
Abstract
Before a cell enters mitosis, the Golgi apparatus undergoes extensive fragmentation. This is required for the correct partitioning of the Golgi apparatus into daughter cells, and inhibition of this process leads to cell cycle arrest in G2 phase. AMP-activated protein kinase (AMPK) plays critical roles in regulating growth and reprogramming metabolism. Recent studies have suggested that AMPK promotes mitotic progression and Golgi disassembly, and that this seems independent of the cellular energy status. However, the molecular mechanism underlying these events is not well understood. Here, we show that both treatment with compound C and depletion of AMPKα2 (but not AMPKα1) delays the G2/M transition in synchronized HeLa cells, as evidenced by flow cytometry and mitotic index analysis. Furthermore, knockdown of AMPKα2 specifically delays further fragmentation of isolated Golgi stacks. Interestingly, pAMPKαThr172 signals transiently appear in the perinuclear region of late G2/early prophase cells, partially co-localizing with the Golgi matrix protein, GM-130. These Golgi pAMPKαThr172 signals were also specifically abolished by AMPKα2 knockdown, indicating specific spatio-temporal activation of AMPKα2 at Golgi complex during late G2/early prophases. We also found that the specific CaMKKβ inhibitor, STO-609, reduces the pAMPKα Thr172 signals in the perinuclear region of G2 phase cells and delays mitotic Golgi fragmentation. Taken together, these data suggest that AMPKα2 is the major catalytic subunit of AMPKα which regulates Golgi fragmentation and G2/M transition, and that the CaMKKβ activates AMPKα2 during late G2 phase.
Abbreviations
| AMP | = | Adenosine monophosphate |
| AMPK | = | AMP-activated protein kinase |
| CaMKKβ | = | Ca2+/Calmodulin-dependent protein kinase kinase β |
| DTB | = | double-thymidine block |
| MRLC | = | myosin regulatory light chain |
| ACC | = | acetyl-CoA carboxylase |
Introduction
The AMP-activated protein kinase (AMPK), a serine/threonine kinase, is a highly conserved energy sensor that plays a central role in maintaining energy homeostasis by balancing catabolic and anabolic processes.Citation1 AMPK is activated under conditions of energy stress, primarily when the intracellular ATP:AMP ratio decreases (e.g., during nutrient deprivation or hypoxia).Citation2 AMPK exists as heterotrimeric complexes composed of a catalytic (α) subunit and 2 regulatory (β and γ) subunits, each of which occurs as 2 or more isoforms encoded by distinct genes (α1, α2, β1, β2, γ1, γ2, γ3). The expression of some of these isoforms is tissue-restricted, and functional distinctions have been reported for the 2 catalytic α subunits; i.e. AMPKα2 has a greater dependence on AMP than AMPKα1, and AMPKα2, but not AMPKα1, is localized to the nucleus.Citation3
Activation of AMPK requires phosphorylation of threonine 172 (Thr172) within the T loop of the catalytic α subunit. To date, 2 AMPK kinases have been characterized: the tumor suppressor, LKB1, and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ).Citation4 LKB1 is ubiquitously expressed and appears to be the principal kinase responsible for activating AMPK in response to metabolic stress in liver and skeletal muscle.Citation5-Citation7 In contrast, the activation of AMPK by CaMKKβ does not require alteration of the ATP:AMP ratio, but rather occurs in response to various cues, including increased intracellular Ca2+.Citation8-Citation10 CaMKKβ seems to be particularly involved in activating AMPK in neurons and T cells.Citation11,12
Recent studies have revealed that AMPK contributes to phenotypes other than maintaining energy homeostasis such as regulating cell polarity, mitotic progression, and cytokinesis. For example, studies in Drosophila have shown that disruption of AMPKα enhances cell proliferation and increases gross mitotic defects, such as lagging chromosome during anaphase, failure of cytokinesis and the acquisition of polyploidy.Citation13 In flies, AMPK mediates the Ser22 phosphorylation of non-muscle myosin regulatory light chain (MRLC), which interacts with actin to regulate the changes in the actin cytoskeleton that are required to maintain cell polarity.Citation13 In mammals, AMPK activity progressively increases as cells move from G1 phase toward the G2/M phases of the cell cycle.Citation14 During the mitotic phase, active AMPK has been observed to be transiently associated with several components of the mitotic apparatus, including spindle poles and the midbody.Citation15 In addition, a recent study identified novel targets of AMPK, including PAK2, APC1, CDC27, and 3 of the regulatory subunits (PPP1R12A, PPP1R12B, and PPP1R12C) that influence the activity of mammalian MRLC.Citation16 Thus, it is not surprising that depletion of AMPK causes mitotic delay, mis-orientation of the spindles, and reductions in the numbers and lengths of the astral microtubules.Citation17
In interphase mammalian cells, the Golgi apparatus is organized into a continuous membranous system; known as the Golgi ribbon, it is formed of individual Golgi stacks that are laterally connected by tubular bridges.Citation18 During mitosis, the Golgi ribbon undergoes extensive fragmentation through a multistage process that is required for its correct partitioning into daughter cells.Citation19 Unlinking of the Golgi stacks occur during G2 phase, and it is controlled by various kinases, including Raf1, MEK1, ERK2 and Plk3.Citation20-Citation23 These kinases can phosphorylate the Golgi reassembly stacking proteins, GRASP65 and GRASP55, which form a complex matrix that is involved in organizing the structure and function of the Golgi apparatus.Citation24,25 Failure of Golgi fragmentation induces cell cycle arrest at G2 phase, which we call “Golgi mitotic checkpoint."Citation26 After the cell enters mitosis, the Golgi stacks undergo further disassembly through the actions of Plk1 and Cdk1, which phosphorylate the golgins (e.g., GM-130), and proteins involved in membrane fusion.Citation27-Citation29 Interestingly, a recent study demonstrated that Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1 (GBF1) is a novel AMPK substrate, and that the AMPK-mediated phosphorylation of GBF1 at Thr1337 plays a critical role (presumably by attenuating the function of GBF1) in the Golgi disassembly that is induced by stress conditions that lower the intracellular concentration of ATP.Citation30 Furthermore, another recent study showed that AMPK phosphorylates GBF1 for mitotic Golgi disassembly, and phosphorylation-defective mutant of GBF1 induces G2/M arrest, suggesting the involvement of AMPK in both Golgi disassembly and G2/M transition.Citation31 However, the precise mechanism has yet to be clarified.
In the present study, we explored the roles of AMPK during mitotic entry in HeLa cells. We show that AMPK is activated at late G2 phase, and that pAMPKαThr172 signals increase in the perinuclear region, partially colocalizing with the Golgi marker, GM-130. Inhibition of AMPK or (more specifically) AMPKα2 depletion transiently arrests mitotic entry and delays mitotic Golgi fragmentation. Furthermore, CaMKKβ inhibition decreases the pAMPKαThr172 signals in the Golgi region and delays mitotic Golgi fragmentation. Our results collectively suggest that the CaMKKβ-AMPKα2 signaling pathway is involved in regulating Golgi fragmentation during the G2/M transition.
