
Abstract
T-cell acute lymphoblastic leukemia (T-ALL) is a cancer of immature T cells that exhibits heterogeneity of oncogenic lesions, providing an obstacle for development of more effective and less toxic therapies. Inherited deficiency of ATM, a regulator of the cellular DNA damage response, predisposes young humans and mice to T-ALLs with clonal chromosome translocations. While acquired ATM mutation or deletion occurs in pediatric T-ALLs, the role of somatic ATM alterations in T-ALL pathogenesis remains unknown. We demonstrate here that somatic Atm inactivation in haematopoietic cells starting as these cells differentiate in utero predisposes mice to T-ALL at similar young ages and harboring analogous translocations as germline Atm-deficient mice. However, some T-ALLs from haematopoietic cell specific deletion of Atm were of more mature thymocytes, revealing that the developmental timing and celluar origin of Atm inactivation influences the phenotype of ATM-deficient T-ALLs. Although it has been hypothesized that ATM suppresses cancer by preventing deletion and inactivation of TP53, we find that Atm inhibits T-ALL independent of Tp53 deletion. Finally, we demonstrate that the Cyclin D3 protein that drives immature T cell proliferation is essential for transformation of Atm-deficient thymocytes. Our study establishes a pre-clinical model for pediatric T-ALLs with acquired ATM inactivation and identifies the cell cycle machinery as a therapeutic target for this aggressive childhood T-ALL subtype.
Introduction
T-ALL is a cancer of immature T cells that exhibits genetic heterogeneity among patients with respect to oncogenic lesions that drive disease.Citation1-3 For example, while NOTCH1 mutations are the most common oncogenic lesions in T-ALL, many T-ALLs lacks these or other mutations that activate NOTCH signals.Citation1,3,4 Combinations of cytotoxic and genotoxic chemotherapeutic drugs that damage organelles and DNA and thus kill proliferating cells are used to treat T-ALL.Citation2,5,6 Although most T-ALLs are curable, more effective T-ALL therapies are needed since drug-resistant and relapsed T-ALLs are significant causes of human cancer morbidity and mortality.Citation2,5,7 Less toxic therapies are also needed because survivors of childhood T-ALL develop life-threatening health issues from side effects of chemotherapy.Citation8 Obstacles to developing novel therapies for pediatric T-ALLs include the heterogeneity of oncogenic lesions, evolution of malignancies with genomic instability, and paucity of knowledge of pathological mechanisms and cellular etiology underlying these cancers.Citation2,5,7
Oncogenic lesions arising in immature T cells cause pediatric T-ALL.Citation1–3 T cells mature via an intra-thymic differentiation program that links assembly and expression of T cell receptor (TCR) genes with cellular survival, proliferation, and continued differentiation.Citation9 Bone marrow progenitors seed the thymus and differentiate into CD4−CD8− (DN) thymocytes, which develop into DN2 thymocytes that proliferate and differentiate into non-proliferating DN3 cells.Citation10 Assembly and expression of TCRγ and TCRδ genes in DN3 thymocytes leads to formation of γδ TCRs that signal cellular survival and differentiation into mature γδ T cells.Citation10 In contrast, assembly and expression of functional TCRβ genes in DN3 cells leads to TCRβ chains that pair with pTα chains to form pre-T cell receptors (pre-TCRs), which signal cellular survival, proliferation through transcriptional activation of Cyclin D3, and differentiation into CD4+CD8+ (DP) thymocytes.Citation10,11 Assembly and expression of TCRα genes in DP cells leads to αβ TCRs that signal differentiation into CD4+ or CD8+ (SP) thymocytes that exit the thymus as mature αβ T cells.Citation10 Genetic changes that block development, prevent apoptosis, and drive proliferation of immature T cells cause pediatric T-ALL.Citation1–3 While all childhood T-ALLs contain mutations that activate oncogenes or inactivate tumor suppressor genes, half of these cancers harbor genomic instability including TCR translocations that target and activate expression of oncogenes.Citation1,2 Such genomic instability arises from aberrant repair of DNA double strand breaks (DSBs) induced during TCR recombination in thymocytes and/or DNA replication in thymocytes or in embryronic cells that differentiate into thymocytes.Citation12
Immature T cells exhibit a response to DSBs that coordinates DNA repair with activation of cell cycle checkpoints to inhibit formation of oncogenic translocations.Citation12 The Ataxia Telangiectasia mutated (ATM) protein kinase is the master regulator of this response.Citation13 DSBs activate ATM, which induces phosphorylation of proteins to control their function.Citation13 ATM facilitates DSB repair via phosphorylation of the H2AX histone and activates the G1/S checkpoint via phosphorylation of the TP53 transcription factor.Citation14-17 ATM deficiency causes immunodeficiency due to lymphopenia from impaired repair of DSBs induced during TCR recombination and TCRβ-dependent thymocyte proliferation.Citation18-25 Due to combined defects in DSB repair and cell cycle checkpoints, ATM inactivation also predisposes to T-ALLs with clonal TCRα/δ translocations.Citation22-26
Although inherited ATM deficiency predisposes children and young mice to T-ALL, it remains unproven whether somatic inactivation of ATM causes pediatric T-ALL. Acquired ATM mutations or homozygous deletions of the 11q23 cytogenetic region spanning ATM have been found in 10–25% of human pediatric T-ALLs and more frequently in drug-resistant and relapsed disease.Citation27-29 However, whether such sporadic ATM mutations are oncogenic and if ATM is a relevant 11q23 tumor suppressor gene are not known. In addition, some ATM alterations found in pediatric T-ALLs have been argued to be germline variants of no biological significance.Citation30 The developmental timing and cellular origin of acquired ATM mutations that cause childhood T-ALL have not been reported. ATM suppresses genomic instability in embryonic cells prior to differentiation of haematopoietic cells.Citation31 In addition, germline versus somatic haematopoietic cell specific inactivation of ATM substrates has distinct effects on T-ALL with respect to age-of-onset, stage of development, and ocogenic genomic lesions.Citation32,33 Therefore, it is important to determine whether acquired inactivation of ATM in haematopoietic cells can cause pediatric T-ALL. We demonstrate here that somatic Atm deletion in haematopoietic cells starting as these cells arise in utero predisposes mice to T-ALL at similar young ages and harboring analogous translocations as germline Atm-deficient mice. However, some T-ALLs from haematopoietic cell specific deletion of Atm were of more mature thymocytes, revealing that the developmental timing and cellular origin of Atm inactivation influences the phenotype of ATM-deficient T-ALLs. Although it has been hypothesized that ATM suppresses cancer by preventing deletion and inactivation of TP53,Citation34,35 we find that Atm inhibits T-ALL independent of Tp53 deletion. Finally, we demonstrate that the Cyclin D3 protein that drives immature T cell proliferation is essential for malignant transformation of Atm-deficient thymocytes. Our study establishes a pre-clinical model for pediatric T-ALLs arising from acquired ATM inactivation and identifies the cell cycle machinery as a therapeutic target for this aggressive childhood T-ALL subtype.
