
Abstract
The epithelial to mesenchymal transition (EMT) is an essential process during development and during tumor progression. Here, we observed the accumulation of the selective autophagy receptor and signaling adaptor sequestosome-1 (SQSTM1/p62) during growth factor-induced EMT in immortalized and tumor-derived epithelial cell lines. Modulation of the p62 level regulated the expression of junctional proteins. This effect was dependent on the ubiquitin-associated domain of p62, which stabilized the TGFβ/Smad signaling co-activator Smad4 and the EMT transcription factor Twist. This study highlights a novel function of p62 in a major epithelial phenotypic alteration.
Abbreviations list
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| MDCK | = | Madin Darby Canine Kidney |
| NMuMG | = | Normal Murine Mammary Gland |
| SQSTM1 | = | Sequestosome 1 |
| TGFβ | = | Transforming Growth Factor β |
| IGF-II | = | insulin growth factor II |
| EGF | = | epidermal growth factor |
Introduction
Epithelial to mesenchymal transition (EMT) plays a fundamental role in embryonic development and tissue repair.Citation1 Several lines of evidence suggest that EMT is necessary for the emergence of cancer stem cells and for tumor progression.Citation2 EMT is characterized by a loss of the apical-basal polarity of epithelial cells, the emergence of a mesenchymal phenotype, and increased cell mobility.Citation1 The proteins that form tight junctions and adherent junctions such as occludin, claudin, and E-cadherin disappear during EMT, and mesenchymal markers begin to be expressed. EMT can be initiated in response to a variety of signals, such as transforming growth factor-β (TGFβ), insulin growth factor II (IGF-II), or epidermal growth factor (EGF).Citation1 These EMT inducers lead to the activation of transcription factors, including Snail and Slug, ZEB1/2, and Twist.Citation2
The selective autophagy receptor and signaling adaptor sequestosome-1 (SQSTM1/p62, hereafter referred to as p62) is a multifunctional protein. p62 contains domains that facilitate interaction with many signaling activators.Citation3 The presence of an LC3-interacting region (LIR) enables p62 to bind to the autophagy protein LC3, and the ubiquitin-associated domain (UBA) binds to ubiquitin to mediate the selective degradation of ubiquitinated cargo by autophagy.Citation4 Because p62 is involved in multiple signaling pathways, its cellular level is crucial. Over-expression of p62 can lead to the activation of aberrant signaling and diseases including tumourigenesis.Citation3 In this study, we observed that p62 accumulates during growth factor-induced EMT in normal and tumoral cells. We demonstrated that p62 increases the stability of mediators of TGFβ signaling and transcription factors engaged in EMT. This cytoplasmic activity of p62 was dependent upon its UBA domain but independent of its interaction with the autophagy protein LC3.
Results and Discussion
p62 accumulates in MDCK, NMuMG, and NBT-II cells during EMT
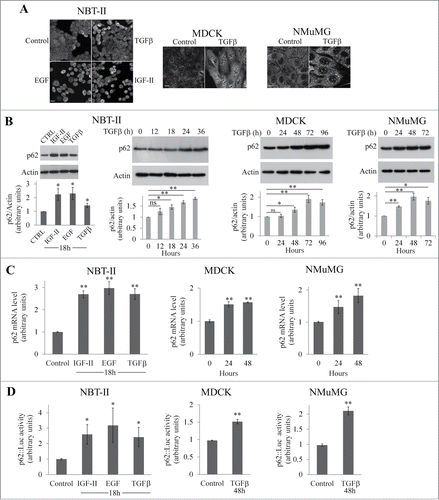
In non-transformed MDCK and NMuMG cells and in NBT-II cells derived from a rat bladder tumor, EMT can be induced by TGFβ, EGF, or IGF-II.Citation5-7 As shown in , p62 staining was increased in all the 3 cell lines in response to growth factors. This accumulation was confirmed by western blot analysis (). Moreover, the accumulation of p62 after TGFβ treatment was time-dependent (). RT-PCR analysis showed that the accumulation of p62 protein response to growth factor treatment was associated with an increase of levels of its mRNA () and with activation of the p62 promoter () in each of the cell lines analyzed.
Figure 1. p62 accumulates in NBT-II, MDCK, and NMuMG cells during EMT. (A) p62 was immunostained in NBT-II cells treated or not treated with TGFβ, EGF, or IGF-II for 18 h and in MDCK and NMuMG treated or not with TGFβ for 72 h. Bar, 10 μm. (B) NBT-II cells were treated with IGF-II, EGF, or TGFβ, and levels of p62 were determined after 18 h (right panel). Cells were treated with TGFβ and p62 levels were analyzed as a function of time. Proteins were analyzed by western immunoblotting with anti-p62 and anti-actin antibodies. (C) Levels of p62 mRNA was quantified in NBT-II cells after an 18-h treatment with indicated growth factors; levels were analyzed in MDCK and NMuMG cells as a function of time after treatment with TGFβ. (D) p62 promoter activity was monitored during growth factor-induced EMT in NBT-II, MDCK and NMuMG cells.
