
Abstract
Identification of functionally relevant differences between induced pluripotent stem cells (iPSC) and reference embryonic stem cells (ESC) remains a central question for therapeutic applications. Differences in gene expression between iPSC and ESC have been examined by microarray and more recently with RNA-SEQ technologies. We here report an in depth analyses of nuclear and cytoplasmic transcriptomes, using the CAGE (cap analysis of gene expression) technology, for 5 iPSC clones derived from mouse lymphocytes B and 3 ESC lines. This approach reveals nuclear transcriptomes significantly more complex in ESC than in iPSC. Hundreds of yet not annotated putative non-coding RNAs and enhancer-associated transcripts specifically transcribed in ESC have been detected and supported with epigenetic and chromatin-chromatin interactions data. We identified super-enhancers transcriptionally active specifically in ESC and associated with genes implicated in the maintenance of pluripotency. Similarly, we detected non-coding transcripts of yet unknown function being regulated by ESC specific super-enhancers. Taken together, these results demonstrate that current protocols of iPSC reprogramming do not trigger activation of numerous cis-regulatory regions. It thus reinforces the need for already suggested deeper monitoring of the non-coding transcriptome when characterizing iPSC clones. Such differences in regulatory transcript expression may indeed impact their potential for clinical applications.
Abbreviations
| CAGE | = | cap analysis of gene expression |
| ENCODE | = | Encyclopedia of DNA Elements |
| eRNA | = | enhancer RNA |
| ESC | = | embryonic stem cells |
| iPSC | = | induced pluripotent stem cells |
| lncRNA | = | long non-coding RNA |
| NAST | = | Non-Annotated Stem Transcripts |
| ncRNA | = | non-coding RNA |
Introduction
Reprogramming of somatic cells into a pluripotent state close to embryonic stem cells (ESC), has been recognized as a major success of modern cell biology with great potential for regenerative medicine.Citation1 Yet, substantial transcriptional variations have been reported among induced pluripotent stem cells (iPSC) and ESC,Citation2 which necessitate better understanding to ensure their utility and clinical safety.
Early gene expression studies, based on microarray technology, have identified hundreds of significantly differentially expressed genes between ESC and iPSC.Citation3–5 Others concluded that specific iPSC clones could not be distinguished from ESC based on their gene expression and DNA methylation profiles, however variable differentiation propensities were measured.Citation2,6 This controversial question of actual differences in gene expression between ESC and iPSC is based on observation for protein coding genes. Indeed, relatively little is known about the differential expression of non-coding regulatory transcripts between ESC and iPSC. With the exception of a report for 105 miRNAs,Citation3 concluding on little differences, to our knowledge, no studies have yet reported systematic investigation for the non-coding fraction of the transcriptome between ESC and iPSC. In addition, transcriptional data on nuclear non-coding transcriptome of ESC and iPSC remain rare.Citation7,8
Here, taking advantage of our expertise in nuclear transcriptome analyses,Citation8 we have deeply analyzed transcriptomes of ESC and iPSC. The CAGE (cap analysis of gene expression) technology, developed in our laboratory,Citation9,10 offers a higher resolution for rare regulatory transcripts than precedent reports. We are reporting 2,501 non-coding loci, putative enhancers and novel lncRNAs, which expression was not properly activated upon reprogramming of mouse lymphocytes B in iPSC.
Results
High nuclear complexity of ESC transcriptomes not mimicked in iPSC
We aim at the identification of protein non-coding genes and regulatory loci transcriptionally active in ESC and not activated upon cells reprogramming. To this end, we have analyzed the nuclear and cytoplasmic transcriptomes of 5 iPSC clones and 3 ESC lines from murine origins (data from Fort et al.Citation8). iPSC lines were derived from primary lymphocytes B using conventional retroviral vectors expressing the 4 Yamanaka's factors (Oct4, Klf4, Sox2, c-Myc). Nuclear enriched and cytoplasmic RNA samples were extracted from cells harvested at similar passage number (from P20 to P31) and analyzed using the CAGE technologyCitation9,10 followed by highthroughput sequencing. CAGE libraries were sequenced at an average depth of 14 millions (+/− 1.4) CAGE-tags (Fig. S1A). CAGE-tags mapping uniquely to the reference genome (genome assembly mm9/NCBI37) were considered to create a strict set of TSSs, using the ParacluCitation11 clustering algorithm. We selected CAGE-tag-clusters detected in at least 2 iPSC clones or 2 ESC lines or in the lymphocytes B (Fig. S1B). For expression threshold, we required CAGE-tag-clusters to be measured at a minimum of 1 tag per million (tpm) in at least one sample. Hierarchical clustering computed with Spearman correlation and Euclidian distance matrixes for all CAGE-tag-clusters groups nuclear from cytoplasmic samples at first and separate ESC from iPSC ( and Fig. S1C-D).