Results
Deletion of Atm in haematopoietic cells predisposes mice to T-ALL
To determine if somatic inactivation of ATM in haematopoietic cells causes T-ALL, we created and analyzed Vav-cre+/−Atmflox/flox (VA) mice since Vav-cre deletes “floxed” genes in haematopoietic cells as these cells begin to develop in fetal livers at ∼10 d of embryogenesis.Citation36-38 Germline Atm-deficient (Atm−/−) mice contain fewer thymocytes than wild-type (WT) mice due mainly to impaired DP-to-SP thymocyte development (-).Citation20 We found a similar phenotype in VA mice (). We aged a cohort of 27 VA mice to assess their cancer predisposition. They survived cancer-free between 53–183 d with a median age of 90 d (), similar to the cancer-free survival of Atm−/− mice and Atm−/− mice expressing an Lck-cre transgene in thymocytes.Citation21,24,25,39,40 All cohort VA mice were euthanized due to thymic malignancies, except for 2 that developed masses of cells in their spleens and/or lymph nodes (). Of the 25 mice that developed thymic cancers, 9 had enlarged spleens, lymph nodes, and/or livers; while the 2 that succumbed to peripheral cancers contained large cell masses in their thymuses. These data indicate that somatic deletion of Atm in haematopoietic cells predisposes young mice to fatal thymic malignancies.
Table 1. Analysis of VA tumor cohort
Figure 1. Inactivation of Atm in HSCs Impairs αβ (T)Cell Development. (A) Representative flow cytometry analysis of CD4 and CD8 expression in thymocytes isolated from WT, Atm−/−, and VA mice. The frequencies of cells in the DN, DP, CD4+ SP, and CD8+ SP gates are indicated. (B) Graph showing the average numbers of total thymocytes from WT (n = 8), Atm−/− (n = 4), and VA (n = 10) mice. Error bars are SD. (C) Graph showing the average numbers of DN, DP, CD4+ SP, and CD8+ SP total thymocytes from WT (n = 8), Atm−/− (n = 4), and VA (n = 4) mice. Error bars are SD. These experiments were each independently performed more than 3 times.
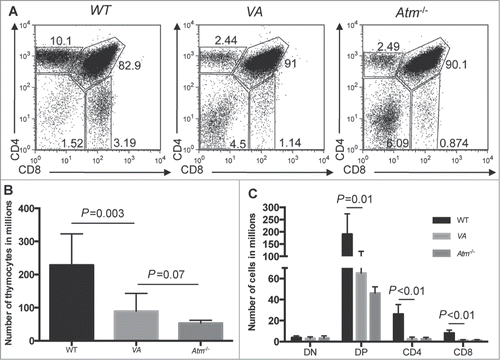
Figure 2. Somatic Inactivation of Atm in HSCs Predisposes Mice to T-ALLs with Oncogenic Translocations. (A) Kaplain-Meier curve depicting the cancer-free survival of 27 cohort VA mice. All cohort mice succumbed to thymic malignancies except for 2 mice that succumbed to cancers in peripheral lymphoid tissues (indicated by asterisks). (B and C) Flow cytometry analysis of VA cancers no. 408 (B) or 656 (C) showing surface expression of TCRβ or CD4 and CD8. Gates were drawn using appropriate non-malignant cells. Shown are the percentages of cells in each gate. (D) Top, schematic of the Tcrβ locus showing locations of the indicated Tcrβ segments, HindIII restriction sites (H3), and 3’Jβ1 and 3’Jβ2 probes. Bottom, Southern blots of HindIII-digested DNA from the indicated VA thymic cancers (and VA splenic cancer 480) or from the kidney of a WT mouse using the 3’Jβ1 or 3’Jβ2 probe. Germline (GL) bands are indicated. The membrane was hybridized with the 3’Jβ1 probe and then stripped and hybridized with the 3’Jβ2 probe, revealing which VA cancers lack 3’Jβ1-hybridizing band(s) due to Vβ-to-Dβ2-Jβ2 rearrangements on both alleles. (E) SKY analysis of a metaphase from VA T-ALLs no. 281 with the clonal translocations indicated. (F) Top, schematic of the Tcrα locus showing locations of indicated Tcrα segments, HindIII restriction sites (H3), and 5’Jα probe. Bottom, Southern blot of HindIII-digested DNA from the indicated VA thymic cancers (and VA splenic cancer 480) or kidney of a WT mouse using the 5’Jα probe. The germline band is indicated. The membrane from D was stripped of the 3’Jβ2 probe and then probed with the 5’Jα probe, revealing which VA cancers lacked 5’Jα-hybridizing band(s) due to Vα-to-Jα rearrangements on both alleles.
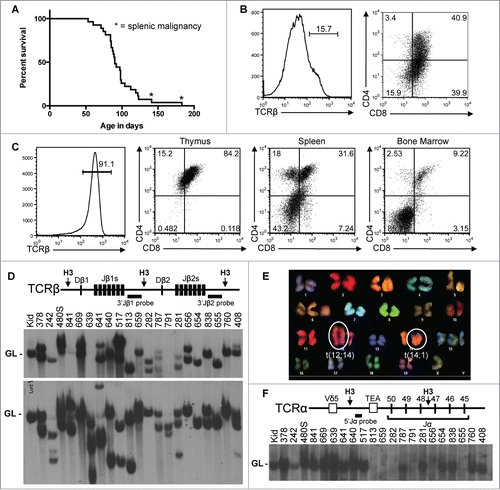
We next analyzed VA malignancies by flow cytometry with antibodies that recognize markers of specific lymphocyte lineages and developmental stages. We assayed expression of the T cell specific TCRβ, CD4, and CD8 proteins and the B cell specific B220, CD43, and IgM proteins. We simplified our classification by denoting a thymic cancer as positive for a marker when >50 % of cells fell in a positive gate established from non-malignant cells of WT mice. Of the 20 VA thymic cancers assayed, 19 were TCRβ− and one was TCRβ+ (; ). Of the 19 TCRβ− malignancies, 8 were CD4+CD8+, 6 were CD8+, one was CD4+, and the others had subpopulations with varying CD4 and/or CD8 expression (; ). The one TCRβ+ malignancy was CD4+CD8+ (), indicating it is an immature αβ T cell cancer. Most mice that succumbed to thymic cancers had cells in their spleens and bone marrow with identical TCRβ, CD4, and CD8 expression as their malignant thymocytes (; data not shown), suggesting dissemination of a single cancer. Analysis of the 2 VA peripheral malignancies showed that one was an immature αβ T cell cancer and the other a B lineage lymphoma (). These data indicate that inactivation of Atm in haematopoietic cells predisposes young mice to disseminated immature αβ T cell cancers from various stages of αβ T cell development.