Results
AMPKα is highly phosphorylated in the G2/M phases and localizes to the mitotic apparatus in HeLa cells
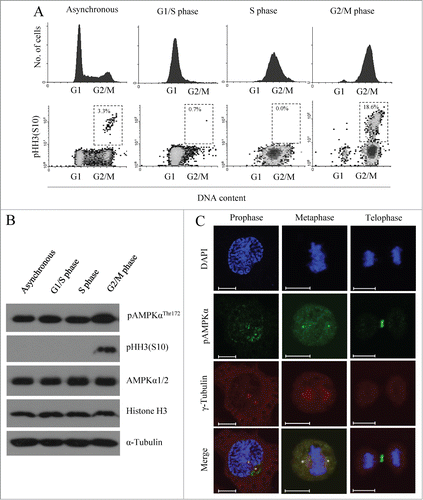
Recent studies have found that AMPK is phosphorylated during mitosis, and that during this time it is localized to components of the mitotic apparatus (e.g., centrosomes, mitotic spindle poles and the midbody), seemingly independent of the cellular energy status.Citation15 Based on these reports, we herein studied the mitotic role of AMPK. We first started from HeLa cells, which harbor a homozygous deletion of LKB1. To assess when AMPK is activated during cell cycle progression, we synchronized HeLa cells at the G1/S border by a double-thymidine block (DTB), and then collected cells that had been released for 0 (G1/S phase), 3 (S phase), or 8 (G2/M phase) hours (h). The cell cycle progression of these cells was monitored by flow cytometry using propidium iodide and pHH3 (S10) staining to monitor the mitotic status (). Consistent with previous reports,Citation14 the basal activity of AMPK measured by either pAMPKαThr172 or the phosphorylation status of its substrate acetyl CoA carboxylase (ACC) and myosin regulatory light chain (MRLC) was high in G2/M phase cells (, and Fig. S1A). Notably, we observed essentially the same findings by using LKB1-intact cells, i.e., U2OS cells and A431 cells (Fig. S1A). In addition, we observed that active pAMPKαThr172 localized on the spindle poles during prophase and metaphase, and on the midbody during telophase (), which was also consistent with previous report.Citation15 The observation that Inhibition of AMPK activity induced multinucleated cells (Fig. S1C) might be related with the localization on the midbody. These data show that AMPK is activated during G2/M phase, when it localizes to various components of the mitotic apparatus.
Figure 1. AMPKα is highly phosphorylated in G2/M phase and localized to mitotic apparatus. (A and B) HeLa cells were synchronized at the G1/S border by DTB and released for 0 h (G1/S phase), 3 h (S phase), and 8 h (G2/M). (A) Cells were fixed, stained with propidium iodide and phospho-histon H3 (S10) to visualize mitotic cells, and analyzed by flow cytometry. The percentages of mitotic cells in G1/S, S, and G2/M phases are indicated in dashed box. (B) Cells were lysed, and phosphorylation of AMPKα at threonine 172 and histone H3 at serine 10 was detected by Western blot analysis using specific antibodies. Detection of α-tubulin served as a loading control. Lysates were also analyzed using AMPKα1/2- and histone H3-specific antibodies to ensure that total protein levels were unchanged. Shown is a representative data from 3 independent experiments. (C) Localization pattern of pAMPKαThr172 signals during mitosis. At 9 h after release from DTB, cells were fixed and stained for pAMPKαThr172 for active AMPK, for γ-tubulin to recognize the spindle poles and with DAPI to visualize the nucleus. Shown is a representative image from 3 independent experiments. Scale bar, 10 μm.
AMPK inhibitor-treated cells spend prolonged periods in G2 phase and enter mitosis at a reduced rate
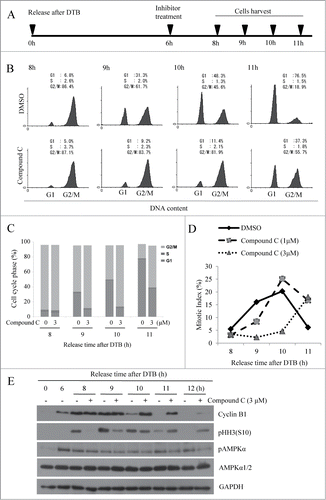
To further ascertain the involvement of AMPK in mitotic entry and progression, we synchronized HeLa cells at the G1/S border using DTB according to the scheme shown in . In brief, HeLa cells were synchronized using DTB and treated with a pharmacological AMPK inhibitor (compound C,32 final concentration 3 μM) at early G2 phase (6 h post-release). The cells were then harvested at various time points, and cell cycle progression was monitored by flow cytometry using propidium iodide staining ( and C). We found that compound C-treated cells progressed through G2/M phase much more slowly than control cells. To dissect whether this was due to a delay in the G2/M transition and/or mitotic progression, we used aceto-orcein staining to analyze the mitotic index. We found that the increase in the mitotic population was dramatically and dose-dependently delayed in compound C-treated cells versus control cells (), indicating that the delay in G2/M progression mainly stemmed from a delay in the G2/M transition. To corroborate this finding, we further analyzed 2 mitotic markers, phosphorylation of histone H3 at serine 10 and degradation of cyclin B1, in control and compound C-treated cells (). In control cells, histone H3 phosphorylation was first detected 8 h post-release; it peaked at 9 h post-release, and decreased at 11 and 12 h post-release. In contrast, compound C-treated cells displayed a weaker and delayed histone H3 phosphorylation, indicating the delay in the G2/M transition. Notably, histone H3 phosphorylation was still observed at 12 h post-release in compound C-treated cells, which is consistent with the observed delay in their mitotic progression. Since cyclin B1 level increases during G2 phase and decreases around anaphase onset,Citation33 delay in G2/M transition would result in normal increase, but delayed decrease of cyclin B1; it was the case as shown in . Inhibition of AMPK activity was confirmed by measuring substrate phosphorylation both in HeLa cells (Fig. S2A) and U2OS cells (Fig. S2B). The delay in mitotic entry was also observed in U2OS cells (Fig. S2C and D) and A431 cells (Fig. S3A and B). These results indicate that pharmacological inhibition of AMPK delayed the G2/M transition, suggesting that AMPK could regulate mitotic entry.
Figure 2. AMPK inhibition interferes with mitotic entry of synchronized HeLa cells. HeLa cells were synchronized at the G1/S border by DTB and released for 6 h (early G2 phase). At that time, cells were treated with various concentrations of compound C (0, 1, or 3 μM) and collected at indicated times after release. (A) Scheme showing the experimental design. (B) Cells were fixed, stained with propidium iodide, and analyzed by flow cytometry. The percentages of cells in G1, S, and G2/M phases are indicated in (C). (D) Cells were stained with aceto-orcein solution to visualize mitotic cells and the percentage of mitotic cells was quantified. More than 300 cells were counted for each condition. (E) Cells were lysed, and pHH3 (S10) and cyclin B1 was detected by Western blot analysis using specific antibodies. Detection of GAPDH served as a loading control. Shown is a representative data from 3 independent experiments.
