially transcribed from the same gene by 2 different promoters.Citation86 PR48 was identified in a yeast 2-hybrid screen using the licensing factor CDC6 as baitCitation87 and it was later found that PR70 is the full length version of this subunit. Very recently, the crystal structures of the holoenzyme harboring the PR70 subunit and the monomeric PR72 subunit have been solved.Citation78 Like the holoenzymes containing the B and B’ subunits, PR70 also contacts the N-terminal HEAT repeats of the scaffold subunit. The binding of PR70 to the scaffold subunit has a unique effect; it “compacts” the scaffold, yet at the same time increases the height of both the scaffold and the holoenzyme. Aside from the EF motifs, PR70 has a hydrophobic N-terminus and a C-terminus. The C terminus makes contact with the catalytic subunit, and since it was previously shown that residues 440-575 contact CDC6,Citation88 it is proposed that the C-terminus is the substrate binding site,Citation78 which is different than that of members of the B and B’ families, which as mentioned earlier, are believed to use an acidic top surface to contact substrates. Another member of this family, PR59, was identified in murine cells and found to bind p107,Citation49 yet this subunit has not been identified in humans. PR59 is 56% identical to PR72, however, PR72 is not known to bind pocket proteins. PR70, on the other hand, which is known to bind pRB,Citation89 has not been identified in mice. Considering PR59 and PR70 both bind pocket proteins, they may share related roles (reviewed in ref.Citation42).
The B″′ family has 3 known members: striatin, S/G2 nuclear autoantigen (SG2NA) and zinedin. The B″′ family are structurally composed of caveolin and calcium-binding domains in their N-termini, which flank a coiled-coil domain. The C-termini are composed of WD-40 repeats. They are believed to be a unique subunit family as PP2A/A and PP2A/C were the only proteins found associated with them with no other B subunits present.Citation90 Later, other interactors were found associated within the PP2A complex, including kinases among others, suggesting that the phosphatase activity is not the only role of this complex. This complex was named STRIPAK.Citation91 STRIPAK complexes have been implicated in vast number of cellular processes, including cell cycle control, cell signaling and migration among others (reviewed in ref.Citation92).
In sum, crystallography studies have revealed that subunits of the B, B′ and B″ families differ in the way they contact the catalytic subunit. B family subunits make little contact with the C subunit with its interaction being limited to Van der Waals contacts.Citation77 The B′ and B″ family members make significantly more contact. The B56γ subunit interacts with PP2A/Cα at 2 major interfaces- the HEAT repeats 6–8 of B56γ and the α5 region of PP2A/Cα and in a surface groove at the interface between PP2A/Aα and B56γ, mainly with hydrogen bonds.Citation79 The PR70 subunit of the B″ family makes 2 main contacts with PP2A/C near its active site and also in a shallow groove in a different region, and these interactions are mainly hydrogen bonds and salt bridges.Citation78 Altogether, the B/C interactions seem to facilitate the formation of distinct pockets to bring phosphorylated substrates in position for catalysis. Hence, the considerable diversity of B family subunits allows the sharing of similar scaffold/catalytic core dimers to target a large variety of unrelated substrates belonging to multiple cellular pathways. Moreover, the B subunits appear also to be responsible for relaying intra and extracellular signals in a variety of pathways. Perhaps better understood is the mitogen-induced recruitment of B55 subunits to PP2A/C-PP2A/A core dimers preassembled with inactive Raf1 and KSR1, which are sequestered by 14-3-3 proteins in the absence of mitogenic input.Citation93
Cell Cycle and Signal Activated Dephosphorylation of Pocket Proteins by PP2A
A dynamic CDK/PP2A equilibrium during the cell cycle
Having shown that PP2A was in a dynamic equilibrium with CDKs that determines the phosphorylation state of pocket proteins during the cell cycle,Citation52 it was necessary to identify the specific B subunit(s) of this PP2A holoenzyme. Using GST pull down assays, it was shown that the B55α subunit binds p107 and p130 to a lesser extent, but did not bind pRB robustly in U-2 OS cell lysates.Citation82 Of note, the B55α/PP2A holoenzyme was pulled down by GST-p107 from whole cell lysates or from a preparation of purified holoenzyme, indicating that no other proteins are required for complex formation. Consistent with these data, endogenous p107 and B55α were found to co-immunoprecipitate. The spacer region of p107, with the help of the C-terminus, was found to mediate the interaction between B55α and by extension the PP2A holoenzyme. In contrast, the PP1 binding site in pRB resides in the C-terminus and overlaps with CDK binding sites.Citation94 Forced modulation of B55α protein levels affected the phosphorylation status of p107, with limited ectopic expression resulting in hypophosphorylation, and B55α knockdown in hyperphosphorylation.Citation82 These findings were also consistent with the ability of the purified B55α/PP2A holoenzyme to dephosphorylate p107 in vitro. Importantly, SV40 small t antigen, which disrupts the binding of B55α to the PP2A core dimer in cells, does not disrupt the interaction between B55α and p107,Citation82 reinforcing the idea that B55α mediates the interaction between p107 and the holoenzyme. Altogether, these results suggested a more complete PP2A/CDK equilibrium model, as depicted in . In G0 or early G1, CDK activity is low and pocket proteins are hypophosphorylated. Upon mitogenic stimulation, activated G1 and G1/S CDKs hyperphosphorylate pocket proteins leading to their inactivation, which allows the cell cycle to proceed. PP2A has the potential to dephosphorylate pocket proteins in all phases of the cell cycle.Citation52 PR70 holoenzymes target Prb,Citation82,89 and B55α holoenzymes target p107 and p130.Citation82 In mitosis, PP1 is also believed to be involved in resetting pRB to its active state.Citation43,44 In this model, CDKs are depicted as the inducible enzymes that determine the phosphorylation state of pocket proteins in equilibrium with PP2A holoenzymes. However, it remained unclear if PP2A could be independently upregulated in this equilibrium by cellular signals to cause rapid cell cycle exit in the absence of CDK inactivation and if this is mediated by regulation of specific B subunits.
PP2A targeting pocket proteins in response to signals and stresses
A handful of studies have described rapid PP2A mediated dephosphorylation of pocket proteins in response to extracellular signals and stresses, suggesting that PP2A activity may be regulated to target pocket proteins even if CDK activity is not concomitantly affected by these signals (). p107 was reported to be rapidly dephosphorylated in response to UV irradiation of NIH-3T3 fibroblasts, which transiently arrested in the G1 phase of the cell cycle 90 minutes post-exposure.Citation51 EMSA assays followed by western blotting showed there was an accumulation of complexed E2F after treatment, and p107 was the predominant pocket protein present. p107 dephosphorylation was blocked in cells pre-treated with concentrations of okadaic acid specific to PP2A, implicating it as the phosphatase responsible. The exact B subunit responsible for the UV induced dephosphorylation of p107 was not identified, but overexpression of PR72, a subunit known to not bind p107, blocked this effect, presumably acting as a dominant negative via sequestration of the PP2A core dimer.Citation51 At the time, a novel subunit designated PR59 was found to target p107,Citation49 but was never directly implicated in this process.
