
Abstract
Approximately 25% of breast cancers overexpress and depend on the receptor tyrosine kinase ERBB2, one of 4 ERBB family members. Targeted therapies directed against ERBB2 have been developed and used clinically, but many patients continue to develop resistance to such therapies. Although much effort has been focused on elucidating the mechanisms of acquired resistance to ERBB2-targeted therapies, the involvement of ERBB4 remains elusive and controversial. We demonstrate that genetic ablation of ERBB4, but not ERBB1-3, led to apoptosis in lapatinib-resistant cells, suggesting that the efficacy of pan-ERBB inhibitors was, at least in part, mediated by the inhibition of ERBB4. Moreover, ERBB4 was upregulated at the protein level in ERBB2+ breast cancer cell lines selected for acquired lapatinib resistance in vitro and in MMTV-Neu mice following prolonged lapatinib treatment. Knockdown of ERBB4 caused a decrease in AKT phosphorylation in resistant cells but not in sensitive cells, suggesting that ERBB4 activated the PI3K/AKT pathway in lapatinib-resistant cells. Importantly, ERBB4 knockdown triggered apoptosis not only in lapatinib-resistant cells but also in trastuzumab-resistant cells. Our results suggest that although ERBB4 is dispensable for naïve ERBB2+ breast cancer cells, it may play a key role in the survival of ERBB2+ cancer cells after they develop resistance to ERBB2 inhibitors, lapatinib and trastuzumab.
Abbreviations
| EGFR | = | epidermal growth factor receptor |
| ERK | = | extracellular regulated kinase |
| FGFR | = | fibroblast growth factor receptor |
| HER | = | human epidermal growth factor receptor |
| MTS | = | 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium |
| PI3K | = | phosphatidylinositol-4,5-bisphosphate 3-kinase |
| Q-VD-OPh | = | quinolyl-valyl-O-methylaspartyl-[2,6-difluoro-phenoxy]-methyl ketone |
| RTK | = | receptor tyrosine kinase |
Introduction
The ERBB family of receptor tyrosine kinases (RTKs) includes 4 members: EGFR (ERBB1/HER1), ERBB2 (HER2/Neu), ERBB3 (HER3), and ERBB4 (HER4).Citation1 Upon ligand binding, these ERBB RTKs form homo- and hetero-dimers to activate downstream signaling cascades, including the ERK1/2 (extracellular regulated kinase) and PI3K (phosphatidylinositol-4,5-bisphosphate 3-kinase) pathways.Citation2 ERBB3 lacks significant intrinsic kinase activity, but can activate downstream signaling pathways, especially the PI3K pathway, through heterodimerization with other ERBB members.Citation3,4 In contrast, ERBB2 is a potent tyrosine kinase, but does not require a ligand for dimerization with ERBB members, including ERBB2 itself. Consequently, ERBB2 overexpression dysregulates ERBB RTK signaling, and is a known oncoprotein.
ERBB2 overexpression is observed in approximately 25% of human breast cancers and is associated with poorer overall survival and decreased time to relapse.Citation5 The development of targeted therapies against ERBB2 represents a major achievement in the treatment of ERBB2+ breast cancers. Several ERBB2-targeted inhibitors, including the humanized monoclonal antibody trastuzumab and the small molecule kinase inhibitor lapatinib, have been approved by the Federal Drug Administration (FDA) and have gone on to demonstrate clinical success. Despite their initial effectiveness, acquired resistance to ERBB2-targeted inhibitors unavoidably develops and poses a significant clinical problem that dramatically affects patient outcomes.Citation6,7 ,Citation8 Several molecular mechanisms underlying acquired resistance to ERBB2 inhibitors have been described, including activation of c-Src tyrosine kinase,Citation9 upregulation of ERBB3,Citation10 activating mutations in the p110α subunit of PI3K (PIK3CA),Citation11 and enhanced ERBB-ligand autocrine signaling.Citation12 Recently, we have also shown that cells with acquired resistance to lapatinib are highly anti-apoptotic due to the alteration of apoptotic pathways.Citation13
In the present study, we report that acquired resistance to ERBB2 inhibitors may be mediated, at least in part, by ERBB4. The role of ERBB4 in breast cancer remains elusive and controversial. A study using a transgenic mouse model clearly demonstrated that ERBB4 is dispensable for the growth of ERBB2+ breast cancer cells. Citation14 Nevertheless, ERBB4 can be an oncoprotein in other cancer types, such as melanoma and Ewing sarcoma.Citation15,16 Our results indicate, for the first time, that ERBB4 may play a critical role in acquired resistance to ERBB2 inhibitors in breast cancer.