We also assessed clonality of VA cancers. For normal DN thymocytes to survive, proliferate, and differentiate, Tcrβ gene rearrangements must occur on one allele.Citation9 Thus, we conducted Southern blotting with 3’Jβ1 and 3’Jβ2 probes on HindIII-digested DNA (). Tcrβ rearrangements delete or alter the size of the 3’Jβ1-hybridizing band and alter the size of the 3’Jβ2-hybridizing band.Citation41 We found that 17 of 21 VA cancers analyzed contained one or 2 rearranged Tcrβ alleles and therefore arose from transformation of a single cell (). The other 4 had 3 Tcrβ rearrangements (), indicating they arose from 2 cells or one cell that continued Tcrβ recombination. In contrast, 3 VA cancers had one or 2 Tcrβ rearrangements (), but they also expressed various levels of CD4 and/or CD8 (), indicating that they arose from a single cell and evolved subpopulations that differentially express CD4 and CD8. These data indicate that deletion of Atm in haematopoietic cells predisposes mice to predominantly clonal immature αβ T cell cancers. Based on our analysis of VA cancers, we conclude that somatic inactivation of ATM causes pediatric T-ALL.
The cellular origin of Atm inactivation influences developmental stage but not genomic instability of T-ALLs
Analogous to humans with inherited ATM deficiency that develop fatal αβ T cell cancers harboring translocations that fuse TCRα segments near oncogenes,Citation22 Atm−/− mice were thought to develop T-ALLs with oncogenic translocations created by aberrant Tcrα recombination.Citation26 Yet, we found Atm−/− T-ALLs harbor clonal Tcrδ translocations and arise from thymocytes that have not initiated Tcrα rearrangements.Citation40 Consistent with our data, others showed that non-malignant αβ T lineage cells of Atm−/− mice harbor translocations formed by aberrant recombination of Tcrδ and/or immunoglobulin heavy chain (Igh) loci in DN thymocytes.Citation42,43 The most frequent of these lesions in non-malignant and malignant cells is a t(12;14) translocation in which Tcrδ genes on chromosome 14 are fused to chromosome 12 sequences, deleting the Bcl11b haploinsufficient tumor suppressor gene.Citation40,42 Other common lesions are t(14;15) and t(12;15) translocations in which Tcrδ or Igh loci on chromosomes 14 and 12, respectively, are fused adjacent to the c-Myc oncogene on chromosome 15.Citation26,39,40 To identify translocations caused by somatic inactivation of Atm, we conducted Spectral Karyotyping (SKY) on 11 VA T-ALLs. SKY is a molecular cytogenetic approach used to visualize all chromosomes and identify translocations.Citation44 Nine T-ALLs harbored a clonal translocation involving chromosomes 12 and/or 14 with 5 containing the t(12;14) translocation or Atm−/− T-ALLs (; ). Of the other 2 VA T-ALLs, one had a clonal t(1;X) translocation; the other one, which was the peripheral T-ALL, lacked a clonal translocation yet had non-clonal translocations (). Analogous to Atm−/− T-ALLs,Citation26 all VA T-ALLs that contained clonal translocations carried non-clonal translocations (). To test if VA T-ALLs arise from cells that have initiated Tcrα recombination, we conducted Southern blots with a 5’Jα probe on HindIII-digested DNA of the 21 VA cancers analyzed for Tcrβ recombination (). Tcrα recombination occurs through Tcrα rearrangements on both alleles,Citation45 causing deletion of sequences to which the 5’Jα probe hybridizes.Citation46 We found that 7 of the 21 VA T-ALLs lacked a 5’Jα-hybridizing band (), consistent with their arising from cells that had initiated Tcrα recombination. Three VA T-ALLs that harbored clonal t(12;14) translocations lacked Tcrα rearrangements, consistent with their translocations arising from Tcrδ recombination errors. These data indicate that Atm deletion in haematopoietic cells predisposes young mice to T-ALLs with similar translocations, but frequently from more mature developing T cells, than germline Atm inactivation.
Table 2. VA SKY summary
Tp53 heterozygosity accelerates malignant transformation of Atm-deficient thymocytes independent of Tp53 deletion
ATM has been hypothesized to suppress transformation by preventing translocations and deletions that promote loss-of-heterozygosity (LOH) and inactivation of TP53.Citation34,35 Yet, Tp53 deletions are rare in Atm−/− T-ALLs,Citation40 suggesting that preventing Tp53 inactivation by deletion is not the major mechanism by which ATM protects immature αβ T cells from transformation. To rigorously test this notion, we created and analyzed a cohort of 28 Vav-cre+/−Atmflox/floxp53WT/flox (VAp) mice. They survived cancer-free between 55–122 d with a median age of mortality of 78 d (), which is 12 d younger than that of VA mice (; p = 0.003). All VAp mice were euthanized due to thymic cancers, except for one that was euthanized because of an enlarged spleen (). Of the 22 VAp mice with thymic cancers analyzed, 7 had cancer cells disseminated to their spleens; while the mouse that had a large spleen also had cancer cells in its thymus. These results show that bi-allelic deletion of Atm and mono-allelic deletion of Tp53 initiating in haematopoietic cells predisposes mice to more rapid onset of fatal thymic malignancies than inactivation of Atm alone.
