
Abstract
Fanconi Anemia (FA) is an inherited multi-gene cancer predisposition syndrome that is characterized on the cellular level by a hypersensitivity to DNA interstrand crosslinks (ICLs). To repair these lesions, the FA pathway proteins are thought to act in a linear hierarchy: Following ICL detection, an upstream FA core complex monoubiquitinates the central FA pathway members FANCD2 and FANCI, followed by their recruitment to chromatin. Chromatin-bound monoubiquitinated FANCD2 and FANCI subsequently coordinate DNA repair factors including the downstream FA pathway members FANCJ and FANCD1/BRCA2 to repair the DNA ICL. Importantly, we recently showed that FANCD2 has additional independent roles: it binds chromatin and acts in concert with the BLM helicase complex to promote the restart of aphidicolin (APH)-stalled replication forks, while suppressing the firing of new replication origins. Here, we show that FANCD2 fulfills these roles independently of the FA core complex-mediated monoubiquitination step. Following APH treatment, nonubiquitinated FANCD2 accumulates on chromatin, recruits the BLM complex, and promotes robust replication fork recovery regardless of the absence or presence of a functional FA core complex. In contrast, the downstream FA pathway members FANCJ and BRCA2 share FANCD2's role in replication fork restart and the suppression of new origin firing. Our results support a non-linear FA pathway model at stalled replication forks, where the nonubiquitinated FANCD2 isoform – in concert with FANCJ and BRCA2 – fulfills a specific function in promoting efficient replication fork recovery independently of the FA core complex.
Introduction
Fanconi Anemia (FA) is a recessively inherited genomic instability syndrome caused by mutation in any one of 16 known FA genes. Clinically, FA patients exhibit congenital abnormalities, progressive bone marrow failure and a strong predisposition to cancers such as acute myeloid leukemia and squamous cell carcinomas.Citation1,2 Cells from FA patients are hypersensitive to DNA interstrand crosslinks (ICLs) and show spontaneous chromosomal aberrations that are further exacerbated upon treatment with replication-inhibiting agents such as hydroxyurea (HU) or aphidicolin (APH).Citation3-5 To promote the repair of DNA ICLs, the 16 FA pathway members are thought to function in a linear hierarchy that can be broadly divided into 3 tiers: an upstream FA core complex, a central FANCD2-FANCI protein heterodimer and several downstream FA pathway members including breast cancer-associated proteins FANCD1/BRCA2 (breast cancer associated 2), FANCN/PALB2 (partner and localizer of BRCA2) and FANCJ/BRIP1 (BRCA1-interacting protein 1).Citation1,6 DNA ICLs are mostly repaired in S-phase when they block the progression of replication forks.Citation7-9 Detection of an ICL triggers the recruitment of the FA core complex [FANCA, -B, -C, -E, -F, -G, -L]. Once recruited, the FA core complex – with FANCL as the catalytic subunit – functions as an E3 ubiquitin ligase that monoubiquitinates both members of the central FANCD2-FANCI heterodimer. Monoubiquitinated FANCD2 and FANCI are then recruited to chromatin and into DNA repair foci at DNA ICLs.Citation10,13 Subsequently, chromatin-bound FANCD2 and FANCI are thought to coordinate downstream FA nucleases like SLX4/FANCP and XPF(FANCQ)/ERCC1 with FANCJ, BRCA2 and FANCN to mediate incisions at the DNA ICL followed by homologous recombination (HR) repair of the newly generated DNA double strand break (DSB).Citation14-20
Recent studies from our laboratory and others revealed novel FA pathway functions at replication forks that are stalled in the presence of HU or APH. The FA core complex, FANCD2 and BRCA2 function to protect nascent DNA strands at stalled replication forks from nucleolytic degradation.Citation5,21,21 Importantly, FANCD2 also regulates the BLM helicase pathway to mediate restart of the stalled replication forks, while simultaneously suppressing the firing of new replication origins.Citation21,23 Intriguingly, our previous findings also showed that FANCD2 can recruit and assemble BLM pathway members such as BLM and Topoisomerase IIIα (Top3A) on chromatin independently of FANCI, indicating a separation of function between these 2 central FA pathway members.Citation21 Consequently, these results raised the question of whether FANCD2 fulfills its new role in replication fork recovery in concert with – or independently of – the upstream FA core complex and the downstream FA pathway members.
In the current study, we show that – unlike FANCD2 – the FA core complex members FANCA, FANCC, FANCG and FANCL are dispensable for the restart of APH-stalled replication forks and the suppression of new origin firing. FA core complex-deficient cells recruit nonubiquitinated FANCD2 to chromatin after APH treatment, followed by FANCD2-dependent assembly of fork restart factors and robust replication fork recovery. In contrast, cells lacking downstream FA pathway members FANCJ or BRCA2 fail to restart APH-stalled replication forks, and show a significant increase in new replication origin firing. Our results strongly support a novel, non-linear FA pathway model where nonubiquitinated FANCD2, FANCJ and BRCA2 form a team that acts completely independently of the FA core complex to coordinate molecular actions of the replication fork restart machinery.
Results
The FA core complex is dispensable for the restart of APH-stalled replication forks
Using a dual labeling DNA fiber assay, we recently showed that FANCD2 promotes the restart of APH-stalled replication forks, while simultaneously suppressing new origin firing.Citation21,23 In this assay, replication tracts are first labeled with DigUTP (red label) for 25 min, treated without or with 30 μM APH for 6 hours to cause replication fork arrest, followed by second labeling with BioUTP (green label) for 40 min (). Reproducibly, FANCD2-deficient patient cells show an approximately 2.5-fold reduction in replication fork restart efficiency (), accompanied by an approximately 4.5-fold increase in new origin firing events (). To investigate if these mechanisms depend on the FA core complex that is thought to act upstream of FANCD2, we considered that this complex is composed of 3 subcomplexes: 2 ancillary modules, FANCA-FANCG and FANCC-FANCE-FANCF, and the actual catalytic module FANCB-FANCL ().Citation24-27 We chose FA patient-derived cell lines deficient for FANCA, FANCC and FANCL, each lacking one of the 3 FA core modules, and their isogenic counterparts corrected with the respective wild type gene (PD220 and PD220+A, PD331 and PD331+C, 913/1T and 913/1T+L). We observed robust APH-induced FANCD2 monoubiquitination in the PD220+A, PD331+C and 913/1T+L cells, confirming efficient gene correction (). Using the same DNA fiber assay described in , we first analyzed FANCA- and FANCC-deficient cells for fork restart after APH treatment. We found that the proportion of replication forks competent for restart was essentially unaffected in cells lacking FANCA or FANCC compared to their corrected counterparts (, parts i and ii); moreover FANCA- or FANCC-deficient cells did not show a significant increase in new origin firing (, parts i and ii).