Table 1. PP2 A holoenzymes targeting pocket proteins during the cell cycle and in response to signals
Oxidative stress is another mechanism that triggers PP2A dependent dephosphorylation of pocket proteins.Citation48 Treatment of HUVEC (human umbilical vein endothelial cells) with H2O2 results in dephosphorylation of all 3 pocket proteins within 30 minutes. This effect does not result in the modulation of Cyclin and CDK activity. The H2O2-dependent dephosphorylation of pocket proteins was prevented by okadaic acid at concentrations selective for inhibition of PP2A, and also by the expression of the SV40 small t antigen, which displaces B subunits from the PP2A core dimer.Citation48 Subsequently, PR70 was found to associate with pRB in U2-OS and HUVEC cells.Citation89 Overexpression of PR70 was sufficient to increase the levels of hypophosphorylated pRB in the absence of H2O2. PR70 shRNA prevented H2O2-dependent pRB dephosphorylation and DNA synthesis inhibition in HUVEC cells. Additionally, PR70 EF-hand motifs were found to be important for binding the PP2A core dimer and treatment with an intracellular calcium chelator prevented the dephosphorylation of pRB induced by H2O2, indicating that the dephosphorylation of pRB by PP2A/PR70 holoenzymes is calcium dependent.Citation89 Although it was previously shown that all 3 pocket proteins were sensitive to H2O2 induced dephosphorylation,Citation48 the particular subunits targeting p107 and p130 were not investigated in this study.
PP2A has also been shown to interact with p130 in ovarian carcinoma cells treated with all-trans-retinoic acid (ATRA). ATRA induces upregulation of PP2A activity along with the stabilization of p130 and G1 arrest.Citation50 p130 bound the catalytic subunit of PP2A, and this interaction was dependent on the intact nuclear localization signals on the C-terminus of p130.Citation95 Two specific serine and threonine residues (S1080 and T1097), which lie adjacent to the nuclear localization signals, were specifically found to be targeted by PP2A. Interestingly, importin α binds p130 at this region when these 2 residues are dephosphorylated, resulting in the translocation of p130 to the nucleus, which is followed by cell cycle arrest.Citation96 No B subunit was implicated in this process, yet since it is known that p130 can be targeted by both B55α and PR70, it is conceivable that one of these B subunits is implicated in the process.Citation82
Although not directly investigated, another potential crosstalk between a pocket protein and PP2A is likely to occur in epithelial cells treated with TGF-β, where hypophosphorylated p107 associates with Smads, which are transcriptional regulators induced by TGF-β. Smad3 complexes with E2F4/5, DP1, and p107 in the cytoplasm, and upon TGF-β stimulation, this complex interacts with Smad4, translocates to the nucleus and binds to an adjacent Smad/E2F consensus site at the c-MYC promoter, repressing its transcription and resulting in cell cycle exit.Citation97 PP2A was not implicated in this process, however the B55α holoenzyme is known to be phosphorylated by the TGF-β type 1 receptor,Citation98 so it is possible it could target the holoenzyme to keep p107 hypophosphorylated. Indeed, Smads themselves are targeted by PP2A upon TGF-β/BMP stimulation. The PP2A/B55β holoenzyme was found to dephosphorylate BMPRII, ultimately leading to the same holoenzyme dephosphorylating Smads in their linker region, resulting in Smad complex nuclear translocation.Citation99
PP2A and pocket proteins in development
Perhaps best understood at this point are the PP2A-dependent signaling events leading to rapid dephosphorylation and activation of p107 in response to FGF1 stimulation in chondrocytes, which is associated with chondrocyte maturation and G1 arrest. These events are thought to be critical for endochondral bone formation and are mediated by FGF through the FGFR3 receptor.
The rapid activation of p107 by PP2A in chondrocytes following FGF stimulation is particularly intriguing for 2 reasons: (1) p130 and pRB are not dephosphorylated until several hours later,Citation100 and (2) chondrocytes require p107 and p130 for FGF-induced cell cycle exitCitation101 and for endochondral bone formation in vivo.Citation102 Therefore, it is of great interest to understand the mechanisms of PP2A activation toward p107 and its consequences. Kolupaeva et al. reported that the PP2A/p107 interaction that mediated p107 dephosphorylation and growth arrest were sensitive to the expression of SV40 st antigen and Adenovirus E4ORF4 proteins in Rat Chondrosarcoma (RCS) cells, which suggested that the holoenzyme implicated was defined by a B subunit of the B or B′ family.Citation100 On the other hand, B55α had been identified as the major B subunit targeting p107,Citation82 which met these characteristics. Thus, studies followed to identify the composition of the PP2A holoenzyme regulated by FGF in chondrocytes. However, before these studies are discussed it is important to review the crucial role of p107 and p130 in endochondral bone formation, which was first revealed via targeted inactivation of p107 and p130 in miceCitation102 and its link to FGF signaling.Citation103
Mice that are p107−/− or p130−/− do not have early morbidity issues and develop normally.Citation102,104 When p107 −/+ and p130 −/+ mice were crossed, no live p107/p130 double knockouts were found at birth, and the dead neonates were detected below the Mendelian ratio, however, double knockout embryos were found alive at 18 days in Mendelian frequencies.Citation102 These embryos were ∼30% smaller than their littermates and had drastically shortened limbs. When stained with dyes to specifically detect cartilage or bone, the double knockout mice were found to have smaller rib cages and displayed less bone in the long bones of their limbs than control littermates. Consistently, chondrocyte density was doubled in the epiphyseal centers of the p107−/−;p130−/− mice as a result of increased chondrocyte proliferation, suggesting p107 and p130 specifically play a role in restricting chondrocyte proliferation in epiphyseal centers.Citation102 Significantly, the phenotype of the p107/p130 double knockout mice resembles that of achondroplasia, which is the most common form of dwarfism, as its most striking feature is the failure of long bone formation in the limbs. Achondroplasia in humans results from mutation of FGFR3 that render it constitutively active in the absence of FGF ligand (reviewed in ref.Citation105). Endochondral bone formation is dependent on the proliferation, maturation, growth arrest, and eventual apoptosis of chondrocytes. This process begins with cartilage formation in the perichondrium, which is eventually replaced by bone. Mesenchymal cells condense and differentiate into 2 subspecies of proliferating chondrocytes, called low- and high- proliferating chondrocytes, which expand in different locations in the perichondrial space. Low proliferating chondrocytes are located toward the distal ends, whereas high-proliferating chondrocytes are organized in columns in the center. Proliferating chondrocytes exit the cell cycle and differentiate (or mature) into pre-hypertrophic and hypertrophic chondrocytes. The expansion of this population of cells is critical in skeletal elongation as it increases the length of what will eventually become the long bones of the limbs. These cells finally undergo apoptosis and are eventually replaced by trabecular bone (reviewed in ref.Citation106).