Figure 3. Atm and Tp53 Cooperate to Suppress T-ALL. (A) Kaplan-Meier curves depicting the cancer-free survival of 28 cohort VAp mice and the 27 cohort VA mice from . All VAp mice succumbed to thymic cancers. (B) Flow cytometry analysis of VAp T-ALL no. 621 showing expression of TCRβ or CD4 and CD8. Gates were drawn using appropriate non-malignant cells. Shown are the percentages of cells in each gate. (C) Top, schematic of the Tcrβ locus showing locations of indicated Tcrβ segments, HindIII restriction sites (H3), and 3’Jβ1 and 3’Jβ2 probes. Bottom, Southern blots of HindIII-digested DNA from the indicated VAp T-ALLs or the kidney of a WT mouse with the 3’Jβ1 or 3’Jβ2 probe. Germline (GL) bands are indicated. The membrane was first hybridized with the 3’Jβ1 probe and then stripped and hybridized with the 3’Jβ2 probe, revealing which VAp cancers lack 3’Jβ1-hybridizing band(s) due to Vβ-to-Dβ2-Jβ2 rearrangements on both alleles. (D) SKY analysis of a metaphase from VAp T-ALL no. 562 with clonal translocations indicated. (E) Top, schematic of the Tcrα locus showing locations of indicated Tcrα segments, HindIII restriction sites (H3), and 5’Jα probe. Bottom, Southern blot of HindIII-digested DNA from the indicated VAp thymic cancers or the kidney of a WT mouse using the 5’Jα probe. The germline (GL) band is indicated. The membrane from C was stripped of the 3’Jβ2 probe and then probed with the 5’Jα probe. (F) Top, schematic of the Tp53 locus showing locations of the Tp53 exons, BglII sites (B2), loxP sites, and 5’Tp53 probe on Tp53WT, Tp53flox, and Tp53Δ alleles. The sizes of BglII fragments from each allele are depicted. Bottom, Southern blots of BglII-digested DNA from the indicated VAp T-ALLs using the 5’Tp53 probe. Bands corresponding to Tp53WT, Tp53flox, and Tp53Δ alleles are indicated. Asterisks indicate tumors with LOH at Tp53.
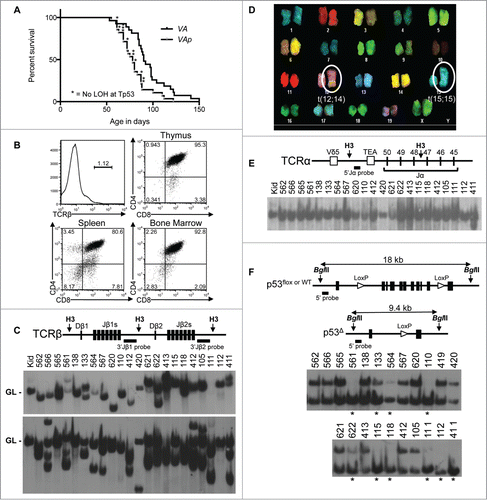
We characterized VAp cancers to determine their lineage, developmental stage, dissemination, clonality, and karyotype. All 21 analyzed by flow cytometry were TCRβ− (; ). Of these, 7 were CD4+CD8+, 2 were CD8+, and the rest had subpopulations with varying expression of CD4 and/or CD8 (, ). The one splenic cancer expressed IgM and B220 (), revealing that it is B cell malignancy. Most mice that succumbed to thymic cancers had cells in their spleens and bone marrow with identical TCRβ, CD4, and CD8 expression as their malignant thymocytes (; data not shown), suggesting dissemination of a single cancer. Analysis of Tcrβ rearrangements showed that 16 of 21 VAp thymic cancers had one or 2 rearranged Tcrβ alleles and thus arose from a single cell (). The other 5 contained 3 or 4 Tcrβ rearrangements (), indicating that arose from 2 thymocytes or one cell that continued Tcrβ recombination. All 7 VAp thymic cancers analyzed by SKY harbored a clonal translocation involving chromosomes 12 and/or 14 with 4 containing the t(12;14) translocation found in Atm−/− and VA T-ALLs (; ). Analysis of Tcrα rearrangements revealed that all 22 VAp thymic cancers retained a 5’Jα-hybridizing band () and thus arose from cells that had not started Tcrα recombination. One of these VAp cancers also contained a non-germline 5’Jα-hybridizing band indicative of aberrant Tcrδ or Tcrα rearrangements, which occur in Atm−/− thymocytes.Citation47,48 These data indicate that bi-allelic Atm deletion and mono-allelic Tp53 deletion in haematopoietic cells predisposes young mice to T-ALLs with similar translocations but often from thymocytes of a less advanced developmental stage as compared to haematopoietic cell specific inactivation of Atm alone.
Table 3. Analysis of VAp tumor cohort
Table 4. VAp SKY summary
To determine whether Tp53 LOH from deletion of the wild-type copy of Tp53 is the mechanism by which Tp53 heterozygosity accelerates transformation of Atm-deficient thymocytes, we analyzed the Tp53 locus in VAp T-ALLs. We conducted Southern blots with a 5’Tp53 probe on BglII-digested DNA from the same 22 VAp T-ALLs analyzed above. This probe hybridizes to an 18 kb BglII fragment from Tp53WT and Tp53flox alleles and to a 9.6 kb BglII fragment from Cre-deleted Tp53flox (Tp53Δ) alleles (). We saw similar intensities of the Tp53WT/flox and Tp53Δ band in 12 cancers (). In contrast, we detected the Tp53Δ band and complete or partial loss of the Tp53WT/flox band in the other 10 malignancies (). The less intense Tp53WT/flox bands in 3 of these VAp T-ALLs arise from contaminating non-malignant cells, as reflected in Southerns for Tcrβ rearrangements (). The 2 T-ALLs with Tp53 LOH analyzed by SKY had clonal translocations of chromosome 11 where Tp53 resides, consistent with deletion of Tp53WT alleles by these lesions. Yet, we had insufficient material to confirm this by cytogenetics. We did not observe loss of the Tp53WT/flox band and appearance of a new band in any VAp T-ALLs tumors, indicative of internal Tp53 deletions. We also did not see this pattern with a 3’Tp53 probe (data not shown). While we cannot rule out deletions spanning the entire Tp53 locus in VAp T-ALLs with Tp53 LOH, there was no correlation between Tp53 LOH and age-of-onset of T-ALL (). Therefore, we conclude that protecting haematopoietic cells from Tp53 deletion is not a major mechanism by which ATM suppresses malignant transformation of immature αβ T cells.