Figure 1. (A) Hierarchical clustering based on Euclidian distance matrix for expression values of all CAGE-tag-clusters (B) Fold-change values (FC) density for the differential expression analyses on the nuclear (Nu) and cytoplasmic (Cy) datasets. CAGE-clusters found differentially expressed in both analyses (Nu/Cy) are marked in blue. (C) Comparison of transcriptional complexity in the nuclear and cytoplasmic compartments of ESC and iPSC cells, calculating the number of CAGE clusters detected in each sample after sub-sampling of CAGE tags (richness). Red bars represent mean values. P-values from 2-sided t-test are shown. (D) Annotation of CAGE-tag-clusters significantly up-regulated in ESC (edgeR, FDR< 0.05, FC>8). (E) Histone marks (ENCODE ChIP-seq dataCitation7) based classification of CAGE tag clusters significantly upregulated in ESC.
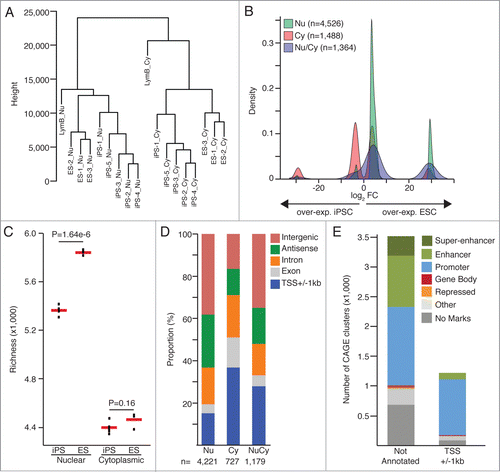
A total of 78,714 CAGE-tag-clusters fulfilled the above-mentioned criteria and were included in differential expression analyses comparing the 5 iPSC clones with the 3 ESC lines for the nuclear and cytoplasmic datasets independently. 526 CAGE-tag-clusters were identified as significantly (FDR < 0.05 and FC > 8, calculated with edgeRCitation12) differentially expressed in the nuclear compartment, 1,488 in the cytoplasm and 1,364 in both sub-cellular compartments. When analyzing the nuclear data sets, a larger proportion of CAGE-tag-clusters over-expressed in ESC were identified relative to the fraction overexpressed in iPSC (). This observation is further supported by a significant increased complexity (calculated with Vegan R package) of the ESC nuclear transcriptome when compared with iPSC (); while their respective cytoplasmic transcriptomes do not show significant differences in complexity levels. In addition, we found 73% (847 out of 1,161) of the differentially expressed protein coding genes expressed at lower levels in iPSC, similarly than reported in Chin et al.Citation3
Novel lncRNAs and actively transcribed enhancers not activated upon reprogramming process
We then aim at identifying yet not annotated (annotation procedure in Methods) non-coding RNAs which expression was not properly induced upon iPSC reprogramming process. For this purpose, we extracted 3,515 CAGE-tag-clusters significantly over-expressed in ESC and residing in intergenic regions or being in an antisense orientation to annotated genes (). Not annotated CAGE-tag-clusters, significantly (FDR < 0.05 and FC > 8, calculated with edgeRCitation12) overexpressed in ESC count for 64% of the transcripts exclusively identified in the nuclear compartment and represent 29% and 52% for the clusters identified in the cytoplasm or in both cellular compartments respectively.