Results
Previously, we generated lapatinib-resistant ERBB2-positive breast cancer cell lines through chronic lapatinib exposure in previously responsive lines and investigated the molecular mechanisms of acquired resistance to lapatinib.Citation13 Unlike resistance to other tyrosine kinase inhibitors, which is often associated with the emergence of a kinase variant that can no longer be successfully inhibited by that drug (e.g., imatinib-resistance mutations in BCR-ABL), activating mutations in ERBB2 are rarely found in breast cancer.Citation17 In support of this notion, we have found that lapatinib completely inhibits autophosphorylation of ERBB2 in lapatinib-resistant cells,Citation13 suggesting that acquired resistance is likely not attributable to ERBB2 insensitivity. Since lapatinib is a dual inhibitor of EGFR and ERBB2 (IC50 = 10.8 and 9.2 nM, respectively),Citation18 it suggests that cells with acquired resistance to lapatinib do not depend on the 2 kinases. We hypothesized that lapatinib-resistant cells might utilize an alternative kinase pathway(s) through which cells maintain survival signaling even in the presence of the inhibition of EGFR and ERBB2 by lapatinib.
To test this idea and to identify such a kinase pathway, we treated lapatinib-sensitive and -resistant cells (BT474 and BT474-LR cells, respectively) with a panel of kinase inhibitors and examined the affects on proliferation using a colorimetric MTS assay. As previously reported, ERBB2+ BT474 cells were sensitive not only to lapatinib (1 μM) but also to gefitinib (1 μM) and erlotinib (1 μM), both of which can inhibit ERBB2 (ref. 19 and 20) (). In contrast, BT474-LR cells showed marked resistance to this class of kinase inhibitors, indicating that upon acquiring resistance to lapatinib, the cells are also resistant to other ERBB2 inhibitors (). Of note, the proliferation of BT474-LR cells, but not BT474 cells, was attenuated in the presence of the fibroblast growth factor receptor (FGFR) inhibitor LY2874455 (). Although we did not quantitate expression levels of FGFR-2 in BT474 and BT474-LR cells, amplification of FGFR-2 has been implicated in the mechanism of acquired resistance to lapatinib in breast cancer cells.Citation21 Interestingly, inhibitors of the ERK-activating kinase MEK had no effects on proliferation of both BT474 and BT474-LR (see PD184352, PD98659, PD325901 and U0126 in ).
Figure 1. Pan-ERBB inhibitors cause apoptosis in BT474 cells with acquired resistance to lapatinib. (a) MTS assays reveal a panel of kinase inhibitors that attenuate proliferation of lapatinib-sensitive (left) and -resistant (right) BT474 cells (BT474 and BT474-LR, respectively). Cells were plated at a density of 1.0 × 104 per well on a 96 well plate, 18-24 hours before treatment. Cells were then treated with 1 μM of the indicated kinase inhibitors (except PD98059, U0126, and KU55933 at 10 μM). *P < 0.05 by t-test (n = 4, error bars indicate SD). (b) Lapatinib-resistant BT474 cells were treated with DMSO, lapatinib, afatinib, canertinib, dacomitinib, neratinib, or varlitinib (1 μM). Forty-eight hours later, cellular apoptosis was analyzed by Annexin V staining.
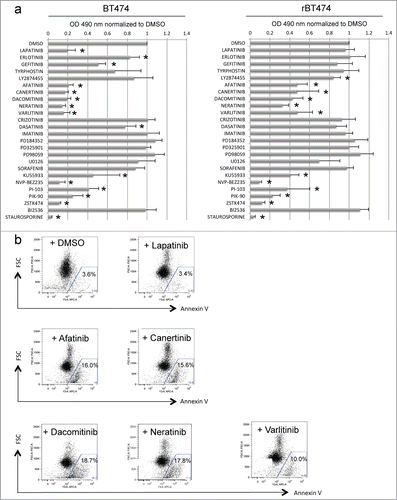
Importantly, both BT474 and BT474-LR were sensitive to a group of inhibitors that target PI3K family members (see KU55933, NVP-BEZ235, PI-103, PIK-90, and ZSTK474 in ), strongly suggesting the importance of the PI3K/AKT pathway for survival in these cells as described by other studies.Citation22,23 We tested whether overexpression of a constitutively active form of PIK3CA could confer lapatinib resistance to BT474 cells. We created BT474 cells stably expressing wild type or a constitutively active mutant (E545K or H1047) of PIK3CA (PIK3CA-WT, PIK3CA-E545K, and PIK3CA-H1047R, respectively). In cells expressing PIK3CA-E545K or PIK3CA-H1047R, but not PIK3CA-WT, AKT and ERK1/2 were markedly phosphorylated even in the presence of lapatinib (). When colony-forming assays were performed, cells expressing PIK3CA-E545K or PIK3CA-H1047R formed significantly more and larger colonies in the presence of lapatinib (). Taken together, these results strongly suggest that the PI3K/AKT pathway plays a crucial role in the survival of ERBB2+ breast cancer cells after exposure to an ERBB2 inhibitor.