Depletion of AMPKα2, but not AMPKα1, delays mitotic entry
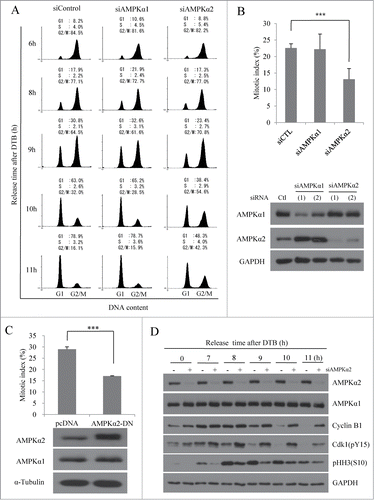
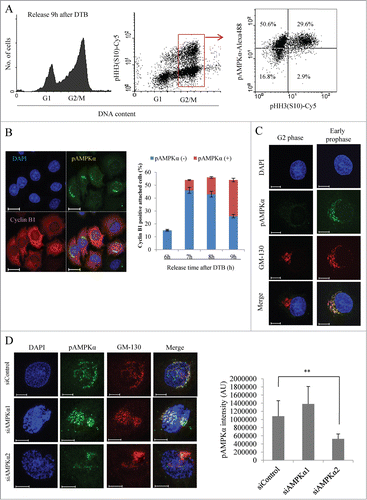
In mammals, there are 2 catalytic subunits of the AMPKα (i.e., AMPKα1 and AMPKα2), which show different intracellular localization, and different responsiveness to AMP and LKB1.Citation3 To further investigate the potential mitotic roles of the 2 catalytic subunits, we used specific siRNAs to deplete AMPKα1 or AMPKα2, and monitored cell cycle progression. We found that the cell cycle progression of AMPKα1-depleted cells was unaltered compared to that of control cells, but AMPKα2-depleted cells (similar to compound C-treated cells) progressed through G2/M phase much more slowly than did control cells (). In a parallel approach, we determined the mitotic index of HeLa cells lacking AMPKα1 or -α2 at 9 h post-release. As shown in , the proportion of mitotic cells was significantly lower in AMPKα2-depleted cells than in control cells. This further confirmed that the slower progression of AMPKα2-depleted cells through G2/M phase stemmed from a delay in mitotic entry. Experiments using additional siRNAs against AMPKα1 and AMPKα2 yielded similar results (), excluding the possibility of off-target effects and proving the specificity of the utilized siRNAs. We also analyzed whether a dominant-negative form of AMPKα2 could delay mitotic entry. In line with the results of our knockdown experiments, we found that mitotic cells were significantly decreased in AMPKα2-DN-transfected cells compared with control cells (), indicating that the activity of AMPKα2 is necessary for mitotic entry. Western blot analysis of cells released from DTB also clearly showed that cells lacking AMPKα2 experienced delays in cyclin B1 degradation and in histone H3 (S10) phosphorylation (), providing additional evidence that the delay in G2/M phase progression was due to a delay in the G2/M transition. Knockdown of AMPKα1 clearly decreased pAMPKα signal and the phosphorylation of its substrates, ACC and MRLC as well, indicating that the extent of AMPKα1 knockdown was enough to significantly reduce the activity of this isoenzyme (Fig. S4A and B). Intriguingly, knockdown of AMPKα2 changed neither pAMPKα signal nor the phosphorylation of its substrates, indicating that pAMPKα2 may be actually a minor form of activated AMPK at the whole cell level comparing to pAMPKα1, which was consistent with the much higher pAMPKα signals from AMPKα1 comparing to AMPKα2 after IP-western blotting of each isoenzyme (Fig. S1B). The delay in G2/M transition by depletion of AMPKα2, but not AMPKα1, was also observed in U2OS cells (Fig. S4C and D). Notably, the inhibitory phosphorylation of cdk1 at Tyr15, which controls mitotic entry and progression, was observed at later time points in AMPKα2-depleted cells compared to control cells (); it is not yet known whether this is a cause or effect of the G2/M transition delay. Collectively, these results clearly suggest that AMPKα2, but not AMPKα1, is the catalytic subunit of AMPK that regulates the G2/M transition in mammalian cells.
Figure 3 (See previous page). Depletion of AMPKα2, but not AMPKα1, delays the mitotic entry. HeLa cells transfected with a control siRNA-, AMPKα1-, or AMPKα2-specific siRNAs were synchronized at the G1/S border by DTB and released for the indicated times. (A) Cells were fixed, stained with propidium iodide, and analyzed by flow cytometry. The percentages of cells in G1, S, and G2/M phases are indicated. (B) At 9 h after the release from DTB, cells were stained with aceto-orcein solution and mitotic cells were counted. Cells were lysed, and expression of AMPKα1 and AMPKα2 was analyzed by Western blot analysis using specific antibodies. Detection of GAPDH served as a loading control. ***, p < 0.001 by one-way ANOVA analysis. (C) HeLa cells transfected with pcDNA or AMPKα2-DN construct were synchronized at the G1/S border by DTB and released for 8 h before analysis. Cells were stained with aceto-orcein solution and mitotic cells were counted. Cells were also lysed, and expression of AMPKα2-DN construct was analyzed by Western blot analysis using specific antibodies. Detection of α-tubulin served as a loading control. ***, P < 0.001 by Student's t-test. (D) HeLa cells transfected with a control siRNA, or AMPKα2-specific siRNAs were synchronized at the G1/S border by DTB and released for the indicated times. Cells were lysed, and subjected to SDS-PAGE and immunoblot analysis using indicated antibodies. Detection of GAPDH served as a loading control.
AMPKα2 depletion delays mitotic Golgi fragmentation
Two checkpoints regulate the G2/M transition: the DNA damage checkpoint, and the Golgi mitotic checkpoint. The former plays a key role in maintaining chromosomal integrity by allowing cells to repair DNA damage before entering mitosis. To exclude the possibility that AMPK inhibition delays mitotic entry by inducing DNA damage, we assessed the activation of DNA damage signaling in our system by measuring the phosphorylation of histone H2A.X at Ser139 (a molecular marker of DNA double-strand breaks). We failed to detect increased H2A.X phosphorylation in either compound C-treated or AMPKα2-depleted cells compared with control cells (Fig. S5), excluding the presence of DNA damage.