Using chromatin histone marks, retrieved from the ENCODE consortium,Citation7 specific of promoter (high levels of H3K4me3 and low levels of H3K4me1) and enhancer regions (low levels of H3K4me3, high levels of H3K4me1 and H3K27ac), we classified 71.2% of the not annotated CAGE-tag-clusters significantly over-expressed in ESC as either associated with putative novel promoters (n = 1 ,313), enhancer associated transcripts (n = 865 ) or as super-enhancers (n = 323 ) (; Fig. S2; Table S1).
In addition to carry enhancer specific histone marks, we required super-enhancers to be bound by 3 core stemness transcription factors (i.e., Nanog, Pou5f1 and Sox2Citation13) and a mediator subunit (Med1 or Med12),Citation14 similarly to criteria considered in Whyte et al.Citation15 Alike previous reports,Citation15,16 super-enhancers regions are associated with higher DNaseI hypersensitivity signal (, ENCODE data for ES-E14Citation7), higher ChIP-seq signal for Mediator subunitsCitation14 () and enhancer associated histone marks (H3K4me1 and H3K27ac, Fig. S3A-B, ENCODE data for ES-E14Citation7). Interestingly, we also observed a stronger ChIP-seq signal for the enhancer associated co-factor p300, the cohesin complex protein Smc1aCitation14 and the loading factor of cohesin NipblCitation14 (Fig. S3C).
Figure 2. (A) Mean DNaseI hypersensitivity signal density (ENCODE data, ES-E14 cellsCitation7) and (B) mean ChIP-Seq signal density for Mediator subunits Med1 and Med12Citation14 for the not annotated super-enhancer, enhancer and annotated TSSs. (C) Normalized nuclear expression (tpm: tags per million) and (D) mean PhastCons score (Euarchontoglires, 30 species, UCSC) for the CAGE tag clusters significantly up-regulated in ESC (edgeR, FDR < 0.05, FC > 8).
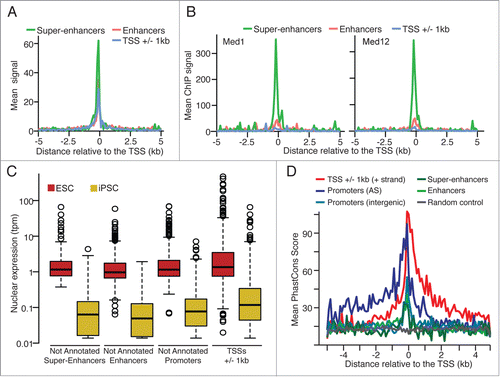
Similarly to our previous report on non-annotated stem transcripts (NAST),Citation8 we found putative non-coding transcripts, associated with promoter, enhancer or super-enhancer regions, expressed at significant lower levels (Mann-Whitney Rank test, P < 0.0023) than annotated protein-coding genes also overexpressed in ESC (). Furthermore, enriched evolutionary conservation scores (PhastConsCitation17 for Euarchontoglires, 30 species) are observed for all not annotated groups () when compared to random genomic locations.
Taken together, these observations support the presence of yet unnoticed ncRNAs and enhancer-RNAs (eRNAs), which transcription is not properly activated upon conventional viral vectors mediated reprogramming process.
Enhancers specifically active in ESC associate with genes lowly expressed in iPSC
To characterize further newly identified ncRNAs and eRNAs, we first look at the association with repeat elements; as others and we have reported implication of repeat derived transcripts in the genetic regulation of pluripotency.Citation8,18,19 Interestingly, we observed significant (Exact Fisher test, P < 2.2 × 10–16) enrichment for ERVK repeat elements overlapping ESC specific super-enhancers, enhancers and promoters (). MaLR elements appear also significantly enriched (Exact Fisher test, P < 0.00077) among super-enhancers and enhancers. In more details, super-enhancers appear to overlap mainly solitary LTR-ERVK elements with RLTR13D6 representing 19.5% (n = 15 out of 77) of the ERVK element associated with super-enhancer. RLTR9D, RLRT9E and the full-length ETnERV2-int LTR elements represent 2 thirds of the ERVKs associated with enhancers (32.5%) and promoters (31.3%) loci resistant to reprogramming. Interestingly, RLTR13D6 is found associated with 8.3% enhancers but with none of the novel promoters.