In addition to the PI3K inhibitors, pan-ERBB inhibitors also significantly suppressed growth of both BT474 and BT474-LR cells (). Pan-ERBB inhibitors are a group of small molecules that broadly antagonize the kinase activity of all ERBB dimers. We tested 5 pan-ERBB inhibitors: afatinib, canertinib, dacomitinib, neratinib and varlitinib (). Among these inhibitors, afatinib, canertinib, dacomitinib, and neratinib “irreversibly” bind to ERBB1-4, as opposed to lapatinib which is a “reversible” inhibitor of EGFR and ERBB2. In contrast, varlitinib is a “reversible” pan-ERBB inhibitor. Both “reversible” and “irreversible” pan-ERBB inhibitors suppressed cellular proliferation () and triggered apoptosis in lapatinib-resistant cells (). Therefore, it is unlikely that the ERBB2-binding potency fully accounts for the efficacy of the pan-ERBB inhibitors. These results suggest that lapatinib-resistant cells are not dependent on EGFR and ERBB2 but still dependent on a kinase(s) that can be inhibited by the pan-ERBB inhibitors.
Since no inhibitors specific for ERBB3 and ERBB4 individually are currently available, we performed siRNA knockdown of each of the ERBB kinases (). We found that siRNA knockdown of ERBB2 or ERBB3 caused apoptosis in BT474 cells, but not in BT474-LR cells (). To our surprise, ERBB4 knockdown caused apoptosis in BT474-LR cells, but not in BT474 cells (). It should be noted that continued lapatinib treatment was not necessary for this effect. Therefore, these results indicate that lapatinib-resistant cells rely on ERBB4 for their survival. These results also suggest that the efficacy of the pan-ERBB inhibitors in killing lapatinib-resistant cells may be mediated by their ability to inhibit ERBB4 efficiently – lapatinib does not inhibit ERBB4 (IC50 = 367 nM) as well as pan-ERBB inhibitors (IC50 = 20–80 nM).Citation18,24,25
Figure 2. siRNA knockdown of each ERBB member shows that ERBB4 is required for breast cancer cells with acquired resistance to ERBB2+ inhibitors. (a) BT474 and BT474-LR cells were plated at a density of 0.4 × 106 per 6 cm plate. Eighteen hours later, cells were treated with control, EGFR, ERBB2, or ERBB3-specific siRNA (20 nM). After 72 hours, cells were harvested and protein levels of EGFR, ERBB2, ERBB3, and actin were analyzed by western blots. (b) Seventy-2 hours following siRNA knockdown, apoptosis was measured by Annexin-V staining. *P < 0.05 by t-test between control and ERBB2 and between control and ERBB3 (n = 4, error bars indicate SD). (c) BT474 and BT474-LR cells were plated at a density of 0.4 × 106 per 6 cm plate. Eighteen hours later, cells were treated with control or one of 2 ERBB4-specific siRNAs (a) or (b) (10 nM). After 72 hours, cells were harvested and protein levels of ERBB4 and actin were analyzed by protein gel blot. (d) Seventy-2 hours following siRNA knockdown in BT474, BT474-LR (lapatinib resistant), and BT474-TR (trastuzumab resistant), apoptosis was measured by Annexin-V staining. *p < 0.05 by t-test between control and ERBB4 (a) or (b) (n = 4, error bars indicate SD). (e) siRNA knockdown was performed using siRNA ERBB4 (b) in SKBR3, SKBR3-LR (lapatinib resistant), SKBR3-TR (trastuzumab resistant), and SKBR3-LTR (lapatinib and trastuzumab resistant) cells. After 72 hours, cells were harvested and protein levels of ERBB4 and actin were analyzed by western blot. (f) Seventy-2 hours after siRNA knockdown in SKBR3, SKBR3-LR, SKBR3-TR, and SKBR3-LTR, apoptosis was measured by Annexin-V staining. *p < 0.05 by t-test between control and ERBB4 (b) (n = 4, error bars indicate SD). Note that these knockdown experiments were carried out in the absence of lapatinib.