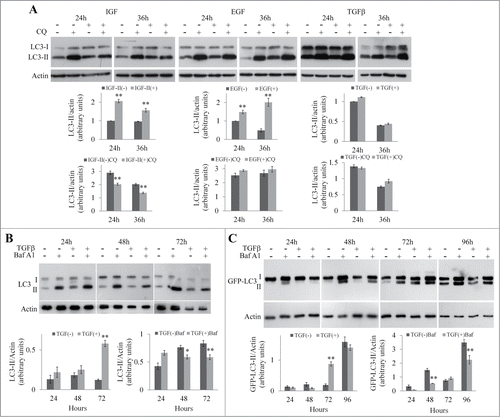
Autophagy plays a major role in the degradation of p62,Citation3,4 and measurement of the level of p62 is a classical readout of autophagic flux.Citation8 In order to investigate whether a blockade in the autophagic flux contributes to accumulation of p62, we measured the autophagic flux with and without growth factor treatment (). The protein LC3 is a marker of the autophagic pathway. When autophagy is induced, LC3-I is converted to LC3-II by a lipid anchor at the C-terminus, and LC3-II becomes associated with the autophagosomal membrane. The level of LC3-II correlates with autophagosome numbers.Citation8 As autophagosomes can accumulate as a result of either a stimulation of autophagy or a blockade of autophagosome consumption by the lysosomal compartment, levels of LC3-II must be analyzed in the absence and in the presence of inhibitors of the lysosomal function such as bafilomycin A1 (Baf A1) or chloroquine (CQ). In NBT-II cells, autophagic flux is impaired in response to IGF-II treatment but not by EGF or TGFβ treatments (). In NMuMG cells, we observed a time-dependent blockade of the autophagic flux culminating after 72 h of TGFβ treatment (). A TGFβ-induced blockade of the autophagic flux was also detected at 48 and 96 h in MDCK cells expressing GFP-LC3 ().
Figure 2. Analysis of autophagic flux in NBT-II, MDCK, and NMuMG cells during growth factors-induced EMT. (A) Expression of LC3 in NBT-II cells during EMT induced by TGFβ, IGF-II, or EGF. Cells were treated with growth factors for 24 or 36 h in the presence or absence of chloroquine (CQ) during the last 4 h. Levels of LC3-I and II and actin were determined by western blotting. The graph represents the LC3-II/actin ratio at the different time points. (B) Expression of LC3 in NMuMG cells during EMT. Cells were treated with TGFβ for 24 to 72 h in the presence or absence of bafilomycin A1 (BafA1) during the last 4 h. Expression of LC3-I, LC3-II, and actin were detected by protein gel blotting. The graph represents the LC3-II/actin ratio at the different time points. (C) MDCK/GFP-LC3 cells were treated with TGFβ for indicated times, and bafilomycin A1 (BafA1) was added for the last 4 h. GFP-LC3-I, GFP-LC3-II, and actin were detected by western blotting using anti-GFP and anti-actin antibodies respectively. The graph represents the GFP-LC3-II/actin ratio at the different time points.
The accumulation of p62 during growth factor-induced EMT is mainly the consequence of the induction of a transcription program whether or not the autophagic flux is blocked.
p62 regulates the expression of junctional proteins during EMT
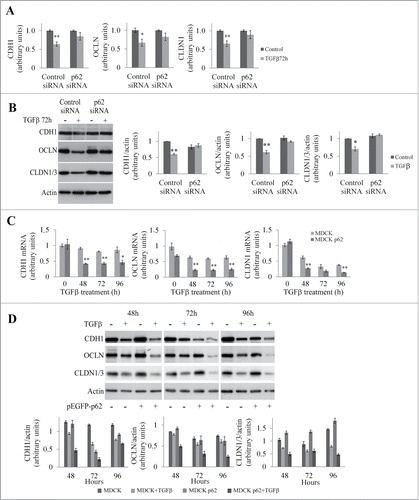
We speculated that accumulation of p62 induced by growth factors is instrumental in execution of the EMT program. The loss of cell polarity during EMT is marked by the disappearance of tight junctions and adherens junctions. To investigate the role of p62 in the expression of junctional proteins, we analyzed the levels of mRNAs of junctional proteins after modulation of p62 levels. We first inhibited expression of p62 using a specific siRNA in MDCK cells (Fig. S1A) and analyzed the mRNA and protein levels of junctional proteins with or without TGFβ treatment ( and ). As expected, after TGFβ treatment we observed that levels of E-cadherin, occludin, and claudin were significantly reduced at the mRNA and protein levels in MDCK cells. However, in p62 depleted cells, no significant reductions in the levels of E-cadherin, occludin, and claudin were observed in response to TGFβ ( and ). Conversely, p62 overexpression in MDCK cells enhanced the reduction in mRNA and protein levels of junctional proteins in response to TGFβ ( and ). Similarly, pronounced reductions in the levels of E-cadherin and occludin were observed in response to TGFβ in NMuMG cells overexpressing p62 (Fig. S1B and C).