Figure 1 (See previous page). The 3 FA core subcomplexes are dispensable for the restart of APH-stalled replication forks. (A) Representative images of DNA fibers accompanied with a schematic of defining sites of replication. Red tracts: DigU; green tracts: BioU. (B) Left panel: Replication fork restart efficiencies were compared between FANCD2-proficient (PD20+FANCD2WT) and -deficient (PD20) cells. Replication restart efficiency was measured as the number of restarted replication forks after APH-mediated fork stalling (DigU-BioU tracts), compared with the total number of DigU-labeled tracts (DigU + DigU-BioU). Right panel: The number of new sites of replication originating during the 40 min recovery period after APH treatment was compared between PD20+FANCD2WT and PD20 cells. New origins of replication were measured as the number of green-only (BioU) tracts per unit length. [N.B. The same DNA fiber assay analysis described in Fig. 1B for analyzing replication fork restart and new origin firing following APH treatment was used throughout this study.] (C) Schematic representation of the 3 FA core subcomplexes: FANCA-FANCG, FANCC-FANCE-FANCF, and FANCB-FANCL. FA patient-derived cell lines lacking FANCA, FANCC, or FANCL (proteins circled in red) were used to represent cells deficient for the respective FA core subcomplex. (D) WCEs from untreated or APH-treated, isogenic cell pairs that were either proficient or deficient for FANCA (lanes 1-4), FANCC (lanes 5-8) or FANCL (lanes 9-12) were analyzed for the presence of FANCD2 and FANCD2Ub. Tubulin, loading control. (E) Replication fork restart efficiencies after APH treatment was compared between isogenic cell pairs proficient or deficient for (i) FANCA, (ii) FANCC or (iii) FANCL. (F) The number of new replication sites originating during BioU labeling after APH treatment was compared between isogenic cell pairs proficient or deficient for (i) FANCA, (ii) FANCC or (iii) FANCL.
![Figure 1 (See previous page). The 3 FA core subcomplexes are dispensable for the restart of APH-stalled replication forks. (A) Representative images of DNA fibers accompanied with a schematic of defining sites of replication. Red tracts: DigU; green tracts: BioU. (B) Left panel: Replication fork restart efficiencies were compared between FANCD2-proficient (PD20+FANCD2WT) and -deficient (PD20) cells. Replication restart efficiency was measured as the number of restarted replication forks after APH-mediated fork stalling (DigU-BioU tracts), compared with the total number of DigU-labeled tracts (DigU + DigU-BioU). Right panel: The number of new sites of replication originating during the 40 min recovery period after APH treatment was compared between PD20+FANCD2WT and PD20 cells. New origins of replication were measured as the number of green-only (BioU) tracts per unit length. [N.B. The same DNA fiber assay analysis described in Fig. 1B for analyzing replication fork restart and new origin firing following APH treatment was used throughout this study.] (C) Schematic representation of the 3 FA core subcomplexes: FANCA-FANCG, FANCC-FANCE-FANCF, and FANCB-FANCL. FA patient-derived cell lines lacking FANCA, FANCC, or FANCL (proteins circled in red) were used to represent cells deficient for the respective FA core subcomplex. (D) WCEs from untreated or APH-treated, isogenic cell pairs that were either proficient or deficient for FANCA (lanes 1-4), FANCC (lanes 5-8) or FANCL (lanes 9-12) were analyzed for the presence of FANCD2 and FANCD2Ub. Tubulin, loading control. (E) Replication fork restart efficiencies after APH treatment was compared between isogenic cell pairs proficient or deficient for (i) FANCA, (ii) FANCC or (iii) FANCL. (F) The number of new replication sites originating during BioU labeling after APH treatment was compared between isogenic cell pairs proficient or deficient for (i) FANCA, (ii) FANCC or (iii) FANCL.](/cms/asset/318caa80-79e6-45a8-93b2-023b6f6197a6/kccy_a_987614_f0001_c.gif)
Importantly, recent studies revealed that cells lacking either one of the ancillary FA core modules still contain very low amounts of FANCD2Ub, whereas cells deficient for the catalytic core module do not.Citation26,27 Thus, we next tested if FANCL-deficient cells are capable of replication fork recovery after APH treatment. Strikingly, the FA patient-derived FANCL-deficient cells exhibited normal replication fork restart efficiencies and robust suppression of new origin firing, comparable to the corrected cells (, part iii; , part iii). Moreover, a genetically engineered human FANCL knockout cell line (HCT116/FANCL−/−) showed no defects in replication fork restart and no increase in new origin firing after APH treatment compared to wild type HCT116 cells (Fig. S1A-C). Lastly, we argued that FANCG may be required for replication fork recovery, since FANCG can act independently of other FA core complex members to regulate formation of a separate protein complex (BRCA2-FANCD2-FANCG-XRCC3) (Fig. S2A).Citation28-30 However, replication fork restart efficiencies and suppression of new origin firing were normal in patient-derived FANCG cells compared to wild type cells (Fig. S2B–D), ruling out that FANCG or FANCG-dependent protein complexes are crucial for these mechanisms. Taken together, our results indicate that the 3 known FA core complex modules – and thus likely the entire FA core complex – are dispensable for the restart of APH-stalled replication forks and the simultaneous suppression of new origin firing.
The nonubiquitinated FANCD2 isoform is crucial for replication fork restart in FA core complex-deficient cells
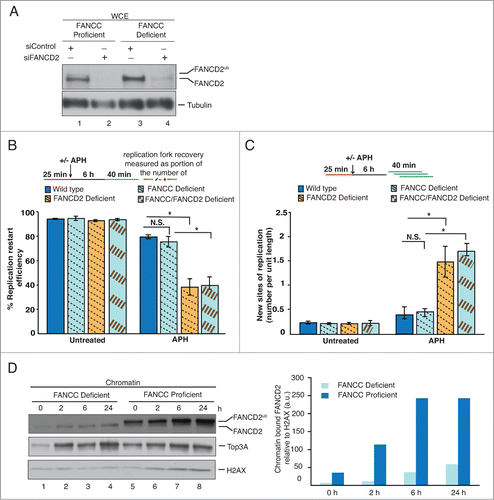
Our finding that the FA core complex is dispensable for the restart of APH-stalled replication forks immediately suggested that the FA core complex-dependent FANCD2Ub formation is dispensable for FANCD2's role during fork recovery. In that case, the nonubiquitinated FANCD2 isoform would be responsible for the successful replication fork restart observed in FA core complex-deficient cells. To test this, we treated PD331+C or PD331 cells with control siRNA or FANCD2 siRNA to generate wild type, FANCC-, FANCD2-, or FANCC/FANCD2 double-deficient cells, followed by dual-label DNA fiber analysis (). As expected, wild type and FANCC-deficient cells exhibited normal replication fork restart efficiencies (, wild type: 78.4%; FANCC-deficient: 74.4%) and were able to suppress new origin firing events () in response to APH. In striking contrast, FANCD2-deficient and FANCC/FANCD2 double-deficient cells showed a significant and comparable decrease in fork restart efficiencies (, FANCD2-deficient: 37.8%; FANCC/FANCD2 double-deficient: 39.3%) and a significant, comparable increase in new origin firing (, ∼3.5 fold increase in FANCD2- and FANCC/FANCD2 double-deficient cells). Thus, depletion of FANCD2 renders cells deficient for replication fork restart and suppression of new origin firing, irrespective of the absence or presence of a functional FA core complex.