Figure 3 (See previous page). (A) Repeat composition of not annotated (super-enhancers, enhancers, promoters) and annotated (TSSs +/-1kb) CAGE-tag-clusters significantly overexpressed in ESC. All CAGE-tag-clusters composition is shown for comparison. (B) Proportion of overlap with Non-Annotates-Stem-Transcripts (NAST) from Fort et al.Citation8 (C and D) Normalized expression values for super-enhancer regions and their associated protein coding genes (C) or lncRNAs (D). Schematic representations of genomic configurations are shown above plots. Error bars, s.d. Indicated P-values are from 2-sided t-tests. iPSC n = 5 , ESC n = 3.
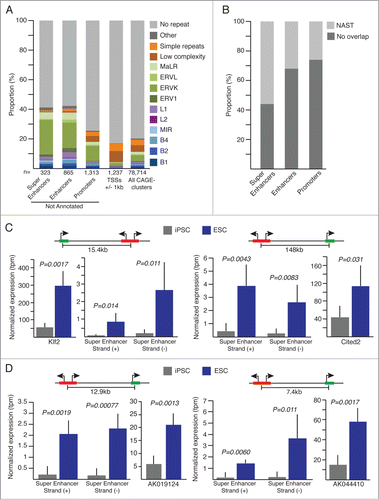
Enhancers are associated with RLTR9D, RLRT9E and RLTR13D6, while novel promoters show also association with RLTR9D, RLRT9E but interestingly a comparable amount of overlap with the ETnERV2-int full-length elements.
Noteworthy, 56.2% of the super-enhancers over-expressed in ESC were previously described by our laboratory as NASTs,Citation8 while the NAST overlapping fraction for annotated enhancer and promoter regions are only 32.1% and 25.9% respectively (). These specific sub-groups of NASTs are indeed expressed at significant higher levels (Mann-Whitney Rank test, P < 2.2 × 10–16) in ESC when compared to iPSC lines originally used for the identification of NASTs in Fort et al.Citation8 (Fig. S4).
We then sought to identify target genes of super-enhancers and enhancers associated with significant higher transcriptional activity in ESC. To this end, we used ChIA-PET dataCitation20 (chromatin interaction analyses by paired-end tags) reporting RNA-polymerase-II mediated chromatin-chromatin interactions in mouse ES-E14 cells. We detected 143 and 46 intra-chromosomal interactions implicating enhancers or super-enhancers, respectively, and promoter regions of known genes. Among interacting annotated genes, 35 show expression fold-change greater than 2 in ESC when compared to iPSC. We provide a list of 13 high-confidence regulatory regions interacting with 4 lncRNAs and 7 protein-coding genes (Table S2). Detailed expression levels for 4 of these candidate regions, distant from 7.4kb to 148kb from their putative targeted gene, 2 protein-coding genes implicated in pluripotency regulation and 2 lncRNA genes of unknown function are shown on . These loci share analogous genomic conformation with no annotated genes localized between regulatory elements and target-genes. As first example, we show a super-enhancer potentially regulating the transcription factor Klf2 (Kruppel-like factor 2, ), which exogenous expression in post-implantation epiblast stem cells (EpiSC) together with Nanog has been shown to trigger ground states ESC in mouseCitation21,22 as well as in humanCitation23 models. Second, a distant super-enhancer interacting with the trans-activator Cited2 (cbp/p300-interacting protein, ) implicated in the maintenance of pluripotency and self-renewal of stem cells via direct regulation of Pou5f1.Citation24,25 In addition, we show examples of super-enhancers associated with 2 lncRNAs (), AK044410 (D230017M19Rik) and AK019124 (2410080I02Rik), suggesting a role for these lncRNAs in the regulation of pluripotency.
In summary, these results suggest that a set of cis-regulatory regions, 40% of them being associated with repeated elements, is not properly activated in iPSC. As a direct consequence, protein coding and lncRNAs implicated in the genetic control of stem state are not properly transcribed.