Figure 3. p62 modulates the expression of junctional proteins in MDCK cells. (A) Quantification of E-cadherin (CDH1), occludin (OCLN), and claudin-1 (CLDN1) mRNA levels in MDCK cells transfected with control or p62 siRNA and treated or not with TGFβ for 72 h. (B) Western blot analysis of CDH1, OCLN, CLDN1/3 in MDCK cells transfected with control or p62 siRNA and treated or not with TGFβ for 72 h. (C) Quantification of CDH1, OCLN, and CLDN1 mRNAs in MDCK and MDCKp62 cells treated or not with TGFβ for 48 to 96 h. (D) MDCK cells and MDCKp62 were treated or not with TGFβ for 48 to 96 h. CDH1, OCLN, and CLDN1/3 were detected by protein gel blotting.
The UBA domain of p62 interferes with the expression of junctional proteins
To identify the domain of p62 responsible for the transcriptional down-regulation of E-cadherin, occludin, and claudin-1, we generated MDCK cell lines expressing p62 mutants with deletions of different domains (Fig. S2A and B). We then investigated whether cells expressing these mutants underwent EMT after being exposed to TGFβ. We used immunofluorescence to investigate the distribution of occludin and E-cadherin in p62 mutant-expressing cells (). After TGFβ treatment, only the cells that expressed the p62ΔUBA and the p62ΔLIR mutants maintained the organization of occludin and E-cadherin characteristic of polarized cells. Although the distribution of the 2 junctional proteins was relatively conserved in MDCK cells expressing the p62ΔLIR mutant, the size of cells was increased, which is one of the morphological changes that accompanies TGFβ-induced EMT.Citation9 To investigate more precisely the role of the LIR domain we stably expressed a p62 mutant carrying a point mutation (W338A) in the LIR motif that abolishes the recognition of LC3 by p62.Citation10 E-cadherin and occludin were disorganized in cells expressing this p62 mutant after TGFβ treatment as shown by immunofluorescence analyses (Fig. S2C).
Figure 4. Effect of p62 mutants on the expression of junctional proteins. (A) MDCK cells and MDCK expressing wild-type p62 or p62 domain-deletion mutants cells were subjected to TGFβ treatment for 72 h. Cells were processed for indirect immunofluorescence analysis of occludin (OCLN), and E-cadherin (CDH1). Bar, 10 μm. (B) MDCK cells and MDCK cells expressing wild-type p62 or domain-deletion mutants were left untreated or treated with TGFβ for 48 h. p62 mutants, CDH1, OCLN, and actin expression were analyzed by western blot.
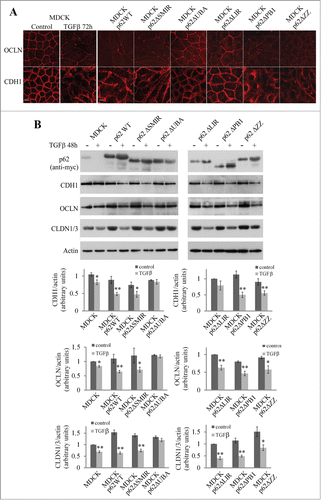
These findings were confirmed by analyzing the junctional protein levels (). We observed a decrease in the expression of E-cadherin and occludin in MDCK cells expressing p62WT, ΔSMIR, ΔPB1, ΔZZ and ΔLIR mutants after TGFβ treatment. In MDCK cells expressing p62ΔUBA no decrease was observed in the levels of junctional proteins. Interestingly, the expression of occludin was also decreased in MDCK cells expressing p62W338A after TGFβ treatment (Fig. S2D). These findings demonstrate that the UBA domain of p62 is required for EMT. In contrast, the LIR motif of p62 is not necessary for EMT.
p62 stabilizes several actors of EMT
Having determined that the UBA domain of p62 regulates EMT, we investigated the pathway that is modulated by p62 and its UBA domain during this process. The expression of junctional proteins is controlled by the transcription factors Snail1/2, ZEB1/2, and Twist.Citation2 We first focused our study on the impact of p62 on Snail. We measured levels of Snail mRNA in control MDCK cells and in MDCK-overexpressing p62 cells (hereafter referred to as MDCKp62) (). In control cells, levels of Snail mRNA increased for the first 48 h of treatment with TGFβ and then slowly decreased. However in MDCKp62, a more robust Snail mRNA level was observed before and after TGFβ treatment. The same conclusion was reached when the expression of Snail was analyzed by protein gel blot (). In contrast, inhibition of p62 expression by siRNA strongly decreased Snail level after TGFβ treatment (). Similarly in NBT-II cells, treatment with p62 siRNA strongly decreased the expression of Snail (Fig. S3A). These data indicated that p62 modulates the expression of the transcription factor Snail. Importantly, the expression of Snail was rescued by expressing a siRNA-resistant form of wt p62 but not by a siRNA-resistant form of p62ΔUBA (Fig. S3B).