Rat Chrondrosarcoma cells (RCS cells), a chondrocyte-like cell line that expresses FGFR3,Citation107 have extensively been used as a cellular model system to study the signaling events initiated by FGF that lead to chondrocyte maturation and cell cycle arrest. Treatment of RCS cells with FGF1 results in the autophosphorylation of FGFR3 and activated downstream targets such Phospholipase Cγ, AKT, ERK and p38 MAP kinases, but also resulted in unexpected growth inhibition, which was also observed in primary mouse chondrocyte cultures. Pharmacological inhibition of MEK and p38 MAPK inhibited growth arrest, possibly implicating these pathways in this process.Citation108,109 Inactivation of the AKT pathway may also play a role in FGF mediated RCS arrest, as FGF2 treatment leads to a gradual reduction in AKT activity, and expression of a constitutively active AKT mutant (myr-AKT) partially inhibits growth arrest.Citation110 Cells expressing myr-AKT were deficient in accumulating p130 upon arrest and retained some Cyclin E/CDK2 activity. However, expression of myr-AKT does not affect the expression of maturation genes induced by FGF treatment.Citation110
The role of pocket proteins was also studied using RCS cells, where FGF1 treatment lead to dephosphorylation of all 3 pocket proteins and growth arrest that could be blocked by the expression of Adenovirus E1A or Large T antigen.Citation100 Importantly, RCS cells expressing a Large T mutant that is capable of binding pRB but not p107 or p130 were able to undergo growth arrest similarly to the parental cells, in complete agreement with the findings in mice, where compound inactivation of p107/p130 but not pRB result in defects in endochondral bone formation.Citation102 Furthermore, micromass chondrocyte cultures that were null for p107 and/or p130 were insensitive to FGF induced cell cycle arrest, while pRB null cultures behaved as the wild type, confirming the importance of p107 and/or p130 and the dispensability of pRB in this process. Of note, the p107 −/+; p130−/− micromass cultures did remain slightly responsive to FGF, suggesting p107 may play a more prominent role.Citation103
On the other hand, endogenous CDK inhibitors have also been implicated in the cell cycle arrest induced by FGF in chondrocytes in both RCS cells and in endochondral bone formation in mice. In both systems, increased expression of CKIs is linked to pocket proteins. In this regard, mice with p107 ablation and knockin of an allele of p27 deficient in CDK binding (p27D51/D5151/D51) exhibit similar defects in endochondral bone formation as those seen in p107/p130 double knockout mice, although the phenotype is not as severe.Citation111 These mice exhibit elevated chondrocyte proliferation and shorter long bones. Thus, if p130 and p27 inactivation are interchangeable in a p107 null background, they may be involved in the same pathway in chondrocyte maturation.Citation111 Moreover, null mice for the CDK inhibitor p57 also exhibit defects in endochondral bone formation, suggesting that this CKI might be an upstream regulator of p107 and p130/p27 in chondrocytes.Citation112
In RCS cells, p21 is upregulated in response to FGF1 stimulation and peaks coinciding with dephosphorylation of pRB (that occurs concomitantly to that of p130), several hours after p107 dephosphorylation. Thus, p21 upregulation, which results in Cyclin E/CDK2 inactivation,Citation113 may activate p130, while the activation of p107 is caused earlier by PP2A.Citation100 Consistently, overexpression of Cyclin D/CDK4 complexes block the dephosphorylation of p107 by FGF1 as well as the G1 cell cycle arrest that results from FGF treatment.Citation100
FGF1 stimulates rapid p107 dephosphorylation by the PP2A/B55α holoenzyme in RCS cells.
FGF1 stimulates a transient p107-PP2A/B55α holoenzyme interaction in chondrocytes
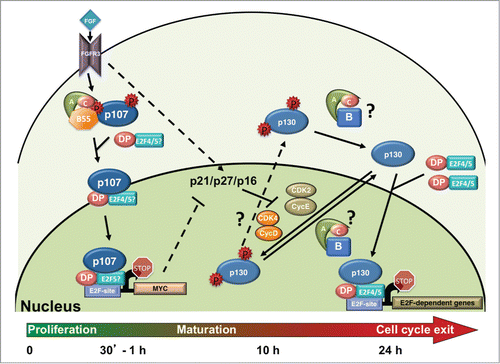
Therefore, a critical step in undestanding activation of p107 was the identification of the B regulatory subunit responsive to FGF1 stimulation. Consistently, it was found that endogenous p107 interacts with PP2A/B55α holoenzymes in RCS cells and that this interaction is transiently enhanced via treatment with FGF1 without an increase in B55α protein expression. Moreover, reciprocal proteomic analyses in these cells showed that B55α, and to a lesser extent B55δ, is found in p107 complexes in addition to PP2A/C and PP2A/A, but no other B regulatory subunits were detected.Citation39 Others used a catalog approach to independently identify B55α as the B subunit targeting p107 upon FGF1 stimulation in RCS cells.Citation114 Consistent with these findings, knockdown of B55α delayed p107 dephosphorylation and cell cycle exit in response to FGF1 stimulation, but did not completely block cell cycle exit.Citation39,114 Furthermore, following dephosphorylation, p107 translocates from the cytoplasm to the nucleus, undergoes changes in complex formation, including complexes with E2F4 and Cyclin/CDK holoenzymes, and enhanced recruitment to the MYC promoter within 1.5 hours of FGF1 treatment.Citation39 The other pocket proteins, p130 and pRB, are not activated until several hours later, indicating that they do not play an initiating role in chondrocyte maturation or cell cycle exit. Since pRB and p130 are not dephosphorylated until much later after FGF1 treatment, it is more likely that FGF1 differentially mediates activation of pocket proteins in RCS cells via 2 separate pathways, as illustrated in and summarized next. Upon treatment with FGF1, p107 is very rapidly activated by PP2A/B55α holoenzymes. This is followed by p107 translocating to the nucleus and binding to the c-MYC promoter and most likely to the promoters of other unidentified genes required for chondrocyte maturation and the initiation of cell cycle exit. Meanwhile, pRB and p130 remain inactive due to phosphorylation by CDK4 and CDK2 complexes. The dephosphorylation of pRB and p130 coincides with an increase in p21 expressionCitation113 several hours later, which cooperate to inactivate Cyclin E/CDK2 complexes. Upregulation of p16 and p27 is also observed after FGF1 treatment,Citation101,108 likely cooperating in the coordinated inactivation of both CDK4 and CDK2 complexes. Differential dephosphorylation of p107 prior to p130 and pRB without the inactivation of CDKs suggests that PP2A/B55α holoenzymes target p107 preferentially over the other pocket proteins. This could be the result of 1) higher affinity for p107, or 2) a consequence of subcellular and/or temporal expression constraints. Differential affinity of PP2A/B55α for p107 over p130 and pRB was already suggested previously.Citation82 It is likely that PP2A/B55α participates in the dephosphorylation of p130 and pRB when CDK4 and CDK2 are inactivated by CKIs (5 hours post stimulation). It is also plausible that cooperation by other phosphatases such as PP2A/PR70 and PP1 also takes place at this point. In any case, the pattern of p130 and pRB dephosphorylation and recruitment of p130 to the promoters of E2F dependent genes is consistent with the repression of E2F genes generally observed during cell cycle exit.
Figure 3. FGF induces rapid activation of B55α/PP2A holoenzymes, p107 dephosphorylation and activation, and cell cycle exit and maturation in chondrocytes. FGF stimulation through the FGFR3 receptor in chondrocytes leads to increased but transient formation of a B55α-δ/PP2A holoenzyme complexes with p107. This results in a shift on p107 localization from predominantly cytoplasmic to nuclear, formation of complexes with E2F4 and likely other E2Fs, and rapid recruitment to the c-MYC promoter, coinciding with its downregulation. p130 remains hyperphosphorylated as CDK4 and CDK2 remain active and may not be actively targeted by B55α/PP2A holoenzymes to switch the equilibrium toward dephosphorylation until CDK activity decreases. p21, p27 and p16 CKI activities increase by different means and appear to trigger inactivation of CDK4 and CDK2 coinciding with p130 and pRB dephosphorylation that occurs 10-15 hours post FGF stimulation. By 24 hrs. chondrocytes have exited the cell cycle and p130 and E2F4 are found at the promoters of cell cycle genes. Because hyperphosphorylated p107 is only detected in the cytoplasmic cellular fractions and this form is rapidly downregulated in the cytoplasm concomitantly with appearance of hypophosphorylated p107 in the nucleus upon FGF stimulation, it seems likely that this dephosphorylation occurs in the cytoplasm. Whether the same is true for p130 is less clear. p130 levels are very low in the absence of FGF stimulation and do not accumulate in the nucleus until several hours post-FGF treatment. While p130 phosphorylation by CDKs is likely occurring in the nucleus, the dephosphorylation step could conceivably occur either in the nucleus or the cytoplasm, hence the question marks for the PP2A reactions and the shuttling of phosphorylated p130 into the cytoplasm.Citation39 See text for additional details.