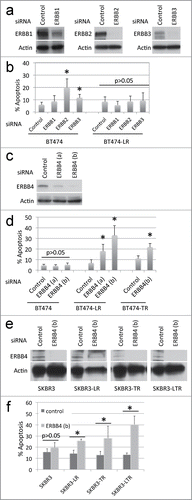
We next asked whether ERBB4 also played a role in acquired resistance to the ERBB2-targeted monoclonal antibody trastuzumab. When BT474 cells that have acquired resistance to trastuzumab (BT474-TR) were treated with ERBB4-specific siRNA, they died by apoptosis (). Trastuzumab was not required for this effect, indicating that cell death occurs independently of trastuzumab and/or ERBB2 activity. We also performed siRNA knockdown of ERBB4 in another ERBB2-positive breast cancer cell line, SKBR3, with acquired resistance to lapatinib (SKBR3-LR), trastuzumab (SKBR3-TR), or both (SKBR3-LTR). ERBB4 knockdown resulted in apoptotic cell death in SKBR3-LR, SKBR3-TR, and SKBR3-LTR cells but not in parental SKBR3 cells (). These results suggest that ERBB4 dependency may result from sustained loss of ERBB2 signaling in ERBB2-amplified breast cancer cells.
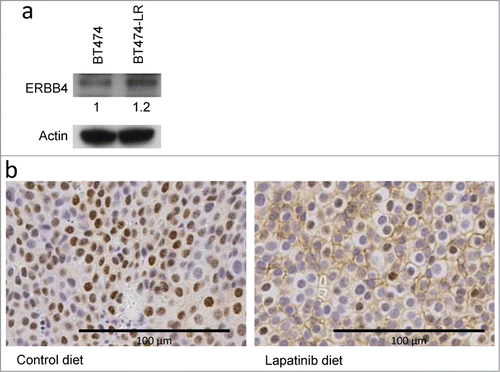
Western blot analysis showed that ERBB4 protein levels are modestly upregulated in lapatinib-resistant cells, compared to sensitive cells (). To investigate ERBB4 protein expression in vivo, we utilized the MMTV-Neu mouse model. MMTV-Neu transgenic mice spontaneously develop mammary tumors driven by activated neu (rat homolog of ERBB2).Citation26 When tumor volumes surpassed 200 mm3, we treated MMTV-Neu mice with lapatinib daily until tumors reached 1000 mm3 (3-8 weeks). The dosage (200 mg/kg) was set in accordance with our previous studies for other inhibitors, including erlotinib,Citation27,28 and tissue concentrations of lapatinib were corroborated by high-pressure liquid chromatography-mass spectrometry analysis (Supplementary Table 1). We harvested the tumors and analyzed ERBB4 expression by immunohistochemistry. Tumors from 57% of control mice (8/14) showed ErbB4 expression on the plasma membrane (). Strikingly, 13 out of 13 lapatinib-treated mice developed tumors with membranous ErbB4 expression () (p = 0.01599 by Fisher's exact test). Nuclear ErbB4 staining was found in both lapatinib-treated and -untreated tumors (). Although nuclear translocation of a cleaved ErbB4 fragment has been documented,Citation29 there was no significant correlation between nuclear ErbB4 and lapatinib treatment in our mouse model.
Figure 3. ERBB4 protein levels are modestly up-regulated in lapatinib-resistant cells, compared to sensitive cells. (a) ERBB4 expression in BT474 and BT474-LR cells were analyzed by protein gel blot with anti-ErbB4 (Ab5721) and actin antibodies. (b) Six to 12 month old MMTV-Neu female mice with mammary tumors (volume 200 mm3) were fed with control or lapatinib-containing diet (200 mg/kg) for 3 to 8 weeks until tumors reach 1000 mm3. Tumors were harvested and stained with the rabbit monoclonal anti-HER4/ErbB4 antibody (clone E200). Six out of 14 tumors from control mice exhibited no membranous ErbB4 staining (left). All (13/13) tumors from lapatinib-treated mice expressed ErbB4 on the plasma membrane (right).