Cyclin D3 drives transformation of Atm-deficient thymocytes
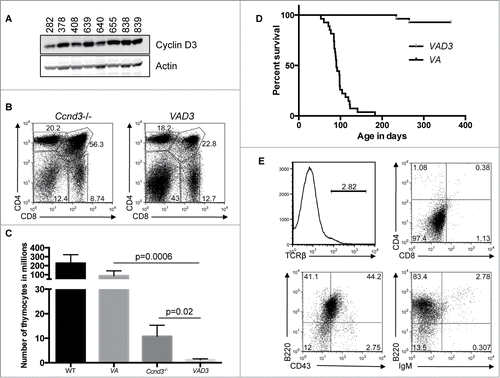
Expression of CD4 and/or CD8 on VA T-ALLs suggests that transformation of Atm-deficient thymocytes depends on sustained proliferation of TCRβ-selected thymocytes. TCRβ drives thymocyte proliferation through transcriptional up-regulation of Ccnd3, which encodes Cyclin D3.Citation11 Cyclin D3 binds and activates cyclin dependent kinase (cdk) 4 and 6 to drive DN cells through multiple cell cycles as they differentiate into non-cycling DP and SP thymocytes, in which Ccnd3 is no longer transcribed and Cyclin D3 is not expressed.Citation11 To determine if VA T-ALLs arise from sustained Cyclin D3 expression, we first conducted Western blots for Cyclin D3 in VA T-ALLs. We detected Cyclin D3 in all VA T-ALLs (). Thus, we made and analyzed Vav-cre+/−Atmflox/floxCcnd3−/− (VAD3) mice. Ccnd3−/− mice exhibit ∼fold20- fewer thymocytes as compared to WT mice because of impaired Tcrβ-mediated proliferation during DN-to-DP differentiation ().Citation11 We observed ∼fold10- and ∼75-fold fewer total thymocytes in VAD3 mice relative to Ccnd3−/− and VA mice, respectively (). These data show that TCRβ-dependent thymocyte expansion during DN-to-DP differentiation is even more substantially impaired in the absence of both Atm and Ccnd3 than either alone. We created and aged a cohort of 28 VAD3 mice for one year to assess their cancer predisposition. Two of these mice succumbed to thymic malignancies around 250 days-of-age, well after 190 days-of-age when VA mice develop T-ALL (; P < 0.0001). The one VAD3 cancer analyzed by flow cytometry expressed B cell markers indicating that it was a B lineage malignancy (). All other VAD3 mice survived cancer free during the one-year study (). Consequently, our data demonstrate that Cyclin D3 is required for transformation of Atm-deficient thymocytes.
Figure 4. Ccnd3 Drives Transformation of Atm-deficient Thymocytes. (A) Western blot analysis of Ccnd3 and actin expression in the indicated VA T-ALLs. (B) Representative flow cytometry analysis of CD4 and CD8 expression on thymocytes from Ccnd3−/− and VAD3 mice. Indicated on the plots are the frequencies of cells in the DN, DP, CD4+ SP, and CD8+ SP gates. (C) Graph showing the average numbers of total thymocytes from WT (n = 8), VA (n = 10), Ccnd3−/− (n = 5), and VAD3 (n = 3) mice. Error bars are SD. (B and C). This analysis was independently performed more than 3 times. The WT and VA data are also shown in . (D) Kaplan-Meier curves depicting the cancer-free survival of the 28 cohort VAD3 mice and the 27 cohort VA mice from . (E) Flow cytometry analysis of TCRβ, CD4 and CD8, B220 and CD43, or B220 and IgM on VAD3 tumor 676. Gates were drawn using normal thymocytes or splenocytes. Shown are the percentages of cells in each gate.
Discussion
We have analyzed VA mice with tissue-specific Atm deletion in haematopoietic cells to determine whether acquired inactivation of ATM can cause pediatric T-ALL. These mice succumbed to clonal disseminated immature αβ T cell cancers of various developmental stages. Based on our analysis of VA malignancies, we conclude that somatic inactivation of ATM in haematopoietic cells contributes to the pathogenesis of pediatric T-ALL. The similar age-of-onset and clonal translocations of T-ALLs arising in VA, Atm−/−, and Lck-cre:Atm−/− miceCitation26,39,40 reveals that ATM function in haematopoietic cells is the predominant driver of T-ALL arising from inherited ATM deficiency. However, in contrast to Atm−/− T-ALLs that arise from thymocytes that have not initiated Tcrα gene recombination,Citation40 one-third of VA T-ALLs had Tcrα rearrangements and arose from thymocytes of a more advanced stage of αβ T cell development. This difference indicates that loss of ATM function in embryonic cells that differentiate into haematopoietic cells can influence the phenotype of T-ALLs caused by inherited ATM deficiency. The acquisition of oncogenic lesions in embryonic ATM-deficient cells prior to their differentiation into haematopoietic cells might decrease the number of such oncogenic lesions in haematopoietic cells necessary for transformation of thymocytes, leading to T-ALLs from cells at earlier stages of αβ T cell development. Alternatively, Atm deletion in a subset of haematopoietic cells that have differentiated beyond the haematopoietic stem cell stage might result in malignant transformation of thymocytes at a later αβ T cell developmental stage than germline Atm inactivation. Distinguishing between these possibilities will require generation and analysis of mice with T lineage specific deletion of Atm initiating at various developmental stages. Although our study indicates that acquired Atm loss in haematopoietic cells starting at day 10 of embryogenesis predisposes young mice to T-ALL, additional models are required to determine whether acquired inactivation of Atm in haematopoietic cells of adult mice also causes T-ALL.
We next analyzed VAp mice with bi-allelic Atm deletion and mono-allelic Tp53 deletion in haematopoietic cells to determine whether ATM protects thymocytes from malignant transformation by preventing Tp53 LOH. VAp mice succumbed to T-ALLs at an earlier median age than VA mice, indicating that Tp53 heterozygosity accelerates transformation of Atm-deficient thymocytes. In contrast to VA T-ALLs that often contain Tcrα rearrangements, all VAp T-ALLs were from thymocytes that had not initiated Tcrα recombination. This difference indicates that both Tp53 copies are needed to fully protect Atm-deficient thymocytes from oncogenic lesions. The wild-type Tp53 allele was retained in half of the VAp T-ALLs analyzed. Tp53 heterozygosity has been shown to accelerate transformation from haploinsufficient Tp53 function or deletion of wild-type Tp53 loci.Citation49-51 We were not able to distinguish loss of wild-type Tp53 loci by translocation or deletion vs. chromosome recombination or disjunction. We also did not assay for Tp53 mutations in VAp T-ALLs that retained the wild-type Tp53 allele since this was beyond the scope of our study. Regardless, the similar frequencies for retention and loss of wild-type Tp53 alleles in VAp T-ALLs indicates that protecting cells from allelic imbalances that generate Tp53 LOH is not a major mechanism by which ATM suppresses malignant transformation of immature αβ T cells.