Insights into the potential mechanism of p107/PP2A/B55α complex formation
While it is established that the PP2A/B55α holoenzyme is responsible for the dephosphorylation of p107 in RCS cells in response to FGF signaling, the mechanism by which the signal leads to the recruitment of PP2A to p107 is still unclear. It has been suggested that one aspect of this mechanism is the dephosphorylation of the B55α subunit upon treatment with FGF1. Evidence of serine phosphorylation regulating B55α holoenzyme assembly as a possible mechanism to inactivate the holoenzyme in mitosis has been reported in the past.Citation115 Kolupaeva et al. have shown through immunoprecipitation of B55α in separate assays that dephosphorylation of B55α, probably on one or more serine residues, occurs around one hour and more clearly at 2 hours after FGF1 treatment, which led to their suggestion that B55α dephosphorylation may activate PP2A to target p107. Using B55α phosphosmimetic mutants, these authors also showed that serine phosphorylation inhibits the binding of the PP2A scaffold subunit and in some cases p107 as well.Citation114 This is not fully consistent with the observation that p107 is dephosphorylated and forms complexes with E2F4 as early as 15-30 minutes post treatment.Citation39 While dephosphorylation may contribute to the stability of the complex, it is more likely the activation event that recruits PP2A/B55α to p107 occurs within minutes rather than hours after FGF1 signaling.
It is known that in RCS cells, treatment with FGF1 activates MAPK pathways. Indeed, ERK 1/2 and p38 are activated very rapidly- within 5 minutes post treatment with FGF1. Treatment with a MEK1/2 inhibitor, which is directly upstream of ERK in this pathway, inhibits the dephosphorylation of p107 one hour after treatment with FGF,Citation109 similar to the knockdown of B55α. This indicates ERK1/2 signaling somehow plays a role in the dephosphorylation of p107 and perhaps the immediate recruitment of the B55α holoenzyme to p107. Also of note, B55α and B55δ holoenzymes dephosphorylate the inhibitory phosphorylation pS259 on Raf1 and pS392 on KSR1,Citation93 which are immediately upstream of MEK1/2 in the activation of the MAPK cascade,Citation116 and these effects are observed within 5-15 minutes of treatment with EGF or PDGF in NIH-3T3 cells. Thus, activation of the MAPK pathway and p107 all involve B55α and seem to occur in a very narrow time scale, which suggest coordination.
Composition of p107 complexes in RCS cells
The composition of p107 complexes has been characterized via mass spectrometric analysis using RCS cell lines stably expressing Flag-tagged p107 at levels that do not preclude their proliferation, nor affect FGF1 induced growth arrest.Citation39 In addition to identification of the components of the B55α and B55δ holoenzymes, the presence of several expected binding partners of p107, broadly classified as E2F/DP proteins, DREAM subunits and Cyclin/CDKs complexes, as well as some potential novel interactors was confirmed. Of note, several E2Fs were detected, including not only the repressor E2Fs and components of the DREAM complex, E2F4 and E2F5, but also the activator E2Fs, E2F1 and E2F3. This suggest that p107 not only forms repressor E2F complexes and or DREAM complexes that are recruited to the nucleus, but also targets activator E2Fs, perhaps to rapidly turn them off in response to FGF signaling. Detection of these p107/activator E2F complexes is consistent with previous identification of endogenous p107/E2F1 and p130/E2F1 complexes in human cells.Citation37 One expected but interesting group of proteins detected were members of the DREAM complex, an evolutionarily conserved protein complex that serves as both a transcriptional regulator and repressor at different phases of the cell cycle (reviewed in ref.Citation117). The mammalian DREAM complex is composed of DP1 or DP2, p107 or p130 (but not pRB), E2F4 or E2F5 and the MuvB core, which contains Lin 9, Lin 37, Lin 52, Lin 54, and RBBP4Citation118,119 (except DP2, all these proteins are detected in the p107/DREAM complex in chondrocytes). The MuvB core can also form complexes with B-Myb and FoxM1, referred to as B-Myb-MuvB.Citation120 The major role of the p130-containing DREAM complex is to repress genes for cell cycle exit and quiescence, while the B-Myb-MuvB complex is recruited in S phase to the promoters of G2/M genes to serve as a transcriptional activator. The “switch” from B-Myb binding to DREAM binding of the MuvB core is the result of phosphorylation of Lin 52 by the DYRK1 kinase (reviewed in refs.Citation117,121). Consistent with these findings, a very recent publication has implicated the DREAM complex in the regulation of chondrocyte proliferation.Citation122 In this study, mice that homozygously express p107 with mutations in the LXCXE binding cleft that render it incapable of binding the MuvB core were crossed with p130 null mice. In MEFs derived from these mice, the DREAM complex failed to assemble, DREAM target genes failed to be repressed, resulting in their increased expression during cell proliferation. Importantly, these mice exhibited neonatal lethality, but were found to be alive at embryonic day 18.5, shortly before birth, which is highly reminiscent of p107/p130 knockout mice.Citation102 When the bones of these mice were analyzed, they were found to have abnormally smaller and underdeveloped endocranial bones that the wild type mice, as well as significantly shorter long bones. This overall demonstrates that similar to p107/p130 null mice, mice deficient in DREAM complex assembly display defective chondrocyte proliferative control and endochondral ossification.Citation122 Further analysis of the dynamics of these complexes in response to FGF signaling is needed to determine if the MuvB core is found with p107 at the c-MYC promoter or other genes whose downregulation is required for maturation and cell cycle exit shortly after treatment with FGF1 in RCS cells. Alternatively, p107 may also promote expression of maturation genes via co-recruitment of other factors yet to be determined upon p107 activation.
Closing Remarks: The CDK/PP2A Equilibrium that Determines the Phosphorylation State of Pocket Proteins Integrates Growth Arrest Signals Through both Inhibition of CDKs and Activation of PP2A
Pocket proteins are targeted by PP2A in 2 manners- in an equilibrium with CDKs throughout the cell cycle, which implies a basal PP2A activity, and inducibly upon extracellular signals or stresses. Throughout the cell cycle, phosphorylation and dephosphorylation of pocket proteins is modulated by Cyclin/CDK complexes and PP2A. When cells are treated with inhibitors that affect the synthesis of unstable Cyclins or CDK activity, all 3 pocket proteins are immediately dephosphorylated.Citation52 In this regard, signals that result in cell cycle arrest or exit that function through the accumulation of CKIs are time-limited by the rate of accumulation of CKIs that are needed to inhibit CDKs. In this scenario, reduction in CDK activity will need a substantial accumulation of CKIs to shift the equilibrium toward pocket protein dephosphorylation by PP2A. This requires protein synthesis and/or stabilization, which are not extremely fast processes. In contrast, recruitment of PP2A holoenzymes upon extracellular signaling or stresses serves as an alternative mechanism of rapidly targeting pocket proteins. For example, FGF1 signaling promotes the rapid dephosphorylation of p107 as a result of complex formation between p107 and PP2A/B55α holoenzymes, and this happens in the presence of CDK activity that maintains the phosphorylation of pRB and p130. Shifts in the PP2A/CDK balance due to the selective recruitment of PP2A to pocket proteins in response to a variety of signaling cues could serve as a general mechanism for rapid activation of pocket proteins. As described earlier, at least 2 other instances of pocket proteins being rapidly activated as the result of stresses are known. UV irradiation results in the dephosphorylation of p107 within 90 minutes, which also results in a G1 cell cycle arrest.Citation51 Oxidative stress also results in the rapid dephosphorylation of all 3 pocket proteins within 30 minutes, and Cyclin/CDK activity is unaffected.Citation48 pRB is targeted by PR70 in this context, and it is likely that also targets p107 and p130 given that the kinetics of dephosphorylation matches that of pRB,Citation89 and that it has been shown that PR70 also associates with p130.Citation82 Moreover, PP2A has also been shown to mediate the localization of pRB on chromatin is response to DNA damage, although the B subunit implicated is unknown.Citation123
It is also important to note that the PP2A activity targeting pocket proteins through the cell cycle is also likely regulated, at least the PP2A activity share mediated by PP2A/B55α and B55δ holoenzymes. The activity of these 2 holoenzymes is required for exit from mitosis, but it is inhibited during mitosis itself (reviewed in refs.Citation54,124,125). This is a result of activation of the Gwl kinase by CDK1, which in turn activates ENSA and ARPP19, which directly inhibit PP2A. Thus, it is very likely that the upregulation of PP2A/B55α and B55δ holoenzymes as cells exit mitosis contributes to the rapid dephosphorylation of the 3 pocket proteins prior to entry into G1. In the case of pRB, PP1 also contributes at this cell cycle stage.