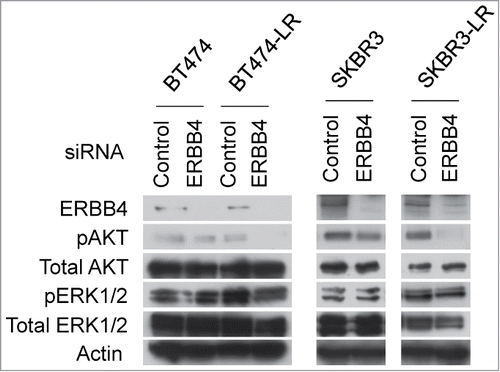
Since PI3K inhibitors significantly attenuated proliferation of BT474-LR cells (), we sought to determine whether apoptosis in BT474-LR cells induced by ERBB4 knockdown is also mediated by inactivation of the PI3K/AKT pathway. Western blots showed that ERBB4 knockdown significantly diminished AKT phosphorylation at Thr308 in lapatinib-resistant cells, but not in sensitive cells (). Interestingly, ERBB4 knockdown did not affect the phosphorylation status of ERK1/2 (Thr202 and Tyr204) (). These results suggest that the PI3K/AKT pathway plays a key role in the survival of breast cancer cells that have acquired resistance to ERBB2 inhibitors and that these resistant cells re-wire the survival signaling pathway by utilizing ERBB4, instead of ERBB2, to activate the PI3K/AKT pathway. Taken together, we postulate that ERBB4 becomes a critical player upon continuous inhibition of ERBB2. ERBB2+ cancer cells may shift their dependency from ERBB2 to ERBB4 when they acquire resistance to ERBB2 inhibitors.
Figure 4. Genetic ablation of ERBB4 results in inhibition of the AKT pathway in lapatinib-resistant cells. siRNA knockdown of ERBB4 was performed in BT474, BT474-LR, SKBR3, and SKBR3-LR. Twenty hours later, the pan-caspase inhibitor Q-VD-OPh (20 μM) was added to prevent apoptosis. Seventy-two hours after siRNA knockdown, cell lysates were prepared and western blotting was performed to detect ERBB4, phospho-AKT (pAKT; phospho-Thr308), total AKT, phospho-ERK1/2 (pERK; phsoho-Thr202 and -Tyr204), total ERK1/2, and actin.
Lastly, we tested whether ERBB4 overexpression is sufficient to promote resistance to ERBB2 inhibitors. We transiently overexpressed 2 constitutively active forms of ERBB4 (E563K and E872K) in BT474 cells and treated the cells with lapatinib. It has been shown that these ERBB4 mutants exhibit enhanced ligand-independent kinase activity, promote cellular transformation, and enhance proliferation in melanoma cells.Citation15 Unexpectedly, lapatinib treatment caused apoptosis in ERBB4-overexpressing cells as efficiently as control cells (), indicating that ERBB4 is critical but not sufficient to render ERBB2+ cells resistant to ERBB2 inhibitors.
Discussion
Acquired resistance to ERBB2 inhibitors is a significant clinical problem that dramatically affects outcomes of patients with ERBB2+ breast cancer. Studies from our laboratory and others suggest that acquired resistance to ERBB2 inhibitors is not due to ERBB2 insensitivity, but rather due to activation of various alternative survival pathways.Citation9-Citation13 In the present study, we provide evidence to support that ERBB4 may play a key role in the survival of breast cancer cells that acquired resistance to ERBB2 inhibitors.
ERBB4 plays an important role in normal mammalian gland development and differentiation.Citation33 However, a role for ERBB4 in cancer has been controversial.Citation14-16,34 Our results show that genetic ablation of ERBB4, but not ERBB1-3, leads to apoptosis in ERBB2+ breast cancer cells that are resistant to ERBB2 inhibitors. Since ERBB4 is not required for the development of ERBB2-driven breast cancer,Citation14 we hypothesize that ERBB2+ cancer cells may shift their dependency from ERBB2 to ERBB4 when they acquire resistance to ERBB2 inhibitors. Therefore, only upon continuous inhibition of ERBB2, ERBB4 becomes a key player in acquiring and maintaining resistance to ERBB2 kinase inhibitors in ERBB2+ cancer cells.
We detected the minimal up-regulation of ERBB4 protein expression in lapatinib-resistant cells and the membrane localization of ERBB4 in lapatinib-treated mouse mammary tumors. However, overexpression of the constitutively active forms of ERBB4 alone did not confer lapatinib resistance in naïve BT474 cells. It is suggested that resistant cells may have acquired an additional mechanism(s) that supports ERBB4 activation and the ERBB4 dependency; such a mechanism may include growth factor-mediated autocrine signaling to ERBB4, a novel post-translational modification of ERBB4, or an unidentified binding partner of ERBB4. These possibilities are not mutually exclusive and need to be fully elucidated.