We also analyzed VAD3 mice with Atm deletion in haematopoietic cells of mice lacking Cyclin D3 to assess the role of TCRβ-mediated proliferation in transforming Atm-deficient thymocytes. VAD3 mice exhibited substantially more impaired cellular expansion during DN-to-DP development than VA or Ccnd3−/− mice. In contrast to VA mice, VAD3 mice did not develop T-ALL, revealing that malignant transformation of Atm-deficient thymocytes depends on Cyclin D3 expression and therefore TCRβ-mediated proliferation. TCRβ-driven cellular proliferation during DN-to-DP thymocyte expansion could transform Atm-deficient cells by creating oncogenic lesions via not mutually exclusive mechanisms. A greater number of thymocytes might increase the cellular population available to acquire oncogenic lesions. DNA replication errors may result in genomic instability and/or mutations that sustain Cyclin D3 expression in DP thymocytes, driving their proliferation and providing more opportunity for DNA replication errors to cause additional oncogenic lesions. Finally, progression of G1 phase DN cells with un-repaired RAG DSBs into S phase could promote formation of oncogenic Tcrδ or Igh translocations.
Our finding that Cyclin D3 is necessary to transform Atm-deficient thymocytes has important implications for pediatric T-ALL patients whose cancer cells harbor inactivating ATM mutations. Cyclin D3 drives cellular proliferation through activating the Cdk4 and Cdk6 kinases.Citation52 Two groups recently showed that a CDK4/CDK6 inhibitor holds promise as a new therapy for T-ALLs driven by the NOTCH1 oncogene.Citation53,54 They found that CDK4/CDK6 inhibition hinders proliferation and induces apoptosis of NOTCH1-driven T-ALLs, and antagonizes ability of NOTCH1 to promote disease over 2 months in chimeric mouse models of human T-ALL.Citation53,54 The rationale for this work was the 2 groups’ prior data that expression of the NOTCH1 oncogene in thymocytes of wild-type but not Ccnd3−/− mice caused T-ALL at young ages.Citation11 They also demonstrated that Cyclin D3 is necessary for initiation of T-ALLs that need pre-TCR signals to develop like those caused by NOTCH1, but is not required for T-ALLs that arise independent of pre-TCR signals like Tp53−/− mouse T-ALLs.Citation11 One prediction of these studies is that Cyclin D3 is not important for the development of pediatric T-ALLs arising from ATM inactivation since Atm−/− T-ALLs arise independent of pre-TCR signaling.Citation24,25 However, our data reveal that Cyclin D3 is essential for the pathogenesis of VA T-ALLs, which lack oncogenic Notch1 mutations (data not shown). In addition to the fundamental importance of this finding for understanding the etiology of ATM-deficient pediatric T-ALLs, our data suggest that CDK4/CDK6 inhibition could be an effective and non-toxic therapy for pediatric T-ALLs harboring bi-allelic ATM inactivation. Therefore, our study should have a high clinical impact by opening a therapeutic avenue for ATM-deficient childhood T-ALLs that was closed by previous publications. In this context, VA mice provide a powerful pre-clinical model to evaluate the long-term efficacy of CDK4/CDK6 inhibition as a more effective and less toxic therapy for this aggressive childhood T-ALL subtype.
Materials and Methods
Mice
Vav-cre,Citation36 Atmflox/flox,Citation31 p53flox/flox,Citation55 and Ccnd3−/− 11 mice of a mixed 129SvEv and C57BL/6 were bred to create animals. Vav-cre+/− males were bred with Atmflox/flox females to generate Vav-cre+/−Atmflox/WT males that were bred with Atmflox/flox females to create Vav-cre+/−Atmflox/flox males, which were bred with Atmflox/flox females to establish experimental Vav-cre+/−Atmflox/flox mice and control Atmflox/flox mice. We used similar breeding strategies to make experimental Vav-cre+/−Atmflox/floxp53flox/WT mice, as well as to create experimental Vav-cre+/−Atmflox/floxCcnd3−/− and control Atmflox/floxCcnd3−/− mice. Mice of both sexes and normal weight were used for experiments and controls. Expect for cohort mice, all mice were analyzed between 4–6 weeks of age. Cohort mice were monitored and euthanized on signs of distress. These studies were conducted under the approval and monitoring of the Children's Hospital of Philadelphia IACUC.
Flow Cytometry
Cells were stained in PBS with 3% FBS using APC-anti-TCRβ, FITC-anti-CD8, PE-anti-CD4, PE-Cy7-anti-B220, FITC-anti-CD43, and APC-anti-IgM antibodies (BD PharMingen). Data were collected using a FACSCalibur with CellQuest software (BD Biosciences) and analyzed by FlowJo software (Tree Star). Statistics were performed in Microsoft Excel or Graphpad Prism 5 using a 2-tailed unpaired Student's t-test.
Kaplan-meier analysis
Curves were generated in Prism 5 and compared using the log-rank (Mantel-Cox) test.
Southern blotting
DNA (20 μg) was digested with 100 units of indicated enzymes (New England Biolabs), separated on 0.8% TAE gels, transferred onto Zeta-probe (BioRad), and hybridized withCitation32P-labeled 3’Jβ1, 3’Jβ2, 5’Jα, 5’Tp53 or 3’Tp53 probes as described.Citation41,46,49
Spectral karyotyping
Metaphase spreads were made as described.Citation32,33 Spectral karyotyping was performed per instructions (Applied Spectral Imaging, ASI). Slides were examined with a BX61 microscope (600x magnification) from Olympus controlled by a LAMBDA 10-B Smart Shutter (Sutter Instrument). Images were captured using a LAMBDA LS light source (Sutter) and a COOL-1300QS camera (ASI), and analyzed by Case Data Manager Version 5.5 (ASI).
Western blots
Lysates were prepared in 0.1% Tween 20 buffer and sonicated. Samples were cleared by centrifugation, denatured by boiling in Laemmli buffer, then run on Tris-glycine gels (Invitrogen), and transferred to Immobilon-FL membrane (Millipore). Membranes were blocked with Odyssey blocking buffer (Licor), incubated with anti-Ccnd3 antibody (C-16, Santa Cruz) in 0.5X Odyssey block, washed, and incubated with IRDye secondary antibody (Licor). Blots were visualized with an infrared scanner (Licor). Membranes were stripped and probed using an anti-actin antibody (AV40173, Sigma).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the University of Pennsylvania Cancer Center Research Training Program 5T32CA009615–22 (LAE); University of Pennsylvania Training Grant GM-07229 and F31 pre-doctoral fellowship CA177092 (AD); Department of Pathology and Laboratory Medicine, Children's Hospital of Philadelphia, a WW Smith Charitable Trust Grant, a Leukemia and Lymphoma Society Scholar Award, and NIH R01 Grants CA125195 and CA136470 (CHB).