Figure 2. The nonubiquitinated FANCD2 isoform promotes the recovery of APH-stalled forks in FA core complex-deficient cells. (A) WCEs showing the efficiency of siRNA-mediated FANCD2 knockdown in FANCC-proficient or -deficient cells. Generated cell types: Wild type (PD331+C, siControl), FANCC-deficient (PD331, siControl), FANCD2-deficient (PD331+C, siFANCD2) and FANCC/FANCD2 double-deficient (PD331, siFANCD2). Tubulin, loading control. (B) Replication fork restart efficiencies after APH treatment were compared between the 4 cell types described in (A). (C) The number of new replication sites originating during BioU labeling after APH treatment was compared between the 4 cell types described in (A). (D) Left panel: FANCC-deficient or -proficient cells were either untreated or treated with APH for the indicated time points. Chromatin fractions isolated from these cells were analyzed for the presence of FANCD2, FANCD2Ub and Top3A. H2AX, loading control. Right panel: Immunoblot signals for FANCD2 shown the in left panel were analyzed by densitometry and normalized against H2AX signals using ImageJ.
Nonubiquitinated FANCD2 is recruited to chromatin in an APH-responsive manner
Our results above indicated that cells utilize selectively the nonubiquitinated FANCD2 isoform to promote the restart of APH-stalled replication forks. Thus, one would expect the nonubiquitinated FANCD2 isoform to accumulate on chromatin in response to APH. To test this, we isolated chromatin fractions from FANCC-proficient (PD331+C) or -deficient (PD331) cells that had been untreated or treated with APH for different lengths of time (2 h, 6 h or 24 h). As expected, FANCC-proficient cells contained chromatin-bound FANCD2 even in absence of APH, and showed a continuous increase in FANCD2 chromatin binding following APH treatment ().Citation31,32 In agreement with previous studies, the chromatin fractions from FANCC-proficient cells contained predominantly the FANCD2Ub isoformCitation33; however a small amount of nonubiquitinated FANCD2 was detectable as well (). Importantly, similar amounts of nonubiquitinated FANCD2 were chromatin-bound in FANCC-deficient cells; moreover the nonubiquitinated FANCD2 isoform accumulated further on chromatin in response to APH treatment (). In addition, we found that Top3A, a member of the BLM complex that binds chromatin and promotes replication fork restart in a FANCD2-dependent manner, exhibited comparable chromatin binding behavior in FANCC-proficient and -deficient cells (). These results demonstrate that nonubiquitinated FANCD2 binds chromatin in an APH-responsive manner and recruits BLM complex members independently of the FA core complex.
A monoubiquitination-dead FANCD2K561R mutant binds chromatin and promotes replication fork restart
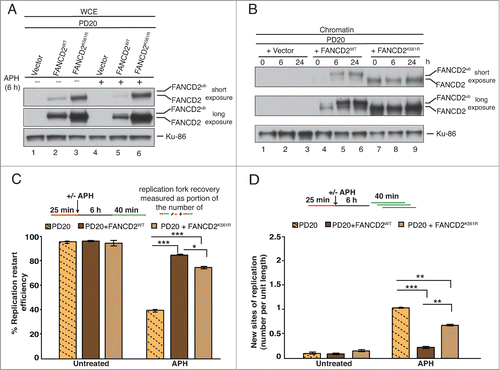
To further test the role of FANCD2Ub formation during replication fork restart, we analyzed FANCD2-deficient patient cells (PD20) that were complemented with either (i) empty vector, (ii) wild type FANCD2 (PD20+FANCD2WT) or (iii) a monoubiquitination-dead FANCD2 mutant (PD20+FANCD2K561R) (). Interestingly, we found that total protein levels of FANCD2K561R were significantly higher than those of FANCD2WT in PD20 cells (, compare lanes 2 and 3, or 5 and 6). Concomitantly, both FANCD2WT and FANCD2K561R were present in chromatin fractions isolated from unperturbed or APH-treated cells, indicating that the FANCD2K561R mutant has residual chromatin binding ability (). In agreement with this, the DNA fiber analysis revealed that the replication fork restart defect in FANCD2-deficient cells (, PD20: 39.3%) was significantly relieved by expressing FANCD2WT (, PD20+FANCD2WT: 84.5%) or the FANCD2K561R mutant (, PD20+FANCD2K561R: 74.8%). Simultaneously, the significant increase in new origin firing observed in PD20 cells (; ∼4.5 fold increase compared to PD20+FANCD2WT) was partially suppressed in PD20+FANCD2K561R cells (; ∼2 fold increase compared to PD20+FANCD2WT). Taken together, these results indicate that the nonubiquitinated FANCD2 isoform plays a crucial role at sites of APH-stalled replication forks to promote fork restart and suppression of new origin firing.
Figure 3. FANCD2K561R binds chromatin and promotes restart of APH-stalled replication forks. (A) FANCD2-deficient cells (PD20) complemented with either empty vector, FANCD2WT or FANCD2K561R were either untreated or treated with APH, and analyzed for FANCD2 and FANCD2Ub. Ku-86, loading control. (B) The 3 cell types described in (A) were either untreated or treated with APH for 6h or 24h, and chromatin fractions from the cells were analyzed for the presence of FANCD2 and FANCD2Ub. Ku-86, loading control. (C) Replication fork restart efficiencies after APH treatment were compared between the 3 cell types described in (A). (D) The number of new replication sites originating during BioU labeling after APH treatment was compared between the 3 cell types described in (A).
The “downstream” FA pathway members FANCJ and BRCA2 cooperate with FANCD2 to promote replication fork restart
The BRCA1 (breast cancer-associated 1) protein is a critical DNA repair factor that is responsible for the recruitment of FANCD2, as well as downstream FA pathway members FANCJ and BRCA2, to sites of DNA damage.Citation6,13,34 Importantly, we recently showed that BRCA1 also functions upstream of FANCD2 to promote the restart of APH-stalled replication forks.Citation23 Thus, we asked if the other BRCA1-dependent DNA repair factors, FANCJ and BRCA2, are involved in FANCD2-dependent replication fork restart as well. We chose a FANCJ-deficient patient cell line (752/1T), and its corrected wild type counterpart (752/1T+J). Furthermore, we treated 752/1T+J and 752/1T cells with control siRNA or FANCD2 siRNA to generate wild type, FANCD2-, FANCJ-, or FANCD2/FANCJ double-deficient cells, followed by dual-label DNA fiber analysis. As expected, APH treatment induced FANCD2Ub formation normally in FANCJ-deficient cells ().Citation35,36 Following APH treatment, the proportion of replication forks competent for restart was severely – and equally – reduced in FANCD2- and FANCJ-deficient cells (, 40.3% and 39.5%). Moreover, this defect in fork restart was not further exacerbated in FANCD2/FANCJ double-depleted cells (, 38.8%). In parallel, the proportion of newly originated replication tracts increased significantly and equally (, ∼5-fold) in FANCD2-, FANCJ- and FANCD2/FANCJ double-deficient cells compared with wild-type cells. Thus, FANCD2 and FANCJ cooperate to promote the recovery of APH-stalled replication forks.