Further insights in the mechanism of activation of PP2A through the B-regulatory subunits that target pocket proteins will help uncover new signals with the ability to rapidly halt the cell cycle. Abrupt dephosphorylation of pocket proteins in response to FGF signaling or oxidative stress are likely paradigms for rapid activation of pocket proteins by other unrelated signals.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Work in the XG lab was supported by a W.W. Smith Charitable Trust to XG.
References
- Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004; 18:2699-711; PMID:15545627; http://dx.doi.org/10.1101/gad.1256504
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501-12; PMID:10385618; http://dx.doi.org/10.1101/gad.13.12.1501
- Pei XH, Xiong Y. Biochemical and cellular mechanisms of mammalian CDK inhibitors: a few unresolved issues. Oncogene 2005; 24:2787-95; PMID:15838515; http://dx.doi.org/10.1038/sj.onc.1208611
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013; 140:3079-93; PMID:23861057; http://dx.doi.org/10.1242/dev.091744
- Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 2013; 14:297-306; PMID:23594950; http://dx.doi.org/10.1038/nrm3567
- MacDonald J, Dick FA. Posttranslational modifications of the retinoblastoma tumor suppressor protein as determinants of function. Genes & Cancer 2013; In Press.
- Viatour P. Bridges between cell cycle regulation and self-renewal maintenance. Genes Cancer 2012; 3:670-7; PMID:23634255
- Knudson AG, Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 1971; 68:820-3; PMID:5279523
- Classon M, Dyson N. p107 and p130: versatile proteins with interesting pockets. Exp Cell Res 2001; 264:135-47; PMID:11237530; http://dx.doi.org/10.1006/excr.2000.5135
- Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet 1998; 14:223-9; PMID:9635405; http://dx.doi.org/10.1016/S0168-9525(98)01470-X
- Wirt SE, Sage J. p107 in the public eye: an Rb understudy and more. Cell Div 2010; 5:9; PMID:20359370; http://dx.doi.org/10.1186/1747-1028-5-9
- Graña X, Garriga J, Mayol X. Role of the retinoblastoma protein family, pRB, p107 and p130 in the negative control of cell growth. Oncogene 1998; 17:3365-83; PMID:9916999; http://dx.doi.org/10.1038/sj.onc.1202575
- Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell 1989; 56:67-75; PMID:2521301; http://dx.doi.org/10.1016/0092-8674(89)90984-7
- Harlow E, Whyte P, Franza BR, Jr., Schley C. Association of adenovirus early-region 1A proteins with cellular polypeptides. Molecular & Cellular Biology 1986; 6:1579-89; PMID:2431282
- Yee SP, Branton PE. Detection of cellular proteins associated with human adenovirus type 5 early region 1A polypeptides. Virology 1985; 147:142-53; PMID:2932846; http://dx.doi.org/10.1016/0042-6822(85)90234-X
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989; 243:934-7; PMID:2537532; http://dx.doi.org/10.1126/science.2537532
- Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO Journal 1989; 8:4099-105; PMID:2556261
- Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998; 391:859-65; PMID:9495340; http://dx.doi.org/10.1038/36038
- Morris EJ, Dyson NJ. Retinoblastoma protein partners. Adv Cancer Res 2001; 82:1-54; PMID:11447760; http://dx.doi.org/10.1016/S0065-230X(01)82001-7
- Woo MS, Sanchez I, Dynlacht BD. p130 and p107 use a conserved domain to inhibit cellular cyclin- dependent kinase activity. Mol Cell Biol 1997; 17:3566-79; PMID:9199292
- Castano E, Kleyner Y, Dynlacht BD. Dual cyclin-binding domains are required for p107 to function as a kinase inhibitor. Mol Cell Biol 1998; 18:5380-91; PMID:9710622
- Pan W, Cox S, Hoess RH, Grafstrom RH. A cyclin D1/cyclin-dependent kinase 4 binding site within the C domain of the retinoblastoma protein. Cancer Res 2001; 61:2885-91; PMID:11306463
- Adams PD, Li X, Sellers WR, Baker KB, Leng X, Harper JW, Taya Y, Kaelin WG Jr Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol Cell Biol 1999; 19:1068-80; PMID:9891042
- Mayol X, Garriga J, Graña X. Cell cycle-dependent phosphorylation of the retinoblastoma-related protein p130. Oncogene 1995; 11:801-8; PMID:7651744
- Mayol X, Garriga J, Graña X. G1 cyclin/CDK-independent phosphorylation and accumulation of p130 during the transition from G1 to G0 lead to its association with E2F-4. Oncogene 1996; 13:237-46; PMID:8710362
- Smith EJ, Leone G, Degregori J, Jakoi L, Nevins JR. The accumulation of an E2F-p130 transcriptional repressor distinguishes a G(0) cell state from a G(1) cell state. Molecular & Cellular Biology 1996; 16:6965-76; PMID:8943352
- Bhattacharya S, Garriga J, Calbo J, Yong T, Haines DS, Graña X. SKP2 associates with p130 and accelerates p130 ubiquitylation and degradation in human cells. Oncogene 2003; 22:2443-51; PMID:12717421; http://dx.doi.org/10.1038/sj.onc.1206339
- Tedesco D, Lukas J, Reed SI. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev 2002; 16:2946-57; PMID:12435635; http://dx.doi.org/10.1101/gad.1011202
- Zhu L, Xie E, Chang LS. Differential roles of two tandem E2F sites in repression of the human p107 promoter by retinoblastoma and p107 proteins. Molecular & Cellular Biology 1995; 15:3552-62; PMID:7791762
- Beijersbergen RL, Carlee L, Kerkhoven RM, Bernards R. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes & Development 1995; 9:1340-53; PMID:7797074; http://dx.doi.org/10.1101/gad.9.11.1340
- Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div 2012; 7:10; PMID:22417103; http://dx.doi.org/10.1186/1747-1028-7-10
- Zhang HS, Postigo AA, Dean DC. Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFbeta, and contact inhibition. Cell 1999; 97:53-61; PMID:10199402; http://dx.doi.org/10.1016/S0092-8674(00)80714-X
- Hurford RK, Jr., Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev 1997; 11:1447-63; PMID:9192872; http://dx.doi.org/10.1101/gad.11.11.1447
- Balciunaite E, Spektor A, Lents NH, Cam H, Te Riele H, Scime A, Rudnicki MA, Young R, Dynlacht BD. Pocket protein complexes are recruited to distinct targets in quiescent and proliferating cells. Mol Cell Biol 2005; 25:8166-78; PMID:16135806; http://dx.doi.org/10.1128/MCB.25.18.8166-8178.2005
- Rowland BD, Bernards R. Re-evaluating cell-cycle regulation by E2Fs. Cell 2006; 127:871-4; PMID:17129771; http://dx.doi.org/10.1016/j.cell.2006.11.019
- Bertoli C, Skotheim JM, de Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 2013; 14:518-28; PMID:23877564; http://dx.doi.org/10.1038/nrm3629
- Calbo J, Parreno M, Sotillo E, Yong T, Mazo A, Garriga J, Graña X. G1 cyclin/cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J Biol Chem 2002; 277:50263-74; PMID:12401786; http://dx.doi.org/10.1074/jbc.M209181200