References
- Paganin M, Ferrando A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev 2011; 25:83-90; PMID:20965628; http://dx.doi.org/10.1016/j.blre.2010.09.004
- Pui CH, Robison LL, Look AT. Acute lympho-blastic leukaemia. Lancet 2008; 371:1030-43; PMID:18358930; http://dx.doi.org/10.1016/S0140-6736(08)60457-2
- Aifantis I, Raetz E, Buonamici S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat Rev Immunol 2008; 8:380-90; PMID:18421304; http://dx.doi.org/10.1038/nri2304
- Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer 2006; 6:347-59; PMID:16612405; http://dx.doi.org/10.1038/nrc1880
- Ko RH, Ji L, Barnette P, Bostrom B, Hutchinson R, Raetz E, Seibel NL, Twist CJ, Eckroth E, Sposto R, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol 2010; 28:648-54; PMID:19841326; http://dx.doi.org/10.1200/JCO.2009.22.2950
- Maloney KW, Giller R, Hunger SP. Recent advances in the understanding and treatment of pediatric leukemias. Adv Pediatr 2012; 59:329-58; PMID:22789585; http://dx.doi.org/10.1016/j.yapd.2012.04.010
- Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, Smith FO, Reaman GH. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 2010; 28:2625-34; PMID:20404250; http://dx.doi.org/10.1200/JCO.2009.27.0421
- Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, Friedman DL, Marina N, Hobbie W, Kadan-Lottick NS, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med 2006; 355:1572-82; PMID:17035650; http://dx.doi.org/10.1056/NEJMsa060185
- Brady BL, Steinel NC, Bassing CH. Antigen receptor allelic exclusion: an update and reappraisal. J Immunol 2010; 185:3801-8; http://dx.doi.org/10.4049/jimmunol.1001158
- Bhandoola A, von Boehmer H, Petrie HT, Zuniga-Pflucker JC. Commitment and developmental potential of extrathymic and intrathymic T cell precursors: plenty to choose from. Immunity 2007; 26:678-89; PMID:17582341; http://dx.doi.org/10.1016/j.immuni.2007.05.009
- Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003; 4:451-61; PMID:14706337; http://dx.doi.org/10.1016/S1535-6108(03)00301-5
- Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair 2004; 3:781-96; PMID:15279764; http://dx.doi.org/10.1016/j.dnarep.2004.06.001
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197-210; http://dx.doi.org/10.1038/nrm3546
- Morgan SE, Kastan MB. p53 and ATM: cell cycle, cell death, and cancer. Adv Cancer Res 1997; 71:1-25; PMID:9111862; http://dx.doi.org/10.1016/S0065-230X(08)60095-0
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, et al. H2AX Prevents DNA Breaks from Progressing to Chromosome Breaks and Translocations. Mol Cell 2006; 21:201-14; PMID:16427010; http://dx.doi.org/10.1016/j.molcel.2006.01.005
- Yin B, Savic V, Juntilla MM, Bredemeyer AL, Yang-Iott KS, Helmink BA, Koretzky GA, Sleckman BP, Bassing CH. Histone H2AX stabilizes broken DNA strands to suppress chromosome breaks and translocations during V(D)J recombination. J Exp Med 2009; 206:2625-39; PMID:19887394; http://dx.doi.org/10.1084/jem.20091320
- Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, Eisenreich TR, Chen J, Dickins RA, Lowe SW, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature 2006; 440:105-9; PMID:16400328; http://dx.doi.org/10.1038/nature04495
- Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A 1996; 93:13084-9; PMID:8917548; http://dx.doi.org/10.1073/pnas.93.23.13084
- Barlow C, Brown KD, Deng CX, Tagle DA, Wynshaw-Boris A. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nature genetics 1997; 17:453-6; PMID:9398849; http://dx.doi.org/10.1038/ng1297-453
- Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci U S A 2000; 97:3336-41; PMID:10716718; http://dx.doi.org/10.1073/pnas.97.7.3336
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 1996; 10:2411-22; PMID:8843194; http://dx.doi.org/10.1101/gad.10.19.2411
- Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood 1996; 87:423-38; PMID:8555463
- Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006; 442:466-70; PMID:16799570; http://dx.doi.org/10.1038/nature04866
- Petiniot LK, Weaver Z, Barlow C, Shen R, Eckhaus M, Steinberg SM, Ried T, Wynshaw-Boris A, Hodes RJ. Recombinase-activating gene (RAG) 2-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Proc Natl Acad Sci U S A 2000; 97:6664-9; PMID:10841564; http://dx.doi.org/10.1073/pnas.97.12.6664
- Petiniot LK, Weaver Z, Vacchio M, Shen R, Wangsa D, Barlow C, Eckhaus M, Steinberg SM, Wynshaw-Boris A, Ried T, et al. RAG-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Mol Cell Biol 2002; 22:3174-7; PMID:11940674; http://dx.doi.org/10.1128/MCB.22.9.3174-3177.2002
- Liyanage M, Weaver Z, Barlow C, Coleman A, Pankratz DG, Anderson S, Wynshaw-Boris A, Ried T. Abnormal rearrangement within the alpha/delta T-cell receptor locus in lymphomas from Atm-deficient mice. Blood 2000; 96:1940-6; PMID:10961898
- Meier M, den Boer ML, Hall AG, Irving JA, Passier M, Minto L, van Wering ER, Janka-Schaub GE, Pieters R. Relation between genetic variants of the ataxia telangiectasia-mutated (ATM) gene, drug resistance, clinical outcome and predisposition to childhood T-lineage acute lymphoblastic leukaemia. Leukemia 2005; 19:1887-95; http://dx.doi.org/10.1038/sj.leu.2403943
- Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell 2002; 1:75-87; PMID:12086890; http://dx.doi.org/10.1016/S1535-6108(02)00018-1
- Gumy Pause F, Wacker P, Maillet P, Betts D, Sappino AP. ATM gene alterations in childhood acute lymphoblastic leukemias. Human Mutation 2003; 21:554; http://dx.doi.org/10.1002/humu.9140
- Gumy-Pause F, Wacker P, Maillet P, Betts DR, Sappino AP. ATM variants and predisposition to childhood T-lineage acute lymphoblastic leukaemia. Leukemia 2006; 20:526-7; author reply 7; PMID:16408093; http://dx.doi.org/10.1038/sj.leu.2404091