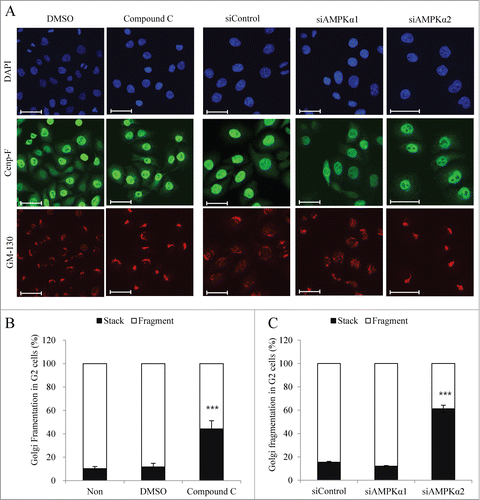
To ensure correct partitioning between daughter cells, Golgi membranes are dispersed by a 2-step fragmentation process that completely vesiculates the Golgi ribbon into small vesicles.Citation19 The first step of this fragmentation process occurs during G2 phase and is characterized by cleavage of the tubular bridges that connect adjacent stacks. The inhibition of proteins involved in ribbon cleavage has been shown to arrest cells in G2 phase,Citation26 and recent studies have demonstrated that GBF1 is a novel AMPK substrate, and that phosphorylation of GBF1 by AMPK is necessary for mitotic Golgi disassembly.Citation30,31 To analyze whether AMPK is involved in mitotic Golgi fragmentation, we analyzed DTB-synchronized HeLa cells that were treated with compound C or transfected with siControl, siAMPKα1, or -α2. At 9 h post-release, the cells were fixed and processed for immunofluorescent analysis using antibodies against Cenp-F (to identify G2 phase cells) and GM-130 (to identify the Golgi apparatus). Inhibition of AMPK by compound C strongly reduced the proportion of G2 cells harboring a fragmented Golgi apparatus (). Furthermore, AMPKα2-depleted cells, but not AMPKα1-depleted cells, showed significant delays in mitotic Golgi fragmentation (), suggesting that AMPKα2, but not AMPKα1, is the catalytic subunit of AMPK that contributes to mitotic Golgi fragmentation during G2 phase.
Figure 4. AMPKα2 knockdown induces the delay of mitotic Golgi fragmentation. HeLa cells were synchronized at the G1/S border by DTB and released for 6h (early G2 phase). After then, cells were treated with DMSO or compound C (final 3 μM) and collected after 3 h. In parallel, HeLa cells transfected with a control siRNA, AMPKα1-, or AMPKα2-specific siRNAs were synchronized at the G1/S border by DTB and collected after release for 9 h. (A) Cells were fixed and processed for immunofluorescence using antibodies against Cenp-F for G2 phase and GM-130 for the Golgi apparatus. Scale bar, 50 μm. (B and C) The structure of the Golgi apparatus was analyzed by immunofluorescence analysis in which we counted the percentage of cells showing a fragmented or stacked Golgi apparatus among Cenp-F-positive G2 phase cells. More than 200 cells were counted for each condition. ***, p < 0.001 by one-way ANOVA analysis.
Active AMPKα signaling is increased during late G2 phase, when it partially localizes to fragmented Golgi vesicles
Since inhibition of AMPK delayed mitotic Golgi fragmentation and induced transient G2/M arrest, we hypothesized that AMPK is activated during both G2 phase and mitosis. To test this, we enriched HeLa cells at G2/M phase (at 8 h post-release from DTB), co-stained them with propidium iodide (for DNA), pAMPKαThr172 (for activated AMPK) and pHH3 (S10) (for mitotic cells), and performed flow cytometric analysis. As expected, >90% [=29.6/(29.6 + 2.9)] of mitotic cells showed active AMPK signal, but, strikingly, activation of AMPK was also observed in 70% [=50.6/(50.6 + 16.8)] of G2 phase cells(). Since this indicated that AMPK is activated in G2 phase cells, we further analyzed the localization of active AMPK in G2 phase cells. HeLa cells were enriched at G2/M phase (at 8 h post-release) and processed for immunofluorescent analysis using antibodies against cyclin B1 (for late G2 phase; we counted cyclin B1-positive attached cells with intact nuclei that may include a minute fraction of early prophase cells) and pAMPKαThr172 (for active AMPK). Notably, the pAMPKαThr172 signals were transiently increased in the perinuclear region of cyclin B1-positive G2 phase cells (), suggesting that it may be localized to the Golgi apparatus. Furthermore, the percentage of G2 phase cells containing pAMPKαThr172 signals was significantly higher at 9 h post-release compared to 6 h post-release, indicating that AMPK is activated during late G2 phase. To further analyze the localization of these pAMPKαThr172 signals, we stained synchronized HeLa cells at G2/M phase, and used specific antibodies against pAMPKαThr172 and GM-130 (to visualize the Golgi apparatus). The pAMPKαThr172 signals partially co-localized with those of GM-130, and were higher in cells containing fragmented Golgi vesicles compared to those containing the stacked form (; we also observed the signal at centrosome). The pAMPKαThr172 signal was also observed at or near Golgi vesicles and centrosome by using different antibody (Santa Cruz, sc-101630), and was disappeared when the antibody was pre-incubated with blocking peptide (Fig. S6), confirming the specificity of our observation. Moreover, depletion of AMPKα2, but not AMPKα1, robustly abolished pAMPKα signal from Golgi fragments (), clearly indicating that the Golgi pAMPKαThr172 signal was mainly derived from AMPKα2. Although pAMPKα2 is a minor form of activated AMPK at the whole cell level during G2/M phases (Fig. S1B, Fig. S4A and B), it seemed to specifically localize at or near Golgi and affect the Golgi fragmentation. On the other hand, centrosomal pAMPKαThr172 signal, that was increased as centrosome matured (Fig. S7A), was partially abolished by depletion of either isoenzyme (Fig. S7B), indicating that both pAMPKα1 and pAMPKα2 are active at the centrosome. Taken together, these results suggest that activation of AMPKα2 increases during late G2/early prophase at or near the fragmented Golgi vesicles, and this spatio-temporal activation of specific isoenzyme seems to contribute to further fragmentation of Golgi vesicles, thus affects G2/M transition.
Figure 5 (See previous page). AMPKα is activated in late G2 phase and transiently localized to the fragmented Golgi vesicles. HeLa cells were synchronized at the G1/S border by DTB and released for the indicated times. (A) At 9 h after the release, cells were fixed, stained with propidium iodide, pAMPKαThr172, and pHH3 (S10), and then analyzed by flow cytometry. Left panel shows DNA profiles. Middle panel shows density plot of DNA contents vs. pHH3(S10). Right panel shows scatter plot of pAMPKαThr172 vs. pHH3(S10) in G2/M phase cells gated by DNA contents. (B) At the indicated time points after the release from DTB, cells were fixed and processed for immunofluorescence using antibodies against pAMPKαThr172 and cyclin B1. The DNA was stained with DAPI. Cyclin B1-positive cells were calculated as percentages of cells with cyclin B1 signal at the indicated times. The percentages of pAMPKα-positive or negative cells among cyclin B1-positive cells were indicated. More than 200 cells were counted for each condition. Scale bar, 20 μm. (C) At 9 h after the release, cells were fixed and processed for immunofluorescence using antibodies against pAMPKαThr172 and GM-130. Cells were also labeled with DAPI to determine mitotic stage. Scale bar, 10 μm. (D) HeLa cells transfected with a control siRNA, AMPKα1-, or AMPKα2-specific siRNAs were synchronized by DTB and collected after release for 9 h. Cells were fixed and processed for immunofluorescence using antibodies against pAMPK αThr172 and GM-130 for the Golgi apparatus (left panel). To calculate pAMPKαThr172 intensity at Golgi apparatus, GM-130 positive area was first selected by using a threshold value, and the mean intensity of pAMPKαThr172 signal on that GM-130 positive area was measured. The pAMPKαThr172 intensity (AU) was calculated by multiplication of mean intensity by area (right panel). More than 10 cells were analyzed. Scale bar, 10 μm. **, P < 0.01 by one-way ANOVA analysis.