Figure 5. p62 regulates the expression of junctional proteins via the Smad/Snail signaling pathway. (A) Snail mRNA levels were quantified by qRT-PCR in MDCK and MDCKp62 cells treated with TGFβ from 0 to 96 h. (B) MDCK, MDCKp62 cells, and MDCK cells treated with p62 siRNA were subjected to TGFβ treatment. Expression of Snail and actin were revealed by protein gel blotting. (C) MDCK and MDCKp62 cells were treated or not with Smad4 siRNA and subjected to TGFβ treatment. Expression of Smad4, Snail, and actin were revealed by western blotting. (D) MDCK and MDCKp62 cells were treated with TGFβ for 72 h. Expression of Smad4 and PCNA were revealed by protein gel blotting of nuclear extracts. (E) MDCK, MDCKp62 cells or siRNA p62-treated MDCK cells were transiently transfected with a Smad reporter construct together with a Renilla luciferase expression vector and treated with TGFβ for the indicated times.
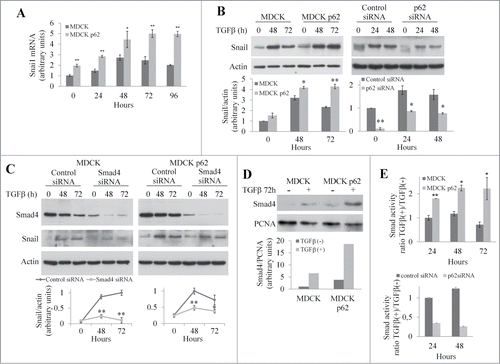
The Smad signaling pathway controls transcription of Snail in response to TGFβ treatment.Citation2 When MDCK or MDCKp62 were treated with a siRNA targeting Smad4, Snail expression was reduced (). Inhibition of Smad4 expression also limited the decrease in junctional proteins in both MDCK and MDCKp62 cells in response to TGFβ treatment (Fig. S4). When TGFβ/Smad pathway is activated, Smad2 and Smad3 are phosphorylated and recruit Smad4 to the cytoplasm before translocating into the nucleus.Citation2 We used western blotting to investigate the nuclear translocation of Smad4 in MDCK and MDCKp62 cells. After 72 h of TGFβ treatment we detected Smad4 in the nuclear fraction of MDCK cells, but in MDCKp62 the level of Smad4 was 3 times higher (). To further analyze the effect of p62 on Smad signaling, we used a Smad activity reporter assay. After TGFβ treatment, we observed that the Smad activity was approximately twice as high in MDCKp62 cells relative to control cells whereas the Smad activity was strongly decreased in p62 depleted MDCK cells (). These results demonstrate that p62 controls the Smad signaling to modulate the expression of junctional proteins.
To elucidate the role of p62 in the activation of the Smad pathway, we investigated the expression of Smad4 during EMT (). We observed that the overexpression of p62 stabilized Smad4 during TGFβ treatment. In contrast, we observed a degradation of Smad4 that was sensitive to the proteasome inhibitor MG132 in MDCK cells (Fig. S5A). No p62-dependent stabilization of Smad2/3 was observed (Fig. S5B). After treatment of cells with p62 siRNA, the expression of Smad4 was lower than in control cells in the absence and in the presence of TGFβ (). These findings suggest that p62 stabilizes Smad4 during TGFβ-induced EMT. Since p62 had no effect on Smad4 transcription (data not shown), we studied the effect of p62 on the stability of Smad4. To do this, we analyzed the levels of Smad4 in the presence of the protein synthesis inhibitor cycloheximide (). The overexpression of p62 increased the half-life of Smad4 from 6 h to 35 h in the presence of TGFβ. This stabilization likely occurs in the cytoplasm because a p62 mutant (4XNLS)Citation11 unable to translocate into the nucleus (Fig. S6A) stabilized Smad4 (Fig. S6B). Moreover cells expressing this mutant underwent EMT in response to TGFβ treatment (Fig. S6B). In contrast, the half-life of Smad4 remained unchanged in cells expressing the p62ΔUBA mutant in the presence of TGFβ () and in cells expressing the p62 UBA domain (Fig. S7). From these results, we conclude that p62 stabilizes Smad4 in the cytoplasm during TGFβ-induced EMT and that the UBA domain is required for this stabilization.
Figure 6. The UBA domain of p62 is required to stabilize Smad4. (A) MDCK, MDCKp62 cells, and MDCK cells treated with p62 siRNA were subjected to TGFβ treatment. p62, Smad4, and actin were detected by immunoblotting (LE: low exposition, HE: high exposition). (B) Immunoblot analysis of Smad4 and actin in control and MDCK cells that overexpress wild-type p62, p62ΔUBA or p62 4XNLS; cells were treated with TGFβ for 48 h and cycloheximide (CHX) for the last 6 or 8 h. (C) MDCK, NMuMG, and NBT-II cells were treated or not with TGFβ for 48 h and 24 h respectively. Immunoprecipitation (IP) was performed using an anti-p62 antibody or a monoclonal control IgG. Smad4, p62 and actin were revealed by immunoblotting.