Figure 4. FA pathway members FANCJ and BRCA2 cooperate with FANCD2 for the recovery of APH-stalled replication forks. (A) WCEs showing the efficiency of siRNA-mediated FANCD2 knockdown in FANCJ-proficient or -deficient cells. Generated cell types: Wild type (752/1T+J, siControl), FANCD2-deficient (752/1T+J, siFANCD2), FANCJ-deficient (752/1T, siControl) and FANCD2/FANCJ double- deficient (752/1T, siFANCD2). All 4 cell lines were untreated or treated with APH, and WCEs from these cells were analyzed for the presence of FANCJ, FANCD2 and FANCD2Ub. Tubulin, loading control. (B) Replication fork restart efficiencies after APH treatment were compared between wild type, FANCD2-, FANCJ- and FANCD2/FANCJ double-deficient cells (C) The number of new replication sites originating during BioU labeling after APH treatment was compared between Wildtype, FANCD2-, FANCJ- and FANCD2/FANCJ double-deficient cells. (D) WCEs showing the efficiency of siRNA-mediated FANCD2 and BRCA2 knockdown in wild type (PD331+C) cells. Generated cell types: wild type (PD331+C, siControl), FANCD2-deficient (PD331+C, siFANCD2), BRCA2-deficient (PD331+C, siBRCA2) and FANCD2/BRCA2 double-deficient (PD331+C, siFANCD2/siBRCA2). All 4 cell lines were untreated or treated with APH, and WCEs from these cells were analyzed for the presence of BRCA2, FANCD2 and FANCD2Ub. Ku-86: loading control. (E) Replication fork restart efficiencies after APH treatment were compared between wild type, FANCD2-, BRCA2- and FANCD2/BRCA2 double-deficient cells. (F) The number of new replication sites originating during BioU labeling after APH treatment was compared between wild type, FANCD2-, BRCA2- and FANCD2/BRCA2 double-deficient cells.
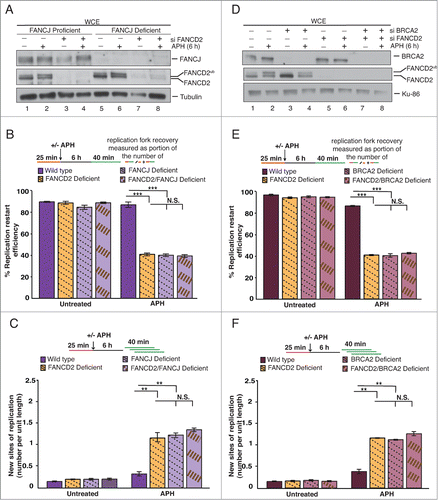
Next, we sought to determine whether BRCA2 promotes replication fork recovery, possibly in concert with FANCD2. To this end, we treated a wild type cell line (PD331+C) with siControl, siFANCD2, siBRCA2 or siFANCD2/BRCA2 to generate wild type, FANCD2-, BRCA2-, or FANCD2/BRCA2 double-deficient cells (). As expected, BRCA2-deficient cells were able to support FANCD2Ub formation in response to APH treatment.Citation37 DNA fiber analysis revealed that FANCD2- and BRCA2-deficient cells exhibited strongly – and equally – reduced replication fork restart efficiencies (, 40.4% and 39.9% that were not further reduced in the FANCD2/BRCA2 double-deficient cells (, 42.1%). Simultaneously, we observed that FANCD2-, BRCA2- and FANCD2/BRCA2 double-deficient cells showed a significant and similar increase in newly fired replication origins (, ∼3-4 fold) compared with the wild type cells.
Our finding that BRCA2 promotes replication fork restart seemingly contradicts 2 previous studies that used cell lines expressing BRCA2 mutants with C-terminal truncations to test the roles of BRCA2 at hydroxyurea (HU)-stalled replication forks. Both studies reported that BRCA2 was crucial to protect HU-stalled replication forks against nuclease-mediated DNA strand degradation, but was not required to mediate replication fork restart.Citation22,38 To address this discrepancy, we first sought to exclude the possibility that our findings were due to off-target effects of the employed BRCA2 siRNA. We repeated the DNA fiber assay in cells treated with a second BRCA2 siRNA (siBRCA2#2) that targets a different BRCA2 exon (Fig. S3A). Following APH-treatment, we observed strongly reduced replication fork restart efficiencies (Fig. S3B, 37.5%) accompanied by a significant increase in newly fired origins (Fig. S3C, 3-4 fold) in the siBRCA2#2-treated cells, supporting our initial results () that BRCA2 promotes replication fork restart and the suppression of new origin firing. Simultaneously, we found that the siBRCA2#2-treated cells exhibited a severe shortening of nascent DNA strands at APH-stalled replication forks compared to the wild type cells (Fig. S3D), confirming the previous findings that BRCA2 protects stalled replication forks from nucleolytic degradation.Citation22,38
Lastly, we wanted to test if BRCA2's role in fork restart extends to HU-stalled replication forks. We treated cells with siControl or siBRCA2 (Fig. S4A), followed by DNA fiber analysis. Following HU-treatment, BRCA2-deficient cells exhibited a severe reduction in replication fork restart efficiencies (Fig. S4B, 35.8%) and showed a significant increase in new origin firing (Fig. S4C, 4-fold), comparable to those phenotypes observed in APH-treated BRCA2-deficient cells (see and ; Fig. S3B and C). Taken together, our results indicate that a complete absence of full length BRCA2 causes severe replication fork recovery defects. Moreover, our findings demonstrate that BRCA2 acts in concert with FANCD2 to promote the restart of APH- or HU-stalled replication forks while simultaneously suppressing the firing of new replication origins.
Discussion
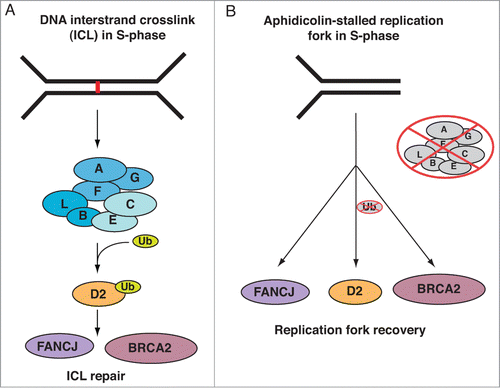
In this study, we expand on our previous findings that FANCD2 functions in the restart of APH-stalled replication forks and the simultaneous suppression of new origin firing. Our new results show that FANCD2 shares this role with FA pathway members FANCJ and BRCA2, but not with the FA core complex proteins. Our data strongly support a non-linear FA pathway model during the recovery of APH-stalled replication forks, via mechanisms that depend selectively on the nonubiquitinated isoform of FANCD2 ().