Discussion
Large efforts have been achieved for the improvement of iPSC reprogramming efficiency and the selection of high quality clones closely resembling to ESC. Obviously, ESC and iPSC share key features of pluripotency, including expression of pluripotency marker genes, cell and colony morphologies as well as differentiation ability into the different germ layers.Citation26-28 However, transcriptional differences between ESC and iPSC have been repeatedly reported, concluding on not equivalent cellular status.Citation3-5 In this study, we have shown that in depth nuclear transcriptome profiling of multiple ESC and iPSC cell lines, based on CAGE technology, identified relevant differentially expressed regulatory transcripts putatively implicated in the genetic regulation of pluripotency.
On the other hand, 2 studies reached the conclusion that most reported changes in gene expression between ESC and iPSC are actually stochastic and caused by lab-specific differences in either reprogramming process, progenitor cells used or aspects of stem cell culture and handling conditions.Citation6,29 If, we cannot formally rule out effects resulting from not fully identical culture conditions for all cell lines, we believe that our results shed new light on relevant transcriptional differences of regulatory non-coding transcripts between iPSC and ESC. These discoveries were made possible applying the CAGE technology, which do not require transcript model for its analyses, to nuclear enriched RNA samples, enhancing the detection of rare nuclear transcripts. We believe this makes it an approach of choice for the identification of novel regulatory non-coding transcripts yet not annotated and residing mainly in the nucleus.Citation30 As shown for a few loci, this approach efficiently identified genes and associated cis-regulatory elements misregulated in iPSC. Notably, this strategy provides hints for functional role of lncRNAs without known functions and their associated cis-regulatory regions, which expression are also found significantly lower in ESC when compared to iPSC.
In conclusion, our analyses reveal higher transcriptional differences between iPSC and ESC than formally estimated.Citation2,6 It suggests that an important part of phenotypic differences observed between different iPSC clones reflect failure in cis-regulatory elements activation and was not detected with previously used technologies. Noteworthy, a striking differentiation-defect in iPSC clones not properly expressing LTR7 associated lncRNA has been reported for human iPSC, highlightening association between ncRNA regulation and iPSC potency.Citation19,31
iPSC technology carries large expectations in the revolution of regenerative medicine and as models for human diseases. However, taken together, previous reports and our observations indicate that the non-coding fraction of the transcriptome should be carefully monitored, with suitable technologies, when evaluating iPSC clones for clinical applications.
Methods
Lymphocyte isolation
CD19+ cells (B lymphocytes) were isolated from freshly dissected spleen (C57BL/6 mice) using MACS beads (Miltenyi Biotech) and were cultured in lymphocyte complete medium (RPMI1640, Sigma) containing 10% FBS, 5 ng/ml interleukin IL-4 (R&D Systems) and 25 mg/ml lipopolysaccharide (LPS; Sigma).
iPSC reprogramming
Retrovirus preparation was carried out as previously described in Takahashi et al.Citation32 Mouse B cells were infected at 1 × 105 cells/ml in the presence of 10 mg/ml polybrene (Sigma), 5 ng/ml IL-4 (R&D Systems) and 25 mg/ml LPS (Sigma). After 24 h, medium was replaced with lymphocyte complete medium, and cells were seeded on mitomycin-treated MEF feeder cells. Seventy-two hours after transduction, medium was replaced with mouse ESC medium, and medium was changed every other day until ESC-like colonies formed. Colonies were isolated, dissociated with trypsin (Invitrogen) and transferred to stem cell medium (DS Pharma Biomedical) maintained with 2,000 U/ml LIF and 0.1 mM 2-mercaptoethanol and kept in culture for further experiments. iPSC injections in blastocysts resulting in chimeric mice confirmed the full reprogramming of our iPS clones. In addition, iPSC colonies were stained by immunostaining for the pluripotent marker proteins Ssea1, Oct-4 and Nanog (D.Y. and H.K., unpublished data ).
Cell culture
ESCs (ES-1: Nanog^(βgeo/+)ES, 129 SV Jae mouse strain, ESR08 passage 21; ES-2: FVB1, FVB mouse strain, passage 21; ES-3: B6G2, C57BL/6 mouse strain, passage 22) were grown under feeder-free conditions in mouse ESC medium containing DMEM (Wako), 1,000 U/ml leukemia inhibitory factor (LIF; Millipore), 15% FBS (Gibco), 2.4 mM L-glutamine (Invitrogen), 0.1 mM non-essential amino acids (NEAA; Invitrogen), 0.1 mM 2-mercaptoethanol (Gibco), 50 U/ml penicillin and 50 μg/ml streptomycin (Gibco). Culture media were changed daily, and cells were passaged every 2–3 d.