- Lee EY, Cam H, Ziebold U, Rayman JB, Lees JA, Dynlacht BD. E2F4 loss suppresses tumorigenesis in Rb mutant mice. Cancer Cell 2002; 2:463-72; PMID:12498715; http://dx.doi.org/10.1016/S1535-6108(02)00207-6
- Kurimchak A, Haines DS, Garriga J, Wu S, De Luca F, Sweredoski MJ, Deshaies RJ, Hess S, Graña X. Activation of p107 by fibroblast growth factor, which is essential for chondrocyte cell cycle exit, is mediated by the protein phosphatase 2A/B55alpha holoenzyme. Mol Cell Biol 2013; 33:3330-42; PMID:23775125; http://dx.doi.org/10.1128/MCB.00082-13
- Gaubatz S, Lees JA, Lindeman GJ, Livingston DM. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol Cell Biol 2001; 21:1384-92; PMID:11158323; http://dx.doi.org/10.1128/MCB.21.4.1384-1392.2001
- Gaubatz S, Lindeman GJ, Ishida S, Jakoi L, Nevins JR, Livingston DM, Rempel RE. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell 2000; 6:729-35; PMID:11030352; http://dx.doi.org/10.1016/S1097-2765(00)00071-X
- Kurimchak A, Graña X. PP2A holoenzymes negatively and positively regulate cell cycle progression by dephosphorylating pocket proteins and multiple CDK substrates. Gene 2012; 499:1-7; PMID:22387205; http://dx.doi.org/10.1016/j.gene.2012.02.015
- Ludlow JW, Glendening CL, Livingston DM, DeCarprio JA. Specific enzymatic dephosphorylation of the retinoblastoma protein. Molecular & Cellular Biology 1993; 13:367-72; PMID:8380224
- Durfee T, Becherer K, Chen PL, Yeh SH, Yang Y, Kilburn AE, Lee WH, Elledge SJ. The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes & Development 1993; 7:555-69; PMID:8384581; http://dx.doi.org/10.1101/gad.7.4.555
- Nelson DA, Ludlow JW. Characterization of the mitotic phase pRb-directed protein phosphatase activity. Oncogene 1997; 14:2407-15; PMID:9188855; http://dx.doi.org/10.1038/sj.onc.1201081
- Nelson DA, Krucher NA, Ludlow JW. High molecular weight protein phosphatase type 1 dephosphorylates the retinoblastoma protein. J Biol Chem 1997; 272:4528-35; PMID:9020179; http://dx.doi.org/10.1074/jbc.272.7.4528
- Dunaief JL, King A, Esumi N, Eagen M, Dentchev T, Sung CH, Chen S, Zack DJ. Protein Phosphatase 1 binds strongly to the retinoblastoma protein but not to p107 or p130 in vitro and in vivo. Curr Eye Res 2002; 24:392-6; PMID:12434308; http://dx.doi.org/10.1076/ceyr.24.5.392.8524
- Cicchillitti L, Fasanaro P, Biglioli P, Capogrossi MC, Martelli F. Oxidative stress induces protein phosphatase 2A-dependent dephosphorylation of the pocket proteins pRb, p107, and p130. J Biol Chem 2003; 278:19509-17; PMID:12621062; http://dx.doi.org/10.1074/jbc.M300511200
- Voorhoeve PM, Hijmans EM, Bernards R. Functional interaction between a novel protein phosphatase 2A regulatory subunit, PR59, and the retinoblastoma-related p107 protein. Oncogene 1999; 18:515-24; PMID:9927208; http://dx.doi.org/10.1038/sj.onc.1202316
- Vuocolo SC, Purev E, Zhang D, Bartek J, Hansen K, Soprano DR, Soprano KJ. Protein phosphatase 2A associates with Rb2/p130 and mediates retinoic acid induced growth suppression of ovariancarcinoma cells. J Biol Chem 2003; 278(43):41881-9;PMID:12915404
- Voorhoeve PM, Watson RJ, Farlie PG, Bernards R, Lam EW. Rapid dephosphorylation of p107 following UV irradiation. Oncogene 1999; 18:679-88; PMID:9989818; http://dx.doi.org/10.1038/sj.onc.1202289
- Garriga J, Jayaraman AL, Limon A, Jayadeva G, Sotillo E, Truongcao M, Patsialou A, Wadzinski BE, Graña X. A Dynamic Equilibrium Between CDKs and PP2A Modulates Phosphorylation of pRB, p107 and p130. Cell Cycle 2004; 3:(10):1320-30; PMID:15467457
- Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta 2008.
- Kurimchak A, Graña X. PP2A Counterbalances Phosphorylation of pRB and Mitotic Proteins by Multiple CDKs: Potential Implications for PP2A Disruption in Cancer. Genes Cancer 2013; 3:739-48; PMID:23634261; http://dx.doi.org/10.1177/1947601912473479
- Stone SR, Hofsteenge J, Hemmings BA. Molecular cloning of cDNAs encoding two isoforms of the catalytic subunit of protein phosphatase 2A. Biochemistry 1987; 26:7215-20; PMID:2827745; http://dx.doi.org/10.1021/bi00397a003
- Gotz J, Probst A, Ehler E, Hemmings B, Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Calpha. Proc Natl Acad Sci U S A 1998; 95:12370-5; PMID:9770493; http://dx.doi.org/10.1073/pnas.95.21.12370
- Ogris E, Gibson DM, Pallas DC. Protein phosphatase 2A subunit assembly: the catalytic subunit carboxy terminus is important for binding cellular B subunit but not polyomavirus middle tumor antigen. Oncogene 1997; 15:911-7; PMID:9285686; http://dx.doi.org/10.1038/sj.onc.1201259
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992; 257:1261-4; PMID:1325671; http://dx.doi.org/10.1126/science.1325671
- Longin S, Zwaenepoel K, Louis JV, Dilworth S, Goris J, Janssens V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J Biol Chem 2007; 282:26971-80; PMID:17635907; http://dx.doi.org/10.1074/jbc.M704059200
- Lee J, Stock J. Protein phosphatase 2A catalytic subunit is methyl-esterified at its carboxyl terminus by a novel methyltransferase. J Biol Chem 1993; 268:19192-5; PMID:8396127
- Guo H, Damuni Z. Autophosphorylation-activated protein kinase phosphorylates and inactivates protein phosphatase 2A. Proc Natl Acad Sci U S A 1993; 90:2500-4; PMID:7681598; http://dx.doi.org/10.1073/pnas.90.6.2500
- Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC. A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A. J Biol Chem 1999; 274:14382-91; PMID:10318862; http://dx.doi.org/10.1074/jbc.274.20.14382
- Lee J, Chen Y, Tolstykh T, Stock J. A specific protein carboxyl methylesterase that demethylates phosphoprotein phosphatase 2A in bovine brain. Proc Natl Acad Sci U S A 1996; 93:6043-7; PMID:8650216; http://dx.doi.org/10.1073/pnas.93.12.6043
- Cayla X, Van Hoof C, Bosch M, Waelkens E, Vandekerckhove J, Peeters B, Merlevede W, Goris J. Molecular cloning, expression, and characterization of PTPA, a protein that activates the tyrosyl phosphatase activity of protein phosphatase 2A. J Biol Chem 1994; 269:15668-75; PMID:8195217
- Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD, Shi Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell 2008; 133:154-63; PMID:18394995; http://dx.doi.org/10.1016/j.cell.2008.02.041
- Longin S, Jordens J, Martens E, Stevens I, Janssens V, Rondelez E, De Baere I, Derua R, Waelkens E, Goris J, Van Hoof C. An inactive protein phosphatase 2A population is associated with methylesterase and can be re-activated by the phosphotyrosyl phosphatase activator. Biochem J 2004; 380:111-9; PMID:14748741; http://dx.doi.org/10.1042/BJ20031643
- Jordens J, Janssens V, Longin S, Stevens I, Martens E, Bultynck G, Engelborghs Y, Lescrinier E, Waelkens E, Goris J, Van Hoof C. The protein phosphatase 2A phosphatase activator is a novel peptidyl-prolyl cis/trans-isomerase. J Biol Chem 2006; 281:6349-57; PMID:16380387; http://dx.doi.org/10.1074/jbc.M507760200