Figure 5. Distinct FA pathway models during DNA ICL repair vs. the restart of an APH-stalled replication fork. (A) Linear FA pathway model during DNA ICL repair. When replication forks converge at a DNA ICL (represented by a red line in the figure), the upstream FA core complex is activated and monoubiquitinates FANCD2, followed by its recruitment to the ICL on chromatin. Chromatin-bound FANCD2Ub then coordinates the actions of downstream DNA repair factors, including FANCJ and BRCA2, to facilitate ICL repair. (B) Non-linear FA pathway model during the recovery of APH-stalled replication forks. When a single moving replication fork is temporarily stalled by APH-treatment, nonubiquitinated FANCD2 is recruited to chromatin independently of the FA core complex. Chromatin-bound nonubiquitinated FANCD2 then functions in concert with FANCJ and BRCA2 to promote replication fork restart.
The idea that FANCD2 does not rely on the FA core complex to recover stalled replication forks is supported by recent findings that FANCD2 binds HU-stalled replication forks, where it interacts with the MCM helicase independently of the FA core complex protein FANCA.Citation39,40 Interestingly, it was also shown that FANCD2 and BRCA2 – but not FANCA or FANCC – promote cellular resistance to topoisomerase II inhibitors that cause DNA DSBs.Citation41,42 Moreover, FANCD2 is known to promote S-phase checkpoint activation in response to ionizing radiation in a manner that depends on ATM-mediated FANCD2 phosphorylation but occurs independently of FA core complex-mediated FANCD2Ub formation.Citation43 These findings indicate a functional separation between FANCD2 and the FA core complex and support the idea that FANCD2Ub formation is dispensable not only for replication fork restart, but for other FANCD2-dependent functions as well.
Importantly, in the light of recent findings that only cells lacking the catalytic FANCB-FANCL module are truly devoid of any E3 ubiquitin ligase activity toward FANCD2, we must assume that the FANCA-, FANCC- and FANCG-deficient cells used in our study still contain trace amounts of FANCD2Ub.Citation26,27 Nevertheless, our combined findings strongly support a model where replication fork restart occurs independently of FANCD2Ub formation: (i) FA patient cells of complementation groups A, C, G and L, as well as a human FANCL knockout cell line, are equally competent for replication fork restart; (ii) FANCD2WT and FANCD2K561R can both promote replication fork restart at similar levels and (iii) nonubiquitinated FANCD2 binds chromatin and recruits other replication fork restart proteins in an APH-inducible manner. Interestingly, a recent study also showed that human cells lacking the FANCA-FANCG and the FANCC-FANCE-FANCF module fail to recruit FANCB-FANCL to chromatin and do not contain any FANCD2Ub, but still exhibit residual chromatin-bound FANCD2.Citation27 Together, these findings strongly indicate that nonubiquitinated FANCD2 is recruited to APH-stalled replication forks independently of the FA core complex to promote fork restart. At the same time, APH treatment does robustly trigger FANCD2Ub formation; moreover chromatin-bound FANCD2 exists predominantly as the FANCD2Ub isoform, even in unperturbed cellular conditions. What then is the role of the FANCD2Ub formation in response to APH? Intriguingly, the Jasin laboratory showed that FANCD2 and FANCA are required to protect nascent DNA strands at HU-stalled replication forks from nucleolytic degradation.Citation5 Thus, the FANCD2Ub isoform may specifically function in replication fork protection, whereas nonubiquitinated FANCD2 mediates fork restart. These 2 mechanisms appear to be functionally uncoupled, since nascent DNA strand degradation at the stalled fork does not interfere with successful restart of DNA synthesis in FA core complex-deficient cells.
It is noteworthy that while the FANCD2K561R mutant can support replication fork restart and suppress new origin firing after APH treatment, it is not as efficient as FANCD2WT (see and ). This indicates that the FANCD2K561R mutant has additional defects that are not related to its inability to become monoubiquitinated. In further support of this idea, we previously found in Xenopus egg extracts that xFANCD2K562R – unlike nonubiquitinated xFANCD2WT – fails to stably associate with late-replicating chromatin.Citation21 Interestingly, several studies including ours observed that FANCD2-deficient cells complemented with the FANCD2WT or FANCD2K561R gene constructs contain much higher protein levels of FANCD2K561R than FANCD2WT (see ), possibly reflecting the cells’ attempt to increase the amount of chromatin-bound FANCD2K561R (see ).Citation44,45 We speculate that transient chromatin interactions of FANCD2K561R are sufficient to partially promote replication fork restart and suppression of new origin firing.
In contrast to the FA core complex, at least 2 of the “downstream” FA pathway members, FANCJ and BRCA2, now join the growing group of replication fork restart proteins including FANCD2, BRCA1, BLM, MRE11, RAD51 and CtIP.Citation21,23,46,47 Intriguingly, each of these proteins has been implicated in HR repair of DNA DSBs, strongly supporting a model where the recovery of APH-stalled replication forks involves HR mechanisms.
FANCJ's role in fork recovery is not surprising, since it interacts with known fork restart factors including BRCA1, FANCD2 and BLM.Citation23,44,48 In fact, since FANCJ – like FANCD2 – stabilizes BLM in human cells, the replication restart defects seen in cells depleted of FANCJ, FANCD2 or both may be partly due to a lack of available BLM protein.Citation21,49 Unexpectedly however, another study reported only mildly delayed fork restart kinetics in FANCJ-deficient chicken DT40 cells.Citation50 However, this discrepancy could be due to differences in molecular mechanisms between human and chicken cells; moreover our study analyzed fork restart after a complete DNA synthesis block, whereas the conditions used by Schwab et al. allowed continuous slow DNA synthesis in presence of HU, which may account for the less severe phenotype observed by these authors.
Our discovery that BRCA2 is required for the restart of APH or HU-stalled replication forks seemingly contradicts 2 previous reports that BRCA2 protects nascent DNA strands at stalled replication forks, but is not required for replication fork restart.Citation22,38 Importantly, both studies used cells that express residual C-terminally truncated BRCA2 proteins: (i) human CAPAN-1 cells (BRCA2 truncated after aa 1981), (ii) mouse embryonic stem cells carrying the BRCA2lex1/lex2 alleles (BRCA2 truncated after aa 3088/3139), and (iii) V-C8 hamster cells (BRCA2 truncated after aa 2494/2567).Citation51-53 In contrast, we observed severe fork restart defects in siBRCA2-depleted cells. This suggests that BRCA2-dependent fork recovery may not require the BRCA2 C-terminus and predicts that separate regions on BRCA2 are responsible for its roles in fork protection versus fork restart.