Established iPS clones (iPS-1: mi44.1B4e passage 21; iPS-2: mi44H2e passage 31; iPS-3: mi55A2 passage 20; iPS-4: mi55G4 passage 28; iPS-5: mi56H1 passage 24) were cultured on MEFs treated with mitomycin (Sigma) in DMEM containing 20% FBS, 2,000 U/ml LIF, 1% NEAA, 0.1 mM 2-mercaptoethanol, 2.4 mM L-glutamine and 3 inhibitors (3i).Citation33
Extraction of nucleus-enriched and cytoplasmic RNAs
For all cell lines nucleus-enriched and cytoplasmic RNA fractions were isolated from 5 to 10 million cells with the procedure detailed in Fort et al.Citation8 Briefly, cells were lysed in chilled lysis buffer (0.8 M sucrose, 150 mM KCl, 5 mM MgCl2, 6 mM 2-mercaptoethanol and 0.5% NP-40) and spin for 5 min at 10,000g (4°C). Supernatants containing cytoplasmic fractions were collected and mixed with 3 volumes of TRIzol-LS Reagent (Life Technologies). Nuclei pellets were washed twice with lysis buffer before resuspension in TRIzol Reagent. A miRNEasy kit (Qiagen) was used according to the manufacturer's protocol to extract both nucleus-enriched and cytoplasmic RNA fractions. During the RNA purification process, samples were treated with DNase I (Qiagen).
CAGE library preparation and data processing
CAGE libraries were prepared starting with 0.5 to 5 μg of RNA, following the protocols developed in our laboratory.Citation9 As detailed in Fort et al.,Citation8 CAGE libraries were sequenced on the Illumina HiSeq 2000 platform with a read length of 50 bases. After discarding sequences with ambiguous base calling, splitting sample reads by barcodes and removing linker sequences and artifactual linker adaptor sequences using TagDust,Citation34 reads were of 26 to 42 bases in length. CAGE reads were mapped to mm9/NCBI37 using Burrows-Wheeler Aligner (BWA) v0.5.6.Citation35 Only reads with MapQ values over 10 and therefore mapping to single loci in the genomes were used in our analyses. Subsequently, reads mapping to rDNA were eliminated. CAGE tag 5′ genomic coordinates were used as input for ParacluCitation11 clustering with the following parameters: (i) a minimum of 5 tags per cluster, (ii) maximum density/baseline density ≥2 and (iii) a maximal cluster length of 200 bp.
CAGE cluster annotation
Annotation of CAGE tag clusters () was performed as described in Fort et al.Citation8 and based on the RefSeq, EnsemblCitation36 and UCSC KnownGenes databases (retrieved from the UCSC browser in January 2012). Repetitive element annotations were retrieved from the UCSC browser, which ran RepeatMaskerCitation37 version open-3–2–7.
Histone mark–based classification of the CAGE-tag clusters was performed using ChIP-seq dataCitation7 for ES-Bruce-4 and ES-E14 cells. Loci carrying a stronger signal for H3K4me1 than for H3K4me3 and carrying H3K27ac were classified as enhancers,Citation38,39 whereas clusters with stronger signal for H3K4me3 than for H3K4me1 and/or carrying H3K9ac marks were considered to be promoters. CAGE clusters carrying H3K9me3 and/or H3K27me3 marks were annotated as repressed. Finally, CAGE-tag clusters presenting trimethylation at lysine 36 of histone H3 (H3K36me3) were annotated as gene body.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Accession Code
All sequencing data have been deposited at the DNA Data Bank of Japan (DDBJ) under accession DRA000914 and DRA002621.