Inhibition of CaMKKβ decreases the pAMPKα signal and delays both Golgi fragmentation and mitotic entry
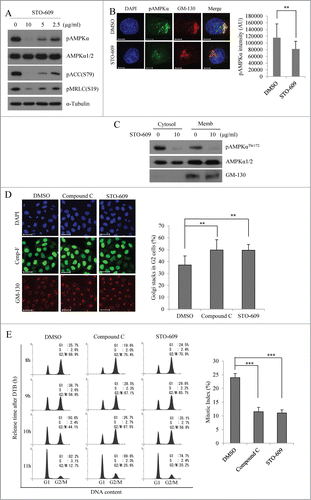
LKB1 and CaMKKβ are known to phosphorylate AMPKα at Thr172.Citation4 Since HeLa cells harbor a homozygous deletion of LKB1,Citation34 and recent studies have shown that CaMKKβ phosphorylates AMPKα during mitosis,Citation17 we investigated whether CaMKKβ is an upstream kinase of active AMPK during the G2/M transition and mitotic Golgi fragmentation. HeLa cells were treated with different concentrations of STO-609 (an inhibitor of CaMKKβ; 0, 2.5, 5, or 10 μg/ml) for 3 h beginning at 6 h post-release, and then subjected to Western blot analysis. As expected, STO-609 treatment significantly and dose-dependently decreased the pAMPKαThr172 signals and the phosphorylation of its downstream targets, pACC and pMRLC (). U2OS cells showed the same results (Fig. S2B). Furthermore, the pAMPKαThr172 signals colocalized with GM-130 signals were clearly abolished by treatment with STO-609 (), which was consistent with a significant decrease of pAMPKαThr172 signals by STO-609 treatment from membrane fraction (), and indicates that activation of AMPK at Golgi apparatus during G2/M phase is dependent on CaMKKβ activity. Similar to our results in compound C-treated cells, STO-609-treated cells showed delayed mitotic Golgi fragmentation (), and progressed through G2/M phase much more slowly ( for HeLa cells; Fig. S2C and D for U2OS cells and Fig. S3 for A431 cells) compared to control cells. The mitotic index of cells treated with 10 μg/ml STO-609 was decreased to a level similar to that seen in cells treated with 3 μM compound C (), indicating a delay in the G2/M transition. Furthermore, CaMKKβ-depleted cells by specific siRNA also showed significant delays in mitotic Golgi fragmentation (Fig. S8). Taken together, these data suggest that CaMKKβ activates AMPK during late G2 phase, and that both mitotic Golgi fragmentation and the G2/M transition are regulated by the CaMKKβ-AMPK signaling pathway.
Figure 6. Inhibition of CaMKKβ reduces the signals of active AMPKα and induces the delay of mitotic Golgi fragmentation as well as mitotic entry. HeLa cells were synchronized at the G1/S border by DTB and treated with different concentration of STO-609 (0, 2.5, 5, or 10 μg/ml) at 6 h after the release from DTB, and then collected after 3 h. (A) At 9 h after the release, cells were lysed and pAMPKαThr172 was detected by Western blot analysis. Detection of AMPKα1/2 served as a loading control to ensure that total protein levels were unchanged. Shown is a representative data from 3 independent experiments. (B) At 6 h after the release, cells were treated with DMSO or STO-609 (10μg/ml) and collected after 3 h. The cells were then fixed and processed for immunofluorescence using antibody against GM-130 or pAMPKαThr172. The calculation of pAMPKαThr172 intensity at Golgi apparatus was performed as described in the legend for . More than 20 cells were analyzed. **, P < 0.01 by Student's t-test. Scale bar, 10 μm. (C) The cytosol or the membrane fraction proteins were isolated from DMSO or STO-609 (10 μg/ml) treated cells and immunoblotted with the indicated antibodies. (D) At 8 h after the release, cells were fixed and processed for immunofluorescence using antibodies against Cenp-F for G2 phase and GM-130 for the Golgi apparatus. The structure of the Golgi apparatus was analyzed by immunofluorescence analysis in which we counted the percentage of cells showing the stacked Golgi apparatus among Cenp-F-positive G2 phase cells. More than 500 cells were counted for each condition. Scale bar, 50 μm. **, P < 0.01 by one-way ANOVA analysis. (E) At 6 h after the release, cells were treated with DMSO, compound C (final 3 μM), or STO-609 (10 μg/ml) and released for the indicated times. Cells were fixed, stained with propidium iodide, and analyzed by flow cytometry. At 9 h after the release, cells were stained with aceto-orcein solution and mitotic cells were counted. ***, P < 0.001 by one-way ANOVA analysis.
Discussion
The AMP-activated protein kinase (AMPK) is a sensor of cellular energy homeostasis and an important mediator of various cellular processes, including autophagy, cell polarity and cell growth.Citation1,35 In particular, recent studies have demonstrated a crucial link between mitotic progression and AMPK activity.Citation14,17 Through a chemical genetic screen, Banko et al. identified novel AMPKα2 substrates, and found that they were enriched in proteins related to cytoskeletal dynamics, mitosis and cytokinesis.Citation16 In mammalian cells, the active form of the catalytic AMPKα subunit has been directly associated with multiple mitotic structures during each stage of mitosis.Citation15 Here, we focused on the role of AMPK during the G2/M transition.
During mitosis, the Golgi apparatus undergoes extensive fragmentation via a multistage process that begins during G2 phase, when the Golgi ribbon is severed into its constituent stacks. Inhibition of mitotic Golgi fragmentation results in cell cycle arrest at G2 phase, suggesting that correct segregation of the Golgi apparatus is monitored by a “Golgi mitotic checkpoint."Citation19 Recent studies have addressed the question of how Golgi fragmentation is functionally connected to regulating mitotic entry. Blockade of Golgi fragmentation was found to impair the recruitment and centrosomal activation of aurora-A, which in turn impaired the first activation of Cdk1-cyclin B at the centrosome during early G2 phase.Citation36 This Golgi-dependent G2 block was not mediated by the known activators of aurora-A (i.e., Pak1, HEF1, Ajuba). Thus, further work is needed to clarify the responsible molecules and mechanisms. Despite the relative lack of knowledge, Golgi fragmentation has been regarded as a preriquisite for mitotic entry.