- Tolstykh T, Lee J, Vafai S, Stock JB. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. Embo j 2000; 19:5682-91; PMID:11060019; http://dx.doi.org/10.1093/emboj/19.21.5682
- Bryant JC, Westphal RS, Wadzinski BE. Methylated C-terminal leucine residue of PP2A catalytic subunit is important for binding of regulatory Balpha subunit. Biochem J 1999; 339(Pt 2):241-6; PMID:10191253; http://dx.doi.org/10.1042/0264-6021:3390241
- Yu XX, Du X, Moreno CS, Green RE, Ogris E, Feng Q, Chou L, McQuoid MJ, Pallas DC. Methylation of the protein phosphatase 2A catalytic subunit is essential for association of Balpha regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol Biol Cell 2001; 12:185-99; PMID:11160832; http://dx.doi.org/10.1091/mbc.12.1.185
- Ikehara T, Ikehara S, Imamura S, Shinjo F, Yasumoto T. Methylation of the C-terminal leucine residue of the PP2A catalytic subunit is unnecessary for the catalytic activity and the binding of regulatory subunit (PR55/B). Biochem Biophys Res Commun 2007; 354:1052-7; PMID:17274953; http://dx.doi.org/10.1016/j.bbrc.2007.01.085
- Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell 2009; 139:468-84; PMID:19879837; http://dx.doi.org/10.1016/j.cell.2009.10.006
- Hemmings BA, Adams-Pearson C, Maurer F, Muller P, Goris J, Merlevede W, Hofsteenge J, Stone SR. α- and β-forms of the 65-kDa subunit of protein phosphatase 2A have a similar 39 amino acid repeating structure. Biochemistry 1990; 29:3166-73; PMID:2159327; http://dx.doi.org/10.1021/bi00465a002
- Zhou J, Pham HT, Ruediger R, Walter G. Characterization of the Aalpha and Abeta subunit isoforms of protein phosphatase 2A: differences in expression, subunit interaction, and evolution. Biochem J 2003; 369:387-98; PMID:12370081; http://dx.doi.org/10.1042/BJ20021244
- Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell 1999; 96:99-110; PMID:9989501; http://dx.doi.org/10.1016/S0092-8674(00)80963-0
- Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2007; 445:53-7; PMID:17086192; http://dx.doi.org/10.1038/nature05351
- Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell 2008; 31:873-85; PMID:18922469; http://dx.doi.org/10.1016/j.molcel.2008.08.006
- Wlodarchak N, Guo F, Satyshur KA, Jiang L, Jeffrey PD, Sun T, Stanevich V, Mumby MC, Xing Y. Structure of the Ca2+-dependent PP2A heterotrimer and insights into Cdc6 dephosphorylation. Cell Res 2013; 23:931-46; PMID:23752926; http://dx.doi.org/10.1038/cr.2013.77
- Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Yu JW, Strack S, Jeffrey PD, Shi Y. Structure of the protein phosphatase 2A holoenzyme. Cell 2006; 127:1239-51; PMID:17174897; http://dx.doi.org/10.1016/j.cell.2006.11.033
- Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell 2009; 33:537-45; PMID:19285938; http://dx.doi.org/10.1016/j.molcel.2009.02.015
- Xing Y, Xu Y, Chen Y, Jeffrey PD, Chao Y, Lin Z, Li Z, Strack S, Stock JB, Shi Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006; 127:341-53; PMID:17055435; http://dx.doi.org/10.1016/j.cell.2006.09.025
- Jayadeva G, Kurimchak A, Garriga J, Sotillo E, Davis AJ, Haines DS, Mumby M, Graña X. B55alpha PP2A holoenzymes modulate the phosphorylation status of the retinoblastoma-related protein p107 and its activation. J Biol Chem 2010; 285:29863-73; PMID:20663872; http://dx.doi.org/10.1074/jbc.M110.162354
- Yang J, Phiel C. Functions of B56-containing PP2As in major developmental and cancer signaling pathways. Life Sci 2010; 87:659-66; PMID:20934435; http://dx.doi.org/10.1016/j.lfs.2010.10.003
- Saraf A, Oberg EA, Strack S. Molecular determinants for PP2A substrate specificity: charged residues mediate dephosphorylation of tyrosine hydroxylase by the PP2A/B' regulatory subunit. Biochemistry 2010; 49:986-95; PMID:20017541; http://dx.doi.org/10.1021/bi902160t
- Janssens V, Jordens J, Stevens I, Van Hoof C, Martens E, De Smedt H, Engelborghs Y, Waelkens E, Goris J. Identification and functional analysis of two Ca2+-binding EF-hand motifs in the B"/PR72 subunit of protein phosphatase 2A. J Biol Chem 2003; 278:10697-706; PMID:12524438; http://dx.doi.org/10.1074/jbc.M211717200
- Hendrix P, Mayer-Jackel RE, Cron P, Goris J, Hofsteenge J, Merlevede W, Hemmings BA. Structure and expression of a 72-kDa regulatory subunit of protein phosphatase 2A. Evidence for different size forms produced by alternative splicing. J Biol Chem 1993; 268:15267-76; PMID:8392071
- Yan Z, Fedorov SA, Mumby MC, Williams RS. PR48, a novel regulatory subunit of protein phosphatase 2A, interacts with Cdc6 and modulates DNA replication in human cells. Mol Cell Biol 2000; 20:1021-9; PMID:10629059; http://dx.doi.org/10.1128/MCB.20.3.1021-1029.2000
- Davis AJ, Yan Z, Martinez B, Mumby MC. Protein phosphatase 2A is targeted to cell division control protein 6 by a calcium-binding regulatory subunit. J Biol Chem 2008; 283:16104-14; PMID:18397887; http://dx.doi.org/10.1074/jbc.M710313200
- Magenta A, Fasanaro P, Romani S, Di Stefano V, Capogrossi MC, Martelli F. Protein phosphatase 2A subunit PR70 interacts with pRb and mediates its dephosphorylation. Mol Cell Biol 2008; 28:873-82; PMID:17991896; http://dx.doi.org/10.1128/MCB.00480-07
- Moreno CS, Park S, Nelson K, Ashby D, Hubalek F, Lane WS, Pallas DC. WD40 repeat proteins striatin and S/G(2) nuclear autoantigen are members of a novel family of calmodulin-binding proteins that associate with protein phosphatase 2A. J Biol Chem 2000; 275:5257-63; PMID:10681496; http://dx.doi.org/10.1074/jbc.275.8.5257
- Goudreault M, D'Ambrosio LM, Kean MJ, Mullin MJ, Larsen BG, Sanchez A, Chaudhry S, Chen GI, Sicheri F, Nesvizhskii AI, Aebersold R, Raught B, Gingras AC. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics 2009; 8:157-71; PMID:18782753; http://dx.doi.org/10.1074/mcp.M800266-MCP200
- Hwang J, Pallas DC. STRIPAK complexes: structure, biological function, and involvement in human diseases. Int J Biochem Cell Biol 2014; 47:118-48; PMID:24333164; http://dx.doi.org/10.1016/j.biocel.2013.11.021
- Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol 2003; 13:1356-64; PMID:12932319
- Hirschi A, Cecchini M, Steinhardt RC, Schamber MR, Dick FA, Rubin SM. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat Struct Mol Biol 2010; 17:1051-7; PMID:20694007; http://dx.doi.org/10.1038/nsmb.1868
- Purev E, Giordano A, Soprano DR, Soprano KJ. Interaction of PP2A catalytic subunit with Rb2/p130 is required for all-trans retinoic acid suppression of ovarian carcinoma cell growth. J Cell Physiol 2006; 206:495-502; PMID:16206244; http://dx.doi.org/10.1002/jcp.20490