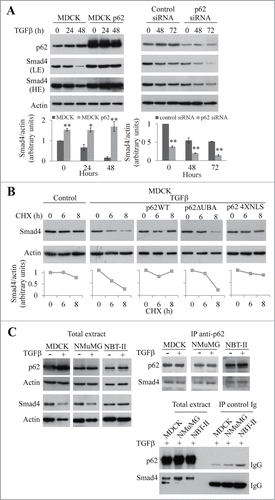
Since the expression of p62 increased in response to EGF and IGF-II, which induce EMT in NBT-II cells in a Smad pathway independent manner (Ref. Citation7 and ), we reasoned that p62 also acts on a factor common to TGFβ, EGF, and IGF-II signaling. The expression of the EMT transcription factor Twist is modulated by p62 in TGFβ treated NBT-II cells (Fig. S3A) and TGFβ treated MDCK cells (Fig. S8A). By treatment of MDCK cells with cycloheximide, we showed that Twist is stabilized by p62, whereas no stabilization of Snail was observed (Fig. S8B). On the basis of the effects of the p62 4XNLS and p62ΔUBA mutants we conclude that, similarly to Smad4, the stabilization of Twist is a cytoplasmic event that depends on the p62 UBA domain (Fig. S8B).
In order to better understand the mechanism by which p62 stabilizes Smad4, we performed a series of co-immunoprecipitation experiments in MDCK, NMuMG, and NBT-II cells. Co-immunoprecipitation experiments indicated that in the 3 cell types studied, Smad4 interacts with p62 (). In agreement with previous studies,Citation12 we showed that Smad4 is post-translationally modified by ubiquitin (Fig. S9A). Ubiquitination of Smad4 regulates positively or negatively its activity depending on which residue is ubiquitinated.Citation12 Deletion of the p62 UBA domain abolished the co-precipitation of Smad4 and p62 in HEK293T cells (Fig. S9C). In this cell line, the interaction between Smad4 and p62 is dependent of the presence of TGFβ (Fig. S9C). Moreover in cells expressing the p62 M404V mutant, which does not bind ubiquitinated proteins,Citation13 we failed to co-immunoprecipitated the ubiquitinated form of Smad4 (Fig. S9D). In addition the absence of detection of ubiquitinated protein other than Smad4 in the immunoprecipitate is not in favor of the noticeable contribution of additional partners to the interaction with p62 (Fig. S9B).
In this study, we report an accumulation of p62 during EMT. We further demonstrated that p62 is responsible for the downregulation of the expression of the junctional proteins E-cadherin, occludin and claudin. The decreased expression of these proteins is crucial during EMT. A recent study showed that the suppression of the expression of claudins 3 and 4 in ovarian cancer cells is sufficient to induce EMT by reducing the level of E-cadherin and up-regulating N-cadherin expression.Citation14 We found that p62 down-regulates the expression of these junctional proteins by increasing the expression of Snail, a major regulator of the expression of the junctional proteins during EMT.Citation15,16 These results are in agreement with the findings of Lv et al. that the level of p62 expression regulates the amount of Snail.Citation17 We show here that p62 regulates the transcription of Snail by acting on the Smad signaling pathway. p62 stabilized Smad4 expression; this effect was dependent on the UBA domain of p62 and independent of its interaction with LC3, indicating that stabilization of Smad4 is likely independent of autophagy. Of note, p62 also stabilizes the expression of another important EMT transcription factor, Twist. Interestingly Qiang et al. recently reported that p62 stabilized Twist in a p62 UBA domain-dependent manner.Citation18 The interaction of p62 with Smad4 and Twist most probably involves the UBA domain since this domain is necessary for the stabilization of these proteins (this work and Ref. Citation18).
The levels of Smad4 and Twist are regulated by the ubiquitin proteasome system.Citation12,19 Moreover, Korolchuk et al. showed that after inhibition of autophagy, p62 accumulates and prevents the delivery of ubiquitinated substrates to the proteasome.Citation20 The effect observed in this study is not a mere blockade of proteosomal degradation because we failed to observe a stabilization of Snail by p62 overexpression in response to EMT inducers (Fig. S8B). We hypothesize that the ubiquitination of Smad4 and Twist in response to growth factor treatment may promote their interaction with the UBA domain of p62 during EMT, thus stabilizing their expression by preventing their degradation. In conclusion, we propose a novel role for p62 during EMT in controlling the stability and activity of several EMT factors. This function requires the UBA domain, but not the LIR domain, of p62. These results suggest that it is important to analyze p62 expression in cancer patients as the loss of cell polarity and differentiation is frequently associated with the aggressiveness of cancer cells.Citation21
Material and Methods
Antibodies
Antibodies directed against HA and Smad4 (Santa Cruz), Smad2/3 (BD Transduction), E-cadherin (BD Transduction and clone rr1, Developmental Studies Hybridoma Bank, University of Iowa), Snail, PCNA, (Cell Signaling), Twist (Aviva Systems Biology), actin (clone C4, Millipore), p62 (Abnova), occludin and claudin1/3 (Invitrogen), Flag (Sigma), GFP (Roche), Myc-tag, LC3B (Sigma), histidine (Cell Signaling), Ubiquitin (Pierce), horseradish peroxidase-linked secondary antibodies (Jackson Immunology and Rockland for TrueBlot ULTRA antibodies) were used for protein gel blot analyses. Antibodies directed against E-cadherin (BD Transduction), occludin (Invitrogen), were used for indirect immunofluorescence. The secondary antibodies used for immunofluorescence analysis were donkey anti-mouse or donkey anti-rabbit TRITC conjugated (Jackson Immunology).