Herein, we, for the first time, showed the involvement of CaMKKβ-AMPKa2 axis in the regulation of Golgi fragmentation. Since we observed the transient activation of AMPKα2 irrespective of the presence of LKB1, it is highly probable that CaMKKβ, but not LKB1, mainly controls Golgi fragmentation through activating AMPKα2, which subsequently regulates G2/M transition of the cell cycle. Since the activity of CaMKKβ is not regulated by energy status,Citation8-Citation10 the regulation of Golgi fragmentation by CaMKKβ-AMPKα2 axis at least partially explains the upstream of the previously proposed energy-independent role of AMPK during mitotic progression.Citation15 Considering the significant decrease of pAMPKα signal by STO-609 ( and C; whole cell level and in both cytosol and membrane fraction), CaMKKβ-AMPKα1 axis might also exist and regulate other mitotic processes reportedly regulated by AMPK.Citation17
Previously, CaMKKβ-mediated phosphorylation of AMPK was shown to occur in response to increased intracellular Ca2+ in LKB1-deficient cells.Citation12 Green et al. demonstrated that CaMKKβ and AMPK associate through their kinase domains; furthermore, CaMKKβ must be in an active conformation to bind AMPK, but this is not required for its association with other substrates, such as CaMKIV.45 These findings suggest that the signals responsible for modifying the activation status of CaMKKβ may act as molecular switches to couple CaMKKβ with AMPK- and/or CaMK-dependent pathways.44 The questions such as how the CaMKKβ is activated during G2/M transition and whether it localizes at Golgi apparatus during this period are waiting to be addressed.
AMPK functions as a heterotrimer that consists of a catalytic α subunit (α1 or α2) and the regulatory subunits, β (β1 or β2) and γ (γ1, γ2, or γ3). Although the 2 AMPKα isoforms are highly homologous, multiple reports indicate that AMPKα1 and AMPKα2 have mutual and exclusive functions.Citation3 Regarding the role of AMPK during mitotic progression, Pinter and colleagues showed that the α2β2γ2 heterotrimeric complex associates with the mitotic apparatus,Citation37 and Banko et al. identified novel AMPKα2 substrates that were enriched for proteins involved in cytoskeletal dynamics, mitosis and cytokinesis,Citation16 suggesting the possible roles of AMPKα2 during mitosis. We observed that both catalytic α subunits (α1 and α2) were activated during G2/M phases, and AMPKα1, but bot AMPKα2, is a major activated form of AMPKα during this period (Fig. S1B). Knockdown experiments further confirmed that AMPKα1 is a major activated form. Whereas depletion of AMPKα1 reduced pAMPKα, pACC and pMRLC significantly in G2/M phase cells, depletion of AMPKα2 did not affect their phosphorylation (Fig. S4A).
Then, how did the depletion of AMPKα2, but not AMPKα1, delay Golgi fragmentation as well as mitotic entry ( and )? Our observation that pAMPKα signals at or near Golgi fragments during early prophase was clearly decreased by knockdown of AMPKα2, but not AMPKα1, () clearly indicates the possibility that AMPKα2 is specifically and transiently activated at Golgi apparatus during late G2/ early prophase, and regulates Golgi fragmentation which affects G2/M transition. In contrast, as previously reported, AMPKα1 seems to regulate later events of mitosis, such as spindle pole formation, cleavage furrow formation, and completion of cytokinesis through phosphorylation of MRLC.Citation13,16,17 We suggest that these 2 isoforms of AMPKα are spatio-temporally and specifically regulated during G2/M phases, and their substrate specificity would contribute to their specific roles during this period of cell cycle.
What would be the mitotic Golgi substrate of AMPKα2? A recent study found that GBF1 is a substrate of AMPK, and that it is involved in regulating mitotic Golgi fragmentation, but this study did not specify the isoenzyme type.Citation31 Furthermore, the chemical genetic screening of Banko et al. identified a number of Golgi-associated proteins as substrates of AMPK,Citation16 suggesting that there are many Golgi substrates of AMPK that have not yet been fully investigated.
In conclusion, we herein propose that AMPKα2 is specifically activated at the Golgi apparatus of late G2/early prophase cells by CaMKKβ, and the CaMKKβ-AMPK complex contributes to mitotic Golgi fragmentation and the G2/M transition.
Materials and Methods
Antibodies, chemicals, and plasmids
The following antibodies were used: mouse monoclonal antibodies to α-tubulin (Abcam, ab7291), AMPKα1/2 (Cell signaling, 2603), AMPKα1 (R&D systems, AF3197), AMPKα2 (R&D systems, AF2850), pAMPKα (Cell signaling, 2535), pAMPKα (Santa Cruz, sc-101630), MRLC (Cell signaling, 3672), pMRLC(S19) (Cell signaling, 3671), pACC(S79) (Cell signaling, 3661), γ-Tubulin (Sigma, T6557), Cyclin B1 (Santa Cruz, sc-245), phospho-histone H3(S10) (Cell signaling, 9706), Cdk1-pY15(Cell signaling, 9111), Histon H3 (Cell signaling, 4499), GM-130 (BD Biosciences, 610822), Cenp-F (Abcam, ab5), GAPDH (Cell signaling, 2118), and pH2A.X-S139 (Millipore, 05-636). Horseradish-peroxidase conjugated secondary antibodies and blocking peptide for pAMPKαThr172 (sc-33524 P) were obtained from Santa Cruz. The following fluorochrome-conjugated secondary antibodies were used: anti-mouse Alexa-488 (Invitrogen, A11059), anti-rabbit Alexa-488 (Invitrogen, A11034), anti-mouse Cy5 (Invitrogen, A10524) and anti-mouse Cy3 (Jackson ImmunoResearch, 715-165-151). The AMPK inhibitor Compound C (171260) was obtained from CalBiochem, and CaMKKβ inhibitor STO-609 (S1313) was from Sigma-Aldrich. Protease inhibitors, phosphatase inhibitors and other reagents were purchased from Sigma-Aldrich. The AMPKα2-DN mutant construct was generously provided by Prof. Joohun Ha (Kyung Hee University, Seoul, Republic of Korea).Citation38
Cell culture and synchronization by double-thymidine treatment
HeLa cells were maintained in MEM (Welgene, LM007-01) supplemented with 10% (v/v) fetal bovine serum (Gibco, 16000-044) in 37°C incubator with 5% CO2 in air. HeLa cells were synchronized at the G1/S border using a double-thymidine block. In brief, cells grown on culture dishes or coverslips were incubated in growth medium containing 1 mM thymidine (Sigma, T9250) for 18 h. Afterward, cells were released from the thymidine block by washing with serum-free medium and were cultured in growth medium for 9h. Subsequently, cells were subjected to the second thymidine block for an additional 16 h. After the second release, cells were collected at distinct time points. For inhibition of AMPK or CaMKKβ, each inhibitor (compound C or STO-609) was treated at 6 h after DTB release (early G2 phase) and collected after 3 h.
Transfection experiment
HeLa cells were transfected with siRNAs using Oligofectamine (Invitrogen, 58303), according to manufacturer's instruction. Plasmid transfection was performed using Lipofectamine 2000 (Invitrogen, 52887), according to manufacturer's instruction. The siRNAs were purchased from Ambion. The sequences of the sense strands of the siRNA were as follows: 2 siRNAs for AMPKα1, 5′-GGUUGGCAAACAUGAAUUCdTdT-3′; 5′-CUGAGUUGCAUAUACUGUA-dTdT-3′, 2 siRNAs for AMPKα2, 5′-GGUUUCUUAAAAACAGCUGdTdT-3′; 5′-GGCCAUAAAGUGGCAGUUAdTdT-3′.