988031_Supplementary_Materials.zip
Download Zip (2.7 MB)988031_Supplementary_Materials.zip
Download Zip (2.8 MB)Funding
This work was supported by a grant to P.C. from the Japan Society for the Promotion of Science (JSPS) through the Funding Program for Next-Generation World-Leading Researchers (NEXT) initiated by the Council for Science and Technology Policy (CSTP), by a grand-in-aid for scientific research from JSPS to P.C. and A.F., and by a research grant from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) to the RIKEN Center for Life Science Technologies. A.F. was supported by a Swiss National Science Foundation (SNSF) Fellowship for Advanced Researchers (PA00P3_142122) and a SNSF Ambizione grant (PZ00P3–154728). K.H. was supported by European Union Framework Program 7 (MODHEP project) for P.C. D.Y. and H.K. were supported by the Japan Science and Technology Agency CREST. The authors thank the RIKEN GeNAS sequencing platform for sequencing of the libraries.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Inoue H, Nagata N, Kurokawa H, Yamanaka S. iPS cells: a game changer for future medicine. EMBO J 2014; 33:409-17; PMID:24500035; http://dx.doi.org/10.1002/embj.201387098.
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 2011; 144:439-52; PMID:21295703; http://dx.doi.org/10.1016/j.cell.2010.12.032.
- Chin MH, Mason MJ, Xie W, Volinia S, Singer M, Peterson C, Ambartsumyan G, Aimiuwu O, Richter L, Zhang J, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell 2009; 5:111-23; PMID:19570518; http://dx.doi.org/10.1016/j.stem.2009.06.008.
- Ghosh Z, Wilson KD, Wu Y, Hu S, Quertermous T, Wu JC. Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PloS One 2010; 5:e8975; PMID:20126639; http://dx.doi.org/10.1371/journal.pone.0008975.
- Marchetto MC, Yeo GW, Kainohana O, Marsala M, Gage FH, Muotri AR. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PloS One 2009; 4:e7076; PMID:19763270; http://dx.doi.org/10.1371/journal.pone.0007076.
- Guenther MG, Frampton GM, Soldner F, Hockemeyer D, Mitalipova M, Jaenisch R, Young RA. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell 2010; 7:249-57; PMID:20682450; http://dx.doi.org/10.1016/j.stem.2010.06.015.
- Consortium EP, Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/10.1038/nature11247.
- Fort A, Hashimoto K, Yamada D, Salimullah M, Keya CA, Saxena A, Bonetti A, Voineagu I, Bertin N, Kratz A, et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat Genet 2014; 46:558-66; PMID:24777452; http://dx.doi.org/10.1038/ng.2965.
- Takahashi H, Lassmann T, Murata M, Carninci P. 5' end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat Protoc 2012; 7:542-61; PMID:22362160; http://dx.doi.org/10.1038/nprot.2012.005.
- Kodzius R, Kojima M, Nishiyori H, Nakamura M, Fukuda S, Tagami M, Sasaki D, Imamura K, Kai C, Harbers M, et al. CAGE: cap analysis of gene expression. Nat Methods 2006; 3:211-22; PMID:16489339; http://dx.doi.org/10.1038/nmeth0306-211.
- Frith MC, Valen E, Krogh A, Hayashizaki Y, Carninci P, Sandelin A. A code for transcription initiation in mammalian genomes. Genome Res 2008; 18:1-12; PMID:18032727; http://dx.doi.org/10.1101/gr.6831208.
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26:139-40; PMID:19910308; http://dx.doi.org/10.1093/bioinformatics/btp616.
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008; 134:521-33; PMID:18692474; http://dx.doi.org/10.1016/j.cell.2008.07.020.
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010; 467:430-5; PMID:20720539; http://dx.doi.org/10.1038/nature09380.
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013; 153:307-19; PMID:23582322; http://dx.doi.org/10.1016/j.cell.2013.03.035.
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013; 153:320-34; PMID:23582323; http://dx.doi.org/10.1016/j.cell.2013.03.036.
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005; 15:1034-50; PMID:16024819; http://dx.doi.org/10.1101/gr.3715005.
- Kunarso G, Chia NY, Jeyakani J, Hwang C, Lu X, Chan YS, Ng HH, Bourque G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 2010; 42:631-4; PMID:20526341; http://dx.doi.org/10.1038/ng.600.
- Lu X, Sachs F, Ramsay L, Jacques PE, Goke J, Bourque G, Ng HH. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat Struct Mol Biol 2014; 21:423-5; PMID:24681886; http://dx.doi.org/10.1038/nsmb.2799.