Plasmids
The GFP-LC3 and GFP-p62 expression vectors were kindly provided by T. Yoshimori (Graduate School of Medicine, Osaka University, Japan) and Z. Yue (Mount Sinai School of Medicine, USA), respectively. The HA-Ub construct was provided by A. Delaunay-Moisan (CEA-Saclay, France). His/Myc-p62 mutants (p62 WT, p62ΔLIR, ΔPB1, ΔSMIR, ΔUBA, ΔZZ) were kind gifts from M. Tanaka (Kyoto Prefectural University of Medicine, Japan). HA-p62, HA-p62ΔUBA, HA-p62W338A, GFP-p62UBA domain, and GFP-p62M404V were described previously.Citation10,22 pDest-eGFP-p62 (R186A/K187A/K264A/R265A) or p62 4XNLS constructs was made site-directed mutagenesis as previously described.Citation11 The p(CAGA)9::Luciferase construct was previously described.Citation23 The p62 promoter construct was kindly provided by A. Bruhat and W. B’chir (INRA, Theix, France). Flag-Smad4 was a gift from A. Mauviel (Institut Curie, Orsay, France) and R. Derynck (University of California San Francisco, United States).
Cell culture
MDCK were grown in MEM Alpha supplemented with 10% foetal calf serum (FCS) in 5% CO2 at 37°C. NBT-II and HEK293T cells were maintained in DMEM containing 10% FCS in 5% CO2 at 37°C. NMuMG cells were cultured in DMEM supplemented with 10% FCS and 0.01 mg/mL bovine insulin (Sigma) in 10% CO2 at 37°C.
Treatment of cells with growth factors and drugs
To induce EMT, MDCK cells were treated with 10 ng/mL TGFβ (Gibco Life Technologies), NMuMG cells were treated with 5 ng/mL TGFβ, and NBT-II cells were treated with 10 ng/mL TGFβ, 100 ng/mL of IGF-II or 30 ng/mL of EGF. Chloroquine (Sigma) was applied at 50 μM for 4 h, bafilomycin A1 (Sigma) at 200 nM for 4 h, MG132 (Sigma) at 10 μM for 6 to 8 h, and cycloheximide (Sigma) at 35 μM for 6 to 8 h.
cDNA and siRNA transfection
MDCK and/or NMuMG cells were transfected with expression vectors encoding GFP-LC3, GFP-p62, HA- or His/Myc-p62 wild-type or mutated. The clones were screened by immunocytochemistry using the appropriate antibody. Transient transfections of MDCK, NMuMG, NBT-II, and HEK293T cells were carried out using FuGENE HD reagent (Roche) according to the manufacturer's protocol.
MDCK and NBT-II cells plated in 24-well plates were transfected with 200 pmol of control or specific siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. The following siRNA sequences were used: dog p62 sense 5′-GCAUCGAAGUGGACAUUGA-3′ and antisense 5′UCAAUGUCCACUUCGAUGC-3′; dog Smad4 sense 5′-GCAGCAGAAUGGAUUUACU-3′ and antisense 5′-AGUAAAUCCAUUCUGCUGC-3′; and rat p62 sense 5′-GCAUCGAGGUUGACAUUGA-3′ and antisense 5′- UCAAUGUCAACCUCGAUGC-3′. All the siRNA sequences were designed and synthesized by Eurogentec.
For siRNA rescue experiment in MDCK cells, we used HA-p62 and HA-p62ΔUBA vectors containing a human p62cDNA that is not recognized by dog p62 siRNA. MDCK cells were transfected with 200 pmol of control or p62 siRNA and 0.5 μg of HA-p62 or HA-p62ΔUBA using Lipofectamine 2000.
Indirect immunofluorescence analyses
For the indirect immunofluorescence studies, the cells were cultured on glass coverslips (24-well plates). Cells were washed with PBS and then fixed with paraformaldehyde 3% in PBS for 15 min. The cells were permeabilized using 0.2% Triton X-100 in PBS for 4 min, washed twice with PBS, and then saturated for 1 h with PBS containing 1% BSA. For immunolocalization, the cells were incubated overnight at 4°C with appropriate primary antibodies diluted in PBS, 1% BSA. The cells were washed 3 times and then incubated for 45 min with appropriate secondary antibodies diluted in the same buffer. After washing, the samples were mounted in Fluorescent Mounting Medium (DAKO). Imaging was carried out on a LSM 510 Zeiss confocal microscope.