Immunoblotting
Conventional immunoblotting was performed as previously described using indicated antibodies.Citation39 Briefly, cells were washed in PBS and lysed in lysis solution [20 mM Tris-HCl (pH7.4), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, 5 mM NaF, 0.5 mM Na3VO4, 1 mg/ml Leupeptin, 1mg/ml Aprotinin]. The lysate was resuspended in Laemmli sample buffer and boiled for 5 min. The samples were separated by SDS-PAGE and then were transferred to PVDF membrane. After blocking for 1 h at room temperature with blocking solution [PBS containing 0.05% (V/V) Tween-20 and 5% (w/v) non-fat milk], the membranes were incubated at 4°C with indicated antibodies. Membranes were then washed 3 times with PBS containing 0.05% Tween-20 and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody for 1 h at room temperature. Detection was carried out using ECL reagents (Amersham Biosciences, RPN2106) and by exposing them to x-ray film.
Preparation of cell membrane fractions
Cell membrane fractions were separated as described previously.Citation40 In brief, cells were lysed in extracting buffer containing 10 mM Hepes, pH 7.4, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.2 mM sodium orthovanadate, 1 mM NaF, and 0.5 mM PMSF, followed by incubation on ice for 15 min. Then, cells were passed through a 26-gauge needle 10 times and the homogenate was centrifuged at 14,000 rpm for 1 min to separate the nuclei from the cytosolic and membrane proteins in the cytoplasmic supernatant. The nuclei-free cytoplasmic supernatant was further centrifuged for 1.5 h at 50,000 rpm at 4°C. The supernatant fractions were saved as the cytosol proteins. The pellets containing membrane proteins were re-extracted with SDS-PAGE sample buffer and subjected to protein gel blot analysis.
Immunofluorescence analysis
HeLa cells were grown on coverslips, washed with PBS, and fixed with ice-cold methanol for 10 min at −20°C. Fixed cells were pre-incubated in blocking solution (1% BSA in PBS) and incubated with primary antibodies diluted in blocking buffer (1:200) for overnight at 4°C. Cells were then washed and probed with fluorescence-conjugated secondary antibody for 1 h at room temperature. For DNA counterstaining, DAPI (Molecular probes, D3571) was used. After washing, cells were mounted in the mounting solution and analyzed on a confocal microscope (LSM710, Carl Zeiss).
Quantification of pAMPKα intensity and statistical analysis
Z-series stacks of images at 0.44 μm intervals through each cell were obtained using a Zeiss LSM-710 confocal microscope. For quantification, all images were acquired at identical settings. Early prophase cells were randomly selected and analyzed for fluorescence intensity. To calculate pAMPKαThr172 intensity, the mean intensity and areas of pAMPKαThr172 signal from projected images with equal threshold were measured. The pAMPKαThr172 intensity (AU) showed was calculated by multiplication of mean intensity by area. More than 20 cells were analyzed per one sample. Statistical analysis was performed by using the unpaired student's t-test in Excel (Microsoft, Redmond, WA) or one-way ANOVA analysis in R software (www.r-project.org).
Cell cycle analysis by flow cytometry
Trypsinized cells were pelleted, washed in cold phosphate-buffered saline (PBS), and fixed in ice-cold 1% paraformaldehyde for 30 min at 4°C. Cells were washed in ice-cold PBS, resuspended in 90% ice-cold methanol, and incubated overnight at −20°C. Cells were subsequently washed, blocked with blocking solution (1% BSA in PBS) for 30 min, and incubated with the primary antibodies diluted in blocking buffer (1:100) for 2h. The cells were washed and incubated with fluorescence-conjugated secondary antibodies diluted in blocking buffer (1:200) for 1 h. The cells were washed and incubated with 50 μg/ml propidium iodide (Sigma, P4170) containing RNase A (Sigma, R4875) for 10 min. The cells were analyzed with a FACSCanto II flow cytometer (Becton Dickinson). Data were plotted with WinMDI software. Ten thousand events were analyzed for each sample.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
991557_Supplementary_Materials.zip
Download Zip (10 MB)Acknowledgments
We thank Dr. Joohun Ha (Kyung-Hee University, Seoul, Korea) for AMPKα2-DN mutant construct.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2011-0030043).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2007; 8:774-85; PMID:17712357; http://dx.doi.org/10.1038/nrm2249
- Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 2008; 32 4:S7-12; PMID:18719601; http://dx.doi.org/10.1038/ijo.2008.116
- Salt I, Celler JW, Hawley SA, Prescott A, Woods A, Carling D, Hardie DG. AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the alpha2 isoform. Biochem J 1998; 334 (Pt 1):177-87; PMID:9693118
- Carling D, Sanders MJ, Woods A. The regulation of AMP-activated protein kinase by upstream kinases. Int J Obes (Lond) 2008; 32 Suppl 4:S55-9; PMID:18719600; http://dx.doi.org/10.1038/ijo.2008.124
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 2003; 13:2004-8; PMID:14614828; http://dx.doi.org/10.1016/j.cub.2003.10.031
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2003; 2:28; PMID:14511394; http://dx.doi.org/10.1186/1475-4924-2-28
- Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J 2005; 24:1810-20; PMID:15889149; http://dx.doi.org/10.1038/sj.emboj.7600667
- Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 2005; 280:29060-6; PMID:15980064; http://dx.doi.org/10.1074/jbc.M503824200
- Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2005; 2:21-33; PMID:16054096; http://dx.doi.org/10.1016/j.cmet.2005.06.005
- Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2005; 2:9-19; PMID:16054095; http://dx.doi.org/10.1016/j.cmet.2005.05.009
- Anderson KA, Ribar TJ, Lin F, Noeldner PK, Green MF, Muehlbauer MJ, Witters LA, Kemp BE, Means AR. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab 2008; 7:377-88; PMID:18460329; http://dx.doi.org/10.1016/j.cmet.2008.02.011
- Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, Means AR. Components of a calmodulin-dependent protein kinase cascade: molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase β. J Biol Chem 1998; 273:31880-9; PMID:9822657; http://dx.doi.org/10.1074/jbc.273.48.31880
- Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE, Lee SH, Shong M, Kim JM, Kim J, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 2007; 447:1017-20; PMID:17486097; http://dx.doi.org/10.1038/nature05828
- Vazquez-Martin A, Lopez-Bonet E, Oliveras-Ferraros C, Perez-Martinez MC, Bernado L, Menendez JA. Mitotic kinase dynamics of the active form of AMPK (phospho-AMPKalphaThr172) in human cancer cells. Cell Cycle 2009; 8:788-91; PMID:19221486; http://dx.doi.org/10.4161/cc.8.5.7787
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle 2009; 8:2385-98; PMID:19556893; http://dx.doi.org/10.4161/cc.8.15.9082
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villen J, Wang B, Kim SR, Sakamoto K, et al. Chemical genetic screen for AMPKalpha2 substrates uncovers a network of proteins involved in mitosis. Mol Cell 2011; 44:878-92; PMID:22137581; http://dx.doi.org/10.1016/j.molcel.2011.11.005
- Thaiparambil JT, Eggers CM, Marcus AI. AMPK Regulates mitotic spindle orientation through phosphorylation of myosin regulatory light chain. Mol Cell Biol 2012; 32:3203-17; PMID:22688514; http://dx.doi.org/10.1128/MCB.00418-12
- Lowe M. Structural organization of the Golgi apparatus. Curr Opin Cell Biol 2011; 23:85-93; PMID:21071196; http://dx.doi.org/10.1016/j.ceb.2010.10.004
- Corda D, Barretta ML, Cervigni RI, Colanzi A. Golgi complex fragmentation in G2/M transition: An organelle-based cell-cycle checkpoint. IUBMB Life 2012; 64:661-70; PMID:22730233; http://dx.doi.org/10.1002/iub.1054
- Hidalgo Carcedo C, Bonazzi M, Spano S, Turacchio G, Colanzi A, Luini A, Corda D. Mitotic golgi partitioning is driven by the membrane-fissioning protein CtBP3/BARS. Science 2004; 305:93-6; PMID:15232108; http://dx.doi.org/10.1126/science.1097775
- Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for golgi fragmentation during mitosis. Cell 1998; 92:183-92; PMID:9458043; http://dx.doi.org/10.1016/S0092-8674(00)80913-7
- Colanzi A, Sutterlin C, Malhotra V. RAF1-activated MEK1 is found on the golgi apparatus in late prophase and is required for golgi complex fragmentation in mitosis. J Cell Biol 2003; 161:27-32; PMID:12695496; http://dx.doi.org/10.1083/jcb.200208099
- Sutterlin C, Lin CY, Feng Y, Ferris DK, Erikson RL, Malhotra V. Polo-like kinase is required for the fragmentation of pericentriolar golgi stacks during mitosis. Proc Natl Acad Sci U S A 2001; 98:9128-32; PMID:11447294; http://dx.doi.org/10.1073/pnas.161283998
- Duran JM, Kinseth M, Bossard C, Rose DW, Polishchuk R, Wu CC, Yates J, Zimmerman T, Malhotra V. The role of GRASP55 in golgi fragmentation and entry of cells into mitosis. Mol Biol Cell 2008; 19:2579-87; PMID:18385516; http://dx.doi.org/10.1091/mbc.E07-10-0998
- Wang Y, Seemann J, Pypaert M, Shorter J, Warren G. A direct role for GRASP65 as a mitotically regulated golgi stacking factor. EMBO J 2003; 22:3279-90; PMID:12839990; http://dx.doi.org/10.1093/emboj/cdg317
- Rabouille C, Kondylis V. Golgi ribbon unlinking: an organelle-based G2/M checkpoint. Cell Cycle 2007; 6:2723-9; PMID:17986866; http://dx.doi.org/10.4161/cc.6.22.4896
- Lowe M, Rabouille C, Nakamura N, Watson R, Jackman M, Jamsa E, Rahman D, Pappin DJ, Warren G. Cdc2 kinase directly phosphorylates the cis-golgi matrix protein GM130 and is required for golgi fragmentation in mitosis. Cell 1998; 94:783-93; PMID:9753325; http://dx.doi.org/10.1016/S0092-8674(00)81737-7
- Preisinger C, Korner R, Wind M, Lehmann WD, Kopajtich R, Barr FA. Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for golgi checkpoint signalling. EMBO J 2005; 24:753-65; PMID:15678101; http://dx.doi.org/10.1038/sj.emboj.7600569
- Uchiyama K, Jokitalo E, Lindman M, Jackman M, Kano F, Murata M, Zhang X, Kondo H. The localization and phosphorylation of p47 are important for golgi disassembly-assembly during the cell cycle. J Cell Biol 2003; 161:1067-79; PMID:12810701; http://dx.doi.org/10.1083/jcb.200303048
- Miyamoto T, Oshiro N, Yoshino K, Nakashima A, Eguchi S, Takahashi M, Ono Y, Kikkawa U, Yonezawa K. AMP-activated protein kinase phosphorylates golgi-specific brefeldin a resistance factor 1 at thr1337 to induce disassembly of golgi apparatus. J Biol Chem 2008; 283:4430-8; PMID:18063581; http://dx.doi.org/10.1074/jbc.M708296200
- Mao L, Li N, Guo Y, Xu X, Gao L, Xu Y, Zhou L, Liu W. AMPK phosphorylates GBF1 for mitotic golgi disassembly. J Cell Sci 2013; 126:1498-505; PMID:23418352; http://dx.doi.org/10.1242/jcs.121954
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Inv 2001; 108:1167-74; PMID:11602624; http://dx.doi.org/10.1172/JCI13505
- Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 2009; 185:193-202; PMID:19364923; http://dx.doi.org/10.1083/jcb.200812045
- McCabe MT, Powell DR, Zhou W, Vertino PM. Homozygous deletion of the STK11/LKB1 locus and the generation of novel fusion transcripts in cervical cancer cells. Cancer Genet Cytogenet 2010; 197:130-41; PMID:20193846; http://dx.doi.org/10.1016/j.cancergencyto.2009.11.017
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13:1016-23; PMID:21892142; http://dx.doi.org/10.1038/ncb2329
- Persico A, Cervigni RI, Barretta ML, Corda D, Colanzi A. Golgi partitioning controls mitotic entry through aurora-A kinase. Mol Biol Cell 2010; 21:3708-21; PMID:20844084; http://dx.doi.org/10.1091/mbc.E10-03-0243
- Pinter K, Jefferson A, Czibik G, Watkins H, Redwood C. Subunit composition of AMPK trimers present in the cytokinetic apparatus: Implications for drug target identification. Cell Cycle 2012; 11:917-21; PMID:22333580; http://dx.doi.org/10.4161/cc.11.5.19412
- Kim MJ, Park IJ, Yun H, Kang I, Choe W, Kim SS, Ha J. AMP-activated protein kinase antagonizes pro-apoptotic extracellular signal-regulated kinase activation by inducing dual-specificity protein phosphatases in response to glucose deprivation in HCT116 carcinoma. J Biol Chem 2010; 285:14617-27; PMID:20220132; http://dx.doi.org/10.1074/jbc.M109.085456
- Nam HJ, Lee IJ, Jang S, Bae CD, Kwak SJ, Lee JH. p90 ribosomal S6 kinase 1 (RSK1) isoenzyme specifically regulates cytokinesis progression. Cell Signal 2014; 26:208-19; PMID:24269382; http://dx.doi.org/10.1016/j.cellsig.2013.11.014
- Cha H, Shapiro P. Tyrosine-phosphorylated extracellular signal–regulated kinase associates with the Golgi complex during G2/M phase of the cell cycle: evidence for regulation of golgi structure. J Cell Biol 2001; 153:1355-67; PMID:11425867; http://dx.doi.org/10.1083/jcb.153.7.1355