Lastly, in the context of BRCA2's new role, it seems counterintuitive that FANCG, and thus the FANCG-dependent BRCA2-FANCD2-FANCG-XRCC3 complex is dispensable for fork restart, while every complex member other than FANCG is crucial for this mechanism.Citation21,47 Additional studies will be necessary to solve this puzzle.
In summary, our study reveals a clear separation of function within the 3-tiered FA pathway and demonstrates that FANCD2 and at least 2 of the “downstream” FA pathway proteins fulfill a crucial function at stalled replication forks that is completely independent of the FA core complex and its E3 ligase activity. The ability of FA core complex-deficient cells to promote fork restart mechanisms that rely heavily on FANCD2 and other FA proteins may contribute to the milder phenotypes typically associated with inherited mutations in FA core complex genes.Citation54-56
Materials and methods
Cell lines and cell culture techniques
PD220 (FANCA-deficient), PD220+A (retrovirally complemented with wild type FANCA)Citation57,58 and PD352i (FANCG-deficient) patient cellsCitation57,59 were obtained from the FA Cell Repository at the Oregon Health & Science University. GM00637 cells, used as control cell line for assays with PD352i cells, were purchased from the Coriell Institute, USA. PD331 (FANCC-deficient) and PD331+C (retrovirally complemented with wild type FANCC) were a gift from Dr. Rosselli.Citation3,60 PD20 (FANCD2-deficient), PD20+FANCD2WT (complemented with wild type FANCD2) and PD20+FANCD2K561R (complemented with monoubiquitination-dead FANCD2 mutant) were a gift from Dr. Kupfer.Citation44 In addition, we generated the following cell lines: 913/1T + S91IN (FANCL-deficient) and 913/1T + S91FLcoIN (retrovirally complemented with wild type FANCL); 752/1T + S91IN (FANCJ-deficient) and 752/1T + S91FJcoIN (retrovirally complemented with wild type FANCJ). The 913/1T cells carry 2 heterozygous FANCL mutations (c.1007_1009delTAT = p.I336_C337delinsS; g.35021C>G = p.K125_L126insNYELINEKEFR).Citation61 The 752/1T cells carry a homozygous FANCJ frameshift mutation (c.308delG = pG103fsS). All cells described above were cultured in DMEM (GIBCO) with 10% FBS and 1% Penicillin-Streptomycin.
HCT116 WT cells and HCT116 FANCL–/– cells were a gift from Dr. Chen.Citation62 These cells were cultured in Mc Coy's media (Corning) enriched with 10% FBS, 1% Pencillin-Streptomycin and 1% Glutamine. Cells were treated with Aphidicolin (Sigma; 30μM for 6 hours or Hydroxyurea (Sigma; 4 mM for 5 h) unless indicated otherwise.
DNA fiber assay
We used a DNA fiber protocol as previously described.Citation21 Moving replication forks were labeled with digoxigenin-dUTPs (DigU) for 25 min and then with biotin-dUTPs (BioU) for 40 min. To allow efficient incorporation of the dUTPs, a hypotonic buffer treatment (10mM HEPES, 30 mM KCl, pH 7.4) preceded each dUTP-labeling step. To visualize labeled fibers, cells were mixed with a 10-fold excess of unlabeled cells, fixed and dropped onto slides. After cell lysis, DNA fibers were released and extended by tilting the slides. Incorporated dUTPs were visualized by immunofluorescence detection using anti-digoxigenin-Rhodamine (Roche) and streptavidin-Alexa-Fluor-488 (Invitrogen). Images were captured using a Deltavision microscope (Applied Precision) and analyzed using Deltavision softWoRx 5.5 software. Replication restart efficiency was measured as the number of restarted replication forks after APH-mediated fork stalling (DigU-BioU tracts), compared with the total number of DigU-labeled tracts (DigU + DigU-BioU). New origins of replication were measured as the number of green-only (BioU) tracts per unit length. All DNA fiber results are the means of 2 or 3 independent experiments (300 DNA fibers per experiment). Error bars represent the standard error of the mean and significance was determined by student's t-test. All values for DNA fiber data analysis following recovery from APH treatment are provided in Table S1. Statistical significance at P < 0.01, P < 0.001 and P < 0.0001 is indicated as *, ** and ***, respectively.
siRNA experiments
iGENOME non-targeting siRNA was used as a control for all siRNA experiments. siRNA duplexes for iGENOME non-targeting siRNA and FANCD2 (sequence- CAACAUACCUCGACUCAUUUU) were purchased from Dharmacon research (Thermo Scientific, MA, USA). Transfections were performed using DharmaFECT1 transfection reagent according to the manufacturer's protocol. siRNA duplex for BRCA2 was purchased from Invitrogen (siBRCA2: BRCA2HSS101095; targets exon 10; siBRCA2#2: BRCA2HSS101097; targets exon 18) and transfections were performed using RNAiMax lipofectamine reagent according to the manufacturer's protocol. All siRNAs were used at a final concentration of 20 nM.
Preparation of whole cell extract (WCE) and chromatin fractions from human cells
To prepare whole cell extracts, cells were washed in PBS, resuspended in lysis buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% Sodium Deoxycholate, 1 mM EDTA, 1 mM DTT, complete protease inhibitor cocktail) and incubated on ice for 10 min. Cell extracts were centrifuged for 5 min at 10000 g, and the supernatant was used for further analysis. Chromatin fractions were prepared as described.63 3 × 106 cells were washed in PBS, pelleted and resuspended in 1 ml cytoskeleton buffer. After 10 min incubation on ice, the suspension was centrifuged at 5000 g for 3 min. The supernatant was discarded and the resultant pellet containing chromatin-bound sample was washed 3 times with CSK buffer. The chromatin pellet was resuspended in 150 μl CSK buffer and mixed with equal volume of 2X LDS Nupage LDS sample buffer. Samples were sonicated briefly prior to PAGE analysis.
Immunoblotting
Protein samples were separated on denaturing gradient gels and transferred to Immobilon P membranes (Millipore). After blocking in 5% milk, membranes were incubated overnight with the primary antibodies. Subsequent incubation with Horseradish peroxidase-conjugated rabbit secondary antibodies (Jackson Labs) or mouse secondary antibodies (Bio-Rad) was carried out for 1 hour. Protein bands were visualized using an ECL Plus system (Millipore).
Antibodies
Commercial antibodies were used against human FANCD2 (Santa Cruz, sc-20022), Tubulin (Abcam, ab7291), H2AX (Bethyl, A300-083A), BRCA2 (Calbiochem, OP95), Top3A (Proteintech 14525-1-A), Ku86 (Santa-Cruz, sc-5280). Antibodies were used at the following dilutions in WB: FANCD2 (1:1000), BRCA2 (1:500), H2AX (1:10000), Tubulin (1:10000), Top3A (1:1000), Ku-86 (1:500).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contributions
MR co-planned and performed the experiments and co-wrote the manuscript. IC performed some of the DNA fiber assays. SLK and HH generated human immortalized FA patient cell lines and complemented FA patient cell lines. AS co-planned the experiments and co-wrote the manuscript.