Quantitative reverse transcription PCR (qRT-PCR)
Total RNA were extracted from cells using a NucleoSpin RNA XS kit from Macherey-Nagel. First-strand cDNA was synthesized using the Superscript II reverse transcription kit (Invitrogen). Real-time PCR was performed in a CFX96 apparatus (BIO-RAD) using the SSoAdvanced SYBR Green Supermix (BIO-RAD). Threshold cycle (Ct) values were calculated by using the second derivative maximum algorithm provided by the Light Cycler software. To compensate for variations in the RNA input and in the efficiency of the qRT-PCR, we used a normalization strategy based on the housekeeping genes GAPDH and β-actin.
The sequences of the oligonucleotide primers used for canine genes were as follows: Sqstm1, sense 5′-AGCACGGAGGACAGGGGAAG-3′ and reverse 5′-CCAGGGCAACCTTGTCCATTTG-3′; E-cadherin, QT00898492 (Qiagen); occludin, QT00897302 (Qiagen); claudin1, QT01446067 (Qiagen); Snail1, QT01344679 (Qiagen); β-actin, QT00895692 (Qiagen); and GAPDH, QT00896756 (Qiagen). For mouse genes the following primers were used: Sqstm1, QT00127855 (Qiagen); Lamp1, QT00112385 (Qiagen); Atg5, QT00114751 (Qiagen); Ocln1, QT00111055 (Qiagen); Cdh1, QT00121163 (Qiagen); Hprt1, QT00166768 (Qiagen); β-actin, sense 5′-TGCGTGACATCAAAGAGAAG-3′ and reverse 5′-GATGCCACAGGATTCCATA-3′; GAPDH, sense 5′-TGGACCACCAACTGCTTA-3′ and reverse 5′-GATGCAGGGATGATGTTC-3′. For rat genes the following primers were used: Sqstm1, QT00446488 (Qiagen) and GAPDH, sense 5′- CAACTCCCTCAAGATTGTCAGCAA-3′ and reverse 5′- GGCATGGACTGTGGTCATGA-3′
Western blot analysis
Protein extracts were separated by denaturing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred onto a polyvinylidene difluoride membrane. For immunoblotting, the membranes were probed overnight with relevant antibodies and then incubated with appropriated horseradish peroxidase-linked secondary antibodies. Bound immunoglobulins were revealed by chemiluminescent detection (Immobilon, Millipore). Quantification was conducted using Image J software.
Nuclear extracts
MDCK and MDCK p62 cells cultured in 6-well plates were treated with TGFβ for 72 h. Cellular extracts were prepared as previously described.Citation24 Briefly, cells were washed twice with PBS and then incubated for 5 min in ice-cold hypotonic lysis buffer (20 mM HEPES, pH 7.6, 20% glycerol, 10 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.1% Triton X-100, 50 mM NaF, 25 mM sodium pyrophosphate, 1 mM Na3VO4, 1 mM dithiothreitol, and protease inhibitors). The cells were scraped off and collected. Nuclei were pelleted by centrifugation at 500 g at 4°C for 5 min and suspended in Laemmli buffer.
Co-immunoprecipitation assays
MDCK, NMuMG and NBT-II cells were lysed in 20 mM Tris, pH 7.5, 50 mM NaCl, 0.5% Triton. After centrifugation, the supernatants were incubated overnight at 4°C with the appropriate antibody (anti-HA or anti-p62) and Protein G-Sepharose beads (Sigma). After washing, the immune complexes were eluted at 95°C in 65 mM Tris, pH 6.8, 4% SDS and 1.5% β-mercaptoethanol and analyzed by SDS-PAGE.
To study the interaction between wild type or mutated p62 and smad4, HEK293T cells cultured in 6-well tissue culture plates were transfected with Flag-Smad4 and the different HA- or GFP- constructs (empty, p62WT, p62ΔUBA, p62M404V) for 48 h and treated with 5 ng/mL TGF β for 24 h. The cells were washed with cold PBS and then lysed at 4°C for 2 h in lysis buffer (20 mM Tris pH7.5, 50 mM NaCl, 0.5% Triton, phosphatase inhibitors, protease inhibitors). The lysates were centrifuged at 16,000 g for 30 min at 4°C to remove cell debris. Supernatants were incubated overnight at 4°C with rabbit polyclonal anti-HA or mouse anti-GFP antibodies and Protein G-Sepharose beads (Sigma). Protein G-Sepharose beads were washed 3 times with lysis buffer and 3 times with 20 mM Tris, pH 7.5, 50 mM NaCl. The immune complexes were eluted at 95°C in 65 mM Tris pH 6.8, 4% SDS and 1.5% β-mercaptoethanol and analyzed by SDS-PAGE.
Luciferase assay
Cells were co-transfected either with p(CAGA)9::LuciferaseCitation23 (to assay the Smad activity) or p62-promoter::Luciferase together with tk-renilla-luciferase (Promega) using Lipofectamine 2000 (Invitrogen). Cells were treated with growth factor for the time indicated. Firefly and Renilla luciferase activities were determined using the Dual Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized against Renilla luciferase activity.