- Zha S, Sekiguchi J, Brush JW, Bassing CH, Alt FW. Complementary functions of ATM and H2AX in development and suppression of genomic instability. Proc Natl Acad Sci U S A 2008; 105:9302-6; PMID:18599436; http://dx.doi.org/10.1073/pnas.0803520105
- Yin B, Yang-Iott KS, Chao LH, Bassing CH. Cellular context-dependent effects of H2ax and p53 deletion on the development of thymic lymphoma. Blood 2011; 117:175-85; PMID:20947684; http://dx.doi.org/10.1182/blood-2010-03-273045
- DeMicco A, Yang-Iott K, Bassing CH. Somatic inactivation of Tp53 in hematopoietic stem cells or thymocytes predisposes mice to thymic lymphomas with clonal translocations. Cell Cycle 2013; 12:3307-16; PMID:24036547; http://dx.doi.org/10.4161/cc.26299
- Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907-13; PMID:15829965; http://dx.doi.org/10.1038/nature03485
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434:864-70; PMID:15829956; http://dx.doi.org/10.1038/nature03482
- Georgiades P, Ogilvy S, Duval H, Licence DR, Charnock-Jones DS, Smith SK, Print CG. VavCre transgenic mice: a tool for mutagenesis in hematopoietic and endothelial lineages. Genesis 2002; 34:251-6; PMID:12434335; http://dx.doi.org/10.1002/gene.10161
- Stadtfeld M, Graf T. Assessing the role of hematopoietic plasticity for endothelial and hepatocyte development by non-invasive lineage tracing. Development 2005; 132:203-13; PMID:15576407; http://dx.doi.org/10.1242/dev.01558
- Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, Adams JM. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 1999; 94:1855-63; PMID:10477714
- Yin B, Lee BS, Yang-Iott KS, Sleckman BP, Bassing CH. Redundant and nonredundant functions of ATM and H2AX in alphabeta T-lineage lymphocytes. J Immunol 2012; 189:1372-9; http://dx.doi.org/10.4049/jimmunol.1200829
- Zha S, Bassing CH, Sanda T, Brush JW, Patel H, Goff PH, Gatti R, Look AT, Alt FW. ATM-deficient thymic lymphoma is associated with aberrant TCRd rearrangement and gene amplification. J Exp Med 2010; 207:1369-80; In Press; PMID:20566716
- Brady BL, Oropallo MA, Yang-Iott KS, Serwold T, Hochedlinger K, Jaenisch R, Weissman IL, Bassing CH. Position-dependent silencing of germline Vss segments on TCRss alleles containing preassembled VssDJssCss1 genes. J Immunol 2010; 185:3564-73; http://dx.doi.org/10.4049/jimmunol.0903098
- Callen E, Bunting S, Huang CY, Difilippantonio MJ, Wong N, Khor B, Mahowald G, Kruhlak MJ, Ried T, Sleckman BP, et al. Chimeric IgH-TCRalpha/delta translocations in T lymphocytes mediated by RAG. Cell Cycle 2009; 8:2408-12; PMID:19556863; http://dx.doi.org/10.4161/cc.8.15.9085
- Isoda T, Takagi M, Piao J, Nakagama S, Sato M, Masuda K, Ikawa T, Azuma M, Morio T, Kawamoto H, et al. Process for immune defect and chromosomal translocation during early thymocyte development lacking ATM. Blood 2012; 120:789-99; PMID:22709691; http://dx.doi.org/10.1182/blood-2012-02-413195
- Liyanage M, Coleman A, du Manoir S, Veldman T, McCormack S, Dickson RB, Barlow C, Wynshaw-Boris A, Janz S, Wienberg J, et al. Multicolour spectral karyotyping of mouse chromosomes. Nat Genet 1996; 14:312-5; PMID:8896561; http://dx.doi.org/10.1038/ng1196-312
- Krangel MS, Carabana J, Abbarategui I, Schlimgen R, Hawwari A. Enforcing order within a complex locus: current perspectives on the control of V(D)J recombination at the murine T-cell receptor alpha/delta locus. Immunol Rev 2004; 200:224-32; PMID:15242408; http://dx.doi.org/10.1111/j.0105-2896.2004.00155.x
- Sleckman BP, Bardon CG, Ferrini R, Davidson L, Alt FW. Function of the TCR alpha enhancer in alphabeta and gammadelta T cells. Immunity 1997; 7:505-15; PMID:9354471; http://dx.doi.org/10.1016/S1074-7613(00)80372-6
- Huang CY, Sharma GG, Walker LM, Bassing CH, Pandita TK, Sleckman BP. Defects in coding joint formation in vivo in developing ATM-deficient B and T lymphocytes. J Exp Med 2007; 204:1371-81; PMID:17502661; http://dx.doi.org/10.1084/jem.20061460
- Matei IR, Gladdy RA, Nutter LM, Canty A, Guidos CJ, Danska JS. ATM deficiency disrupts Tcra locus integrity and the maturation of CD4+CD8+ thymocytes. Blood 2007; 109:1887-96; PMID:17077325; http://dx.doi.org/10.1182/blood-2006-05-020917
- Bassing CH, Ranganath S, Murphy M, Savic V, Gleason M, Alt FW. Aberrant V(D)J recombination is not required for rapid development of H2ax/p53-deficient thymic lymphomas with clonal translocations. Blood 2008; 111:2163-9; PMID:17855626; http://dx.doi.org/10.1182/blood-2007-08-104760
- Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, Donehower LA. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J 1998; 17:4657-67; PMID:9707425; http://dx.doi.org/10.1093/emboj/17.16.4657
- Ide F, Kitada M, Sakashita H, Kusama K, Tanaka K, Ishikawa T. p53 haploinsufficiency profoundly accelerates the onset of tongue tumors in mice lacking the xeroderma pigmentosum group A gene. Am J Pathol 2003; 163:1729-33; PMID:14578172; http://dx.doi.org/10.1016/S0002-9440(10)63531-6
- Choi YJ, Anders L. Signaling through cyclin D-dependent kinases. Oncogene 2014; 33:1890-903; PMID:23644662; http://dx.doi.org/10.1038/onc.2013.137
- Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012; 22:438-51; PMID:23079655; http://dx.doi.org/10.1016/j.ccr.2012.09.015
- Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, Genesca L, Trimarchi T, Kelliher MA, Clark M, et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 2012; 22:452-65; PMID:23079656; http://dx.doi.org/10.1016/j.ccr.2012.09.016
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001; 29:418-25; PMID:11694875; http://dx.doi.org/10.1038/ng747