987614_Supplementary_Materials.zip
Download Zip (2.4 MB)Acknowledgments
We thank the patients with FA and families, the Fanconi Anemia Research Fund, Inc.., and German family organizations “Aktionskreis FA e.V.” and “Deutsche FA Hilfe e.V.” for their support of our research. We are grateful to F. Rosselli, G. Kupfer and J. Chen for sharing human cell lines with us. We are grateful to A. Auerbach (New York, NY) and D. Schindler (Würzburg, Germany) for providing FA fibroblast cells and for sharing the mutation data that confirmed our retroviral complementation results. We would like to thank N. Shima for careful reading of the manuscript and helpful discussions.
Funding
AS was supported by the National Science Foundation (award 1121023) and the American Cancer Society (RSG-13-039-01-DMC). HH was supported by the NIH R01 CA155294-04 and by the BMBF grant “FoneFA.” In addition, this work was also partly supported by a Brainstorm Award from the Masonic Cancer Center at the University of Minnesota.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Kee Y, D'Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest 2012; 122:3799-806; PMID:23114602; http://dx.doi.org/10.1172/JCI58321
- Kupfer GM. Fanconi anemia: a signal transduction and DNA repair pathway. Yale J Biol Med 2013; 86:491-7; PMID:24348213
- Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 2009; 11:761-8; PMID:19465921; http://dx.doi.org/10.1038/ncb1883
- Kee Y, D'Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev 2010; 24:1680-94; PMID:20713514; http://dx.doi.org/10.1101/gad.1955310
- Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012; 22:106-16; PMID:22789542; http://dx.doi.org/10.1016/j.ccr.2012.05.015
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet 2007; 8:735-48; PMID:17768402; http://dx.doi.org/10.1038/nrg2159
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009; 326:1698-701; PMID:19965384; http://dx.doi.org/10.1126/science.1182372
- Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 2008; 134:969-80; PMID:18805090; http://dx.doi.org/10.1016/j.cell.2008.08.030
- Clauson C, Scharer OD, Niedernhofer L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb Perspect Med 2013; 3:a012732; PMID:24224206
- Meetei AR, De Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, Van De Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 2003; 35:165-70; PMID:12973351; http://dx.doi.org/10.1038/ng1241
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007; 129:289-301; PMID:17412408; http://dx.doi.org/10.1016/j.cell.2007.03.009
- Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D'Andrea AD, et al. Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell 2001; 7:241-8; PMID:11239453; http://dx.doi.org/10.1016/S1097-2765(01)00172-1
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 2001; 7:249-62; PMID:11239454; http://dx.doi.org/10.1016/S1097-2765(01)00173-3
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 2011; 43:142-6; PMID:21240275; http://dx.doi.org/10.1038/ng.750
- Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, McIntyre RE, Gallagher F, Kettunen MI, Lewis DY, Brindle K, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet 2011; 43:147-52; PMID:21240276; http://dx.doi.org/10.1038/ng.752
- Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet 2011; 43:138-41; PMID:21240277; http://dx.doi.org/10.1038/ng.751
- Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, Jiricny J, Takeda S, Hirota K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci U S A 2011; 108:6492-6; PMID:21464321; http://dx.doi.org/10.1073/pnas.1018487108
- Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, Knipscheer P. XPF-ERCC1 Acts in Unhooking DNA Interstrand Crosslinks in Cooperation with FANCD2 and FANCP/SLX4. Mol Cell 2014; 54:460-71; PMID:24726325; http://dx.doi.org/10.1016/j.molcel.2014.03.015
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013; 493:356-63; PMID:23325218; http://dx.doi.org/10.1038/nature11863
- Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 2014; 43:257-78; PMID:24773018; http://dx.doi.org/10.1146/annurev-biophys-051013-022737
- Chaudhury I, Sareen A, Raghunandan M, Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res 2013; 41:6444-59; PMID:23658231; http://dx.doi.org/10.1093/nar/gkt348
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 2012; 72:2814-21; PMID:22447567; http://dx.doi.org/10.1158/0008-5472.CAN-11-3417
- Yeo JE, Lee EH, Hendrickson E, Sobeck A. CtIP mediates replication fork recovery in a FANCD2-regulated manner. Hum Mol Genet 2014; 23:3695-705; PMID:24556218; http://dx.doi.org/10.1093/hmg/ddu078
- Hodson C, Walden H. Towards a molecular understanding of the fanconi anemia core complex. Anemia 2012; 2012:926787; PMID:22675617; http://dx.doi.org/10.1155/2012/926787
- Medhurst AL, Laghmani EH, Steltenpool J, Ferrer M, Fontaine C, de Groot J, Rooimans MA, Scheper RJ, Meetei AR, Wang W, et al. Evidence for subcomplexes in the Fanconi anaemia pathway. Blood 2006; PMID:16720839
- Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, Passmore LA. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell 2014; 54:858-69; PMID:24905007; http://dx.doi.org/10.1016/j.molcel.2014.05.001
- Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, Li L. Modularized functions of the fanconi anemia core complex. Cell Rep 2014; 7:1849-57; PMID:24910428; http://dx.doi.org/10.1016/j.celrep.2014.04.029
- Wilson JB, Blom E, Cunningham R, Xiao Y, Kupfer GM, Jones NJ. Several tetratricopeptide repeat (TPR) motifs of FANCG are required for assembly of the BRCA2/D1-D2-G-X3 complex, FANCD2 monoubiquitylation and phleomycin resistance. Mutat Res 2010; 689:12-20; PMID:20450923; http://dx.doi.org/10.1016/j.mrfmmm.2010.04.003
- Wilson JB, Yamamoto K, Marriott AS, Hussain S, Sung P, Hoatlin ME, Mathew CG, Takata M, Thompson LH, Kupfer GM, et al. FANCG promotes formation of a newly identified protein complex containing BRCA2, FANCD2 and XRCC3. Oncogene 2008; 27:3641-52; PMID:18212739; http://dx.doi.org/10.1038/sj.onc.1211034
- Hussain S, Wilson JB, Blom E, Thompson LH, Sung P, Gordon SM, Kupfer GM, Joenje H, Mathew CG, Jones NJ. Tetratricopeptide-motif-mediated interaction of FANCG with recombination proteins XRCC3 and BRCA2. DNA Repair (Amst) 2006; 5:629-40; PMID:16621732; http://dx.doi.org/10.1016/j.dnarep.2006.02.007
- Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, Wallisch M, Akkari Y, Olson S, Wang W, et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol Cell Biol 2006; 26:425-37; PMID:16382135; http://dx.doi.org/10.1128/MCB.26.2.425-437.2006