- Zhang Y, Wong CH, Birnbaum RY, Li G, Favaro R, Ngan CY, Lim J, Tai E, Poh HM, Wong E, et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature 2013; 504:306-10; PMID:24213634; http://dx.doi.org/10.1038/nature12716.
- Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O, Wray J, Yamanaka S, Chambers I, Smith A. Nanog is the gateway to the pluripotent ground state. Cell 2009; 138:722-37; PMID:19703398; http://dx.doi.org/10.1016/j.cell.2009.07.039.
- Hall J, Guo G, Wray J, Eyres I, Nichols J, Grotewold L, Morfopoulou S, Humphreys P, Mansfield W, Walker R, et al. Oct4 and LIF/Stat3 additively induce Kruppel factors to sustain embryonic stem cell self-renewal. Cell Stem Cell 2009; 5:597-609; PMID:19951688; http://dx.doi.org/10.1016/j.stem.2009.11.003.
- Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F, Clarke J, Mansfield W, et al. Resetting Transcription Factor Control Circuitry toward Ground-State Pluripotency in Human. Cell 2014; 158:1254-69; PMID:25215486; http://dx.doi.org/10.1016/j.cell.2014.08.029.
- Li Q, Ramirez-Bergeron DL, Dunwoodie SL, Yang YC. Cited2 gene controls pluripotency and cardiomyocyte differentiation of murine embryonic stem cells through Oct4 gene. J Biol Chem 2012; 287:29088-100; PMID:22761414; http://dx.doi.org/10.1074/jbc.M112.378034.
- Li Q, Hakimi P, Liu X, Yu WM, Ye F, Fujioka H, Raza S, Shankar E, Tang F, Dunwoodie SL, et al. Cited2, a transcriptional modulator protein, regulates metabolism in murine embryonic stem cells. J Biol Chem 2014; 289:251-63; PMID:24265312; http://dx.doi.org/10.1074/jbc.M113.497594.
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature 2007; 448:313-7; PMID:17554338; http://dx.doi.org/10.1038/nature05934.
- Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007; 448:318-24; PMID:17554336; http://dx.doi.org/10.1038/nature05944.
- Boland MJ, Hazen JL, Nazor KL, Rodriguez AR, Gifford W, Martin G, Kupriyanov S, Baldwin KK. Adult mice generated from induced pluripotent stem cells. Nature 2009; 461:91-4; PMID:19672243; http://dx.doi.org/10.1038/nature08310.
- Newman AM, Cooper JB. Lab-specific gene expression signatures in pluripotent stem cells. Cell Stem Cell 2010; 7:258-62; PMID:20682451; http://dx.doi.org/10.1016/j.stem.2010.06.016.
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature 2012; 489:101-8; PMID:22955620; http://dx.doi.org/10.1038/nature11233.
- Ohnuki M, Tanabe K, Sutou K, Teramoto I, Sawamura Y, Narita M, Nakamura M, Tokunaga Y, Nakamura M, Watanabe A, et al. Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proc Natl Acad Sci U S A 2014; 111:12426-31; PMID:25097266; http://dx.doi.org/10.1073/pnas.1413299111.
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663-76; PMID:16904174; http://dx.doi.org/10.1016/j.cell.2006.07.024.
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature 2008; 453:519-23; PMID:18497825; http://dx.doi.org/10.1038/nature06968.
- Lassmann T, Hayashizaki Y, Daub CO. TagDust–a program to eliminate artifacts from next generation sequencing data. Bioinformatics 2009; 25:2839-40; PMID:19737799; http://dx.doi.org/10.1093/bioinformatics/btp527.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; http://dx.doi.org/10.1093/bioinformatics/btp324.
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. Ensembl 2013. Nucleic Acids Res 2013; 41:D48-55; PMID:23203987; http://dx.doi.org/10.1093/nar/gks1236.
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 2005; 110:462-7; PMID:16093699; http://dx.doi.org/10.1159/000084979.
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011; 470:279-83; PMID:21160473; http://dx.doi.org/10.1038/nature09692.
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009; 459:108-12; PMID:19295514; http://dx.doi.org/10.1038/nature07829.