- Purev E, Soprano DR, Soprano KJ. PP2A interaction with Rb2/p130 mediates translocation of Rb2/p130 into the nucleus in all-trans retinoic acid-treated ovarian carcinoma cells. J Cell Physiol 2011; 226:1027-34; PMID:20857408; http://dx.doi.org/10.1002/jcp.22418
- Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002; 110:19-32; PMID:12150994; http://dx.doi.org/10.1016/S0092-8674(02)00801-2
- Griswold-Prenner I, Kamibayashi C, Maruoka EM, Mumby MC, Derynck R. Physical and functional interactions between type I transforming growth factor β receptors and Balpha, a WD-40 repeat subunit of phosphatase 2A. Mol Cell Biol 1998; 18:6595-604; PMID:9774674
- Bengtsson L, Schwappacher R, Roth M, Boergermann JH, Hassel S, Knaus P. PP2A regulates BMP signalling by interacting with BMP receptor complexes and by dephosphorylating both the C-terminus and the linker region of Smad1. J Cell Sci 2009; 122:1248-57; PMID:19339557; http://dx.doi.org/10.1242/jcs.039552
- Kolupaeva V, Laplantine E, Basilico C. PP2A-mediated dephosphorylation of p107 plays a critical role in chondrocyte cell cycle arrest by FGF. PLoS One 2008; 3:e3447; PMID:18927618
- Dailey L, Laplantine E, Priore R, Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol 2003; 161:1053-66; PMID:12821644; http://dx.doi.org/10.1083/jcb.200302075
- Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA, Jacks T. Shared role of the pRb-related p130 and p107 proteins in limb development. Genes & Development 1996; 10:1633-44; PMID:8682294; http://dx.doi.org/10.1101/gad.10.13.1633
- Laplantine E, Rossi F, Sahni M, Basilico C, Cobrinik D. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J Cell Biol 2002; 158:741-50; PMID:12177046; http://dx.doi.org/10.1083/jcb.200205025
- Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Jacks T Targeted disruption of p107: functional overlap between p107 and Rb. Genes & Development 1996; 10:1621-32; PMID:8682293; http://dx.doi.org/10.1101/gad.10.13.1621
- Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 2002; 16:1446-65; PMID:12080084; http://dx.doi.org/10.1101/gad.990702
- Wuelling M, Vortkamp A. Transcriptional networks controlling chondrocyte proliferation and differentiation during endochondral ossification. Pediatr Nephrol 2010; 25:625-31; PMID:19949815; http://dx.doi.org/10.1007/s00467-009-1368-6
- Mukhopadhyay K, Lefebvre V, Zhou G, Garofalo S, Kimura JH, de Crombrugghe B. Use of a new rat chondrosarcoma cell line to delineate a 119-base pair chondrocyte-specific enhancer element and to define active promoter segments in the mouse pro-α 1(II) collagen gene. J Biol Chem 1995; 270:27711-9; PMID:7499238
- Krejci P, Bryja V, Pachernik J, Hampl A, Pogue R, Mekikian P, Wilcox WR. FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Exp Cell Res 2004; 297:152-64; PMID:15194433; http://dx.doi.org/10.1016/j.yexcr.2004.03.011
- Raucci A, Laplantine E, Mansukhani A, Basilico C. Activation of the ERK1/2 and p38 mitogen-activated protein kinase pathways mediates fibroblast growth factor-induced growth arrest of chondrocytes. J Biol Chem 2004; 279:1747-56; PMID:14593093; http://dx.doi.org/10.1074/jbc.M310384200
- Priore R, Dailey L, Basilico C. Downregulation of Akt activity contributes to the growth arrest induced by FGF in chondrocytes. J Cell Physiol 2006; 207:800-8; PMID:16523491; http://dx.doi.org/10.1002/jcp.20620
- Yeh N, Miller JP, Gaur T, Capellini TD, Nikolich-Zugich J, de la Hoz C, Selleri L, Bromage TG, van Wijnen AJ, Stein GS, Lian JB, Vidal A, Koff A. Cooperation between p27 and p107 during endochondral ossification suggests a genetic pathway controlled by p27 and p130. Mol Cell Biol 2007; 27:5161-71; PMID:17502351; http://dx.doi.org/10.1128/MCB.02431-06
- Yan YM, Lee MH, Massague J, Barbacid M. Ablation Of the Cdk Inhibitor P57(Kip2) Results In Increased Apoptosis and Delayed Differentiation During Mouse Development. Genes & Dev 1997; 11:973-83; PMID:9136926; http://dx.doi.org/10.1101/gad.11.8.973
- Aikawa T, Segre GV, Lee K. Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin E-Cdk2. J Biol Chem 2001; 276:29347-52; PMID:11384971; http://dx.doi.org/10.1074/jbc.M101859200
- Kolupaeva V, Daempfling L, Basilico C. The B55alpha regulatory subunit of protein phosphatase 2A mediates fibroblast growth factor-induced p107 dephosphorylation and growth arrest in chondrocytes. Mol Cell Biol 2013; 33:2865-78; PMID:23716589; http://dx.doi.org/10.1128/MCB.01730-12
- Schmitz MH, Held M, Janssens V, Hutchins JR, Hudecz O, Ivanova E, Goris J, Trinkle-Mulcahy L, Lamond AI, Poser I, et al. Live-cell imaging RNAi screen identifies PP2A-B55alpha and importin-beta1 as key mitotic exit regulators in human cells. Nat Cell Biol 2009; 12:886-93; PMID:20711181; http://dx.doi.org/10.1038/ncb20
- Adams DG, Coffee RL, Jr., Zhang H, Pelech S, Strack S, Wadzinski BE. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J Biol Chem 2005; 280:42644-54; PMID:16239230; http://dx.doi.org/10.1074/jbc.M502464200
- Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 2013; 13:585-95; PMID:23842645; http://dx.doi.org/10.1038/nrc3556
- Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, DeCaprio JA. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell 2007; 26:539-51; PMID:17531812; http://dx.doi.org/10.1016/j.molcel.2007.04.015
- Pilkinton M, Sandoval R, Colamonici OR. Mammalian Mip/LIN-9 interacts with either the p107, p130/E2F4 repressor complex or B-Myb in a cell cycle-phase-dependent context distinct from the Drosophila dREAM complex. Oncogene 2007; 26:7535-43; PMID:17563750; http://dx.doi.org/10.1038/sj.onc.1210562
- Sadasivam S, Duan S, DeCaprio JA. The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 2012; 26:474-89; PMID:22391450; http://dx.doi.org/10.1101/gad.181933.111
- Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev 2011; 25:801-13; PMID:21498570; http://dx.doi.org/10.1101/gad.2034211
- Forristal C, Henley SA, Macdonald JI, Bush JR, Ort C, Passos DT, Talluri S, Ishak CA, Thwaites MJ, Norley CJ, et al. Loss of the mammalian DREAM complex deregulates chondrocyte proliferation. Mol Cell Biol 2014; 34(12):2221-34; PMID:24710275; http://dx.doi.org/10.1128/MCB.01523-13
- Avni D, Yang H, Martelli F, Hofmann F, ElShamy WM, Ganesan S,Scully R, Livingston DM. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol Cell 2003; 12:735-46; PMID:14527418; http://dx.doi.org/10.1016/S1097-2765(03)00355-1
- Lorca T, Castro A. Deciphering the New Role of the Greatwall/PP2A Pathway in Cell Cycle Control. Genes Cancer 2012; 3:712-20; PMID:23634258; http://dx.doi.org/10.1177/1947601912473478
- Lorca T, Castro A. The Greatwall kinase: a new pathway in the control of the cell cycle. Oncogene 2012; PMID:22469975