Statistical analyses
Data are expressed as a mean + SEM of at least 3 experiments. The statistical significance was assessed by a Student's t-test (* P-value < 0.05, ** P-value < 0.01).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
987619_Supplementary_Materials.zip
Download Zip (1.2 MB)Acknowledgments
We thank T. Yoshimori, M. Tanaka, and A. Delaunay-Moisan for providing us with the GFP-LC3, the p62 constructs, and the HA-Ub constructs, respectively. We are also grateful to W. B’chir and A. Bruhat for providing us with constructs of p62 promoter and to A. Mauviel and R. Derynck for providing expression vector containing cDNA of Flag-Smad4. We also thank V. Nicolas and C. Delomenie for their assistance in confocal microscopy and in qRT-PCR, respectively (IFR141, University Paris Sud 11).
Funding
This work was supported by institutional funding from INSERM and from University Paris-Sud and by grants from the Ligue Nationale Contre le Cancer (LL) and INCa (LL and PC).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/10.1016/j.cell.2009.11.007
- Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 2013; 19:1438-49; PMID:24202396; http://dx.doi.org/10.1038/nm.3336
- Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res 2012; 66:457-62; PMID:22841931; http://dx.doi.org/10.1016/j.phrs.2012.07.004
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487
- Park SH, Choi MJ, Song IK, Choi SY, Nam JO, Kim CD, Lee BH, Park RW, Park KM, Kim YJ, et al. Erythropoietin decreases renal fibrosis in mice with ureteral obstruction: role of inhibiting TGF-β-induced epithelial-to-mesenchymal transition. J Am Soc Nephrol 2007; 18:1497-507; PMID:17389738; http://dx.doi.org/10.1681/ASN.2005080866
- Xie L, Law BK, Aakre ME, Edgerton M, Shyr Y, Bhowmick NA, Moses HL. Transforming growth factor β-regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res 2003; 5:R187-98; PMID:14580254; http://dx.doi.org/10.1186/bcr640
- Morali OG, Delmas V, Moore R, Jeanney C, Thiery JP, Larue L. IGF-II induces rapid β-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene 2001; 20:4942-50; PMID:11526479; http://dx.doi.org/10.1038/sj.onc.1204660
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Lamouille S, Derynck R. Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 2007; 178:437-51; PMID:17646396; http://dx.doi.org/10.1083/jcb.200611146
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/10.1074/jbc.M702824200
- Pankiv S, Lamark T, Bruun JA, Overvatn A, Bjorkoy G, Johansen T. Nucleocytoplasmic shuttling of p62/SQSTM1 and its role in recruitment of nuclear polyubiquitinated proteins to promyelocytic leukemia bodies. J Biol Chem 2010; 285:5941-53; PMID:20018885; http://dx.doi.org/10.1074/jbc.M109.039925
- Heldin CH, Moustakas A. Role of Smads in TGFbeta signaling. Cell Tissue Res 2012; 347:21-36; PMID:21643690; http://dx.doi.org/10.1007/s00441-011-1190-x
- Hocking LJ, Lucas GJ, Daroszewska A, Cundy T, Nicholson GC, Donath J, Walsh JP, Finlayson C, Cavey JR, Ciani B, et al. Novel UBA domain mutations of SQSTM1 in Paget's disease of bone: genotype phenotype correlation, functional analysis, and structural consequences. J Bone Miner Res 2004; 19:1122-7; PMID:15176995; http://dx.doi.org/10.1359/JBMR.0403015
- Lin X, Shang X, Manorek G, Howell SB. Regulation of the epithelial-mesenchymal transition by Claudin-3 and Claudin-4. PloS One 2013; 8:e67496; PMID:23805314
- Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci 2003; 116:1959-67; PMID:12668723; http://dx.doi.org/10.1242/jcs.00389
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2:76-83; PMID:10655586; http://dx.doi.org/10.1038/35000025
- Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, Yan J, Lv X, Chen X, Hu ZW. DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res 2012; 72:3238-50; PMID:22719072; http://dx.doi.org/10.1158/0008-5472.CAN-11-3832
- Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S, Thorburn A, He YY. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A 2014; 111:9241-6; PMID:24927592; http://dx.doi.org/10.1073/pnas.1322913111
- Lander R, Nordin K, LaBonne C. The F-box protein Ppa is a common regulator of core EMT factors Twist, Snail, Slug, and Sip1. J Cell Biol 2011; 194:17-25; PMID:21727196; http://dx.doi.org/10.1083/jcb.201012085
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517-27; PMID:19250912; http://dx.doi.org/10.1016/j.molcel.2009.01.021
- Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2012; 2:397-413; PMID:22860231
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010; 285:22576-91; PMID:20452972; http://dx.doi.org/10.1074/jbc.M110.118976
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 1998; 17:3091-100; PMID:9606191; http://dx.doi.org/10.1093/emboj/17.11.3091
- Wong C, Rougier-Chapman EM, Frederick JP, Datto MB, Liberati NT, Li JM, Wang XF. Smad3-Smad4 and AP-1 complexes synergize in transcriptional activation of the c-Jun promoter by transforming growth factor β. Mol Cell Biol 1999; 19:1821-30; PMID:10022869