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 2002; 100:2414-20; PMID:12239151; http://dx.doi.org/10.1182/blood-2002-01-0278
- Montes de Oca R, Andreassen PR, Margossian SP, Gregory RC, Taniguchi T, Wang X, Houghtaling S, Grompe M, D'Andrea AD. Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood 2005; 105:1003-9; PMID:15454491; http://dx.doi.org/10.1182/blood-2003-11-3997
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 2006; 20:34-46; PMID:16391231; http://dx.doi.org/10.1101/gad.1381306
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005; 8:255-65; PMID:16153896; http://dx.doi.org/10.1016/j.ccr.2005.08.004
- Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 2005; 37:934-5; PMID:16116423; http://dx.doi.org/10.1038/ng1625
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002; 297:606-9; PMID:12065746
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011; 145:529-42; PMID:21565612; http://dx.doi.org/10.1016/j.cell.2011.03.041
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, Cortez D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J Biol Chem 2013; 288:31458-67; PMID:24047897; http://dx.doi.org/10.1074/jbc.M113.511337
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 2013; 51:678-90; PMID:23993743; http://dx.doi.org/10.1016/j.molcel.2013.07.023
- Kachnic LA, Li L, Fournier L, Ferraiolo N, Dahm-Daphi J, Borgmann K, Willers H. FANCD2 but not FANCA promotes cellular resistance to type II topoisomerase poisons. Cancer Lett 2011; 305:86-93; PMID:21414716; http://dx.doi.org/10.1016/j.canlet.2011.02.030
- Treszezamsky AD, Kachnic LA, Feng Z, Zhang J, Tokadjian C, Powell SN. BRCA1- and BRCA2-deficient cells are sensitive to etoposide-induced DNA double-strand breaks via topoisomerase II. Cancer Res 2007; 67:7078-81; PMID:17671173; http://dx.doi.org/10.1158/0008-5472.CAN-07-0601
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen P, Gregory R, Kim S, Lane W, Kastan M, D'Andrea A. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 2002; 109:459-72; PMID:12086603; http://dx.doi.org/10.1016/S0092-8674(02)00747-X
- Chen X, Wilson JB, McChesney P, Williams SA, Kwon Y, Longerich S, Marriott AS, Sung P, Jones NJ, Kupfer GM. The fanconi anemia proteins FANCD2 and FANCJ interact and regulate each other's chromatin localization. J Biol Chem 2014; 289:25774-82; PMID:25070891; http://dx.doi.org/10.1074/jbc.M114.552570
- Murina O, von Aesch C, Karakus U, Ferretti LP, Bolck HA, Hanggi K, Sartori AA. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep 2014; 7:1030-8; PMID:24794434; http://dx.doi.org/10.1016/j.celrep.2014.03.069
- Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. Embo J 2009; 28:2601-15; PMID:19629035; http://dx.doi.org/10.1038/emboj.2009.206
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010; 37:492-502; PMID:20188668; http://dx.doi.org/10.1016/j.molcel.2010.01.021
- Davies SL, North PS, Hickson ID. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol 2007; 14:677-9; PMID:17603497; http://dx.doi.org/10.1038/nsmb1267
- Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, Mosedale G, North PS, Cantor SB, Hickson ID, Brosh RM, Jr. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom's syndrome. Embo J 2011; 30:692-705; PMID:21240188; http://dx.doi.org/10.1038/emboj.2010.362
- Schwab RA, Nieminuszczy J, Shin-ya K, Niedzwiedz W. FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J Cell Biol 2013; 201:33-48; PMID:23530069; http://dx.doi.org/10.1083/jcb.201208009
- Morimatsu M, Donoho G, Hasty P. Cells deleted for Brca2 COOH terminus exhibit hypersensitivity to gamma-radiation and premature senescence. Cancer Res 1998; 58:3441-7; PMID:9699678
- Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, Yeo CJ, Jackson CE, Lynch HT, Hruban RH, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996; 56:5360-4; PMID:8968085
- Wiegant WW, Overmeer RM, Godthelp BC, van Buul PP, Zdzienicka MZ. Chinese hamster cell mutant, V-C8, a model for analysis of Brca2 function. Mutat Res 2006; 600:79-88; PMID:16643964; http://dx.doi.org/10.1016/j.mrfmmm.2006.03.001
- Soulier J, Leblanc T, Larghero J, Dastot H, Shimamura A, Guardiola P, Esperou H, Ferry C, Jubert C, Feugeas JP, et al. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/BRCA pathway. Blood 2005; 105:1329-36; PMID:15383454; http://dx.doi.org/10.1182/blood-2004-05-1852
- Kalb R, Neveling K, Hoehn H, Schneider H, Linka Y, Batish SD, Hunt C, Berwick M, Callen E, Surralles J, et al. Hypomorphic mutations in the gene encoding a key Fanconi anemia protein, FANCD2, sustain a significant group of FA-D2 patients with severe phenotype. Am J Hum Genet 2007; 80:895-910; PMID:17436244; http://dx.doi.org/10.1086/517616
- Bakker ST, de Winter JP, te Riele H. Learning from a paradox: recent insights into Fanconi anaemia through studying mouse models. Dis Model Mech 2013; 6:40-7; PMID:23268537; http://dx.doi.org/10.1242/dmm.009795
- Jakobs PM, Fiddler-Odell E, Reifsteck C, Olson S, Moses RE, Grompe M. Complementation group assignments in Fanconi anemia fibroblast cell lines from North America. Somat Cell Mol Genet 1997; 23:1-7; PMID:9217996; http://dx.doi.org/10.1007/BF02679950
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, Constantinou A. FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 2013; PMID:23993743; http://dx.doi.org/10.1016/j.molcel.2013.07.023
- Donahue SL, Campbell C. A DNA double strand break repair defect in Fanconi anemia fibroblasts. J Biol Chem 2002; 277:46243-7; PMID:12361951; http://dx.doi.org/10.1074/jbc.M207937200
- Hirano S, Yamamoto K, Ishiai M, Yamazoe M, Seki M, Matsushita N, Ohzeki M, Yamashita YM, Arakawa H, Buerstedde JM, et al. Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. Embo J 2005; 24:418-27; PMID: 15616572; http://dx.doi.org/10.1038/sj.emboj.7600534
- Neveling K. Molecular causes and consequences of genetic instability with respect to the FA/BRCA caretaker pathway. Universität Würzburg, 2012; 176.
- Leung JW, Wang Y, Fong KW, Huen MS, Li L, Chen J. Fanconi anemia (FA) binding protein FAAP20 stabilizes FA complementation group A (FANCA) and participates in interstrand cross-link repair. Proc Natl Acad Sci U S A 2012; 109:4491-6; PMID:22396592; http://dx.doi.org/10.1073/pnas.1118720109
- Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 2007; 21:3331-41; PMID:18079179; http://dx.doi.org/10.1101/gad.457807