Recently, pan-ERBB inhibitors were proved to overcome resistance to ERBB2 kinase inhibitors.Citation12,30,31 Several pan-ERBB inhibitors are being tested in clinical trials for patients with advanced ERBB2-positive breast cancer, including those who have progressed following prior ERBB2-targeted therapy. However, the detailed molecular mechanism of how pan-ERBB inhibitors block proliferation of cancer cells remains elusive except that many, but not all, of pan-ERBB inhibitors irreversibly bind to ERBB kinases. Elucidating the molecular mechanism of acquired ERBB2 inhibitor resistance and how pan-ERBB inhibitors reverse this resistance will significantly contribute to the development of novel and more effective treatment regimens for ERBB2+ cancer, with potential for reduced toxicity. Our results suggest that the efficacy of pan-ERBB inhibitors in killing ERBB2 inhibitor resistant cells is mediated, at least in part, by blocking ERBB4 activity. Thus, our findings provide a rationale for fully elucidating the role of ERBB4 in developing and maintaining resistance to ERBB2 inhibitors in breast cancer.
Materials and Methods
Cell culture
Cell lines used in this study were described previously.Citation13,32 Cell viability was assessed after 48-hour treatment using CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega). DMSO and the apoptosis-inducing pan-kinase inhibitor staurosporine were used as negative and positive controls, respectively. The absorbance at 490 nm (OD 490 nm) was normalized against DMSO.
BT474 cells stably expressing bovine PIK3CA-WT, PIK3CA-E545K, and PIK3CA-H1047R have been described previously.Citation35 For Matrigel colony assays, cells (1 × 103) in single cell suspension were resuspended in ice-cold Matrigel then delivered to wells of 96-well dishes in a volume of 100 μl Matrigel and cultured for 24 h, at which time cells were treated with lapatinib or BKM120 (1 μM each) delivered in 100 μl at 2X concentration, allowing the inhibitor to equilibrate to 1 μM. Inhibitor was delivered in 1X concentration in 100 μl every 4 days through day 14, when images were captured using the ProgRes MF digital camera (Jenoptiks) mounted on the Motic AE31 microscope.
Transient siRNA transfections were performed with Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's instructions. EGFR-specific siRNA (siRNA ID# s563), ERBB2-specific siRNA (siRNA ID# s613), ERBB3-specific siRNA (siRNA ID# s4778), and control siRNA (Catalog # 4390843) were designed and synthesized by Ambion (Silencer Select). Control and ERBB4-specific 27mer siRNAs were purchased from OriGene (catalog # SR301445; sequencing information can be made available upon request).
Antibodies and reagents
Anti-EGFR, ERBB2, ERBB3 AKT, pAKT, ERK1/2, and pERK1/2 antibodies were purchased from Cell Signaling Technology. Anti-actin antibody () was purchased from Sigma. Anti-ERBB2 (), ERBB4, and actin antibodies were purchased from Santa Cruz Biotechnology. The rabbit monoclonal anti-HER4/ErbB4 antibody (clone E200; Novus Biologicals) was used for immunohistochemistry. Anti-ErbB4 antibody (Ab5721) was raised against mouse ErbB4 (amino acids 1036 through 1239). GST-ErbB41036-1239 was used to immunize rabbits (Covance). Antiserum was affinity-purified against the immunogen.
Kinase inhibitors used in were purchased from LC Laboratories (Woburn), except LY2874455 (Active Biochemicals, Hong Kong), dacomitinib, KU55933 (Selleckchem, Houston), and varlitinib (KreBay Biochem, Monmouth Junction). The pan-caspase inhibitor Q-VD-OPh was purchased from MP Biomedicals (Santa Ana). Annexin V (Alexa Fluor® 647 conjugate) was purchased from Life Technologies (Carlsbad).
Mice
MMTV-Neu mice were obtained from the Jackson Laboratory (FVB-Tg(MMTV-Erbb2)NK1Mul/J; Stock number 005038). All animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Dartmouth College.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
994966_Supplementary_Materials.zip
Download Zip (1.2 MB)Acknowledgments
We would like to thank Drs. Scott Gerber, Yolanda Sanchez, Alan Eastman, and Todd Miller for valuable discussions concerning this work.
Funding
This study was supported by NIH grants: R00CA140948 (to MK), P30CA23108 (to the Norris Cotton Cancer Center), P50CA58183 (to RS), and R01CA143126 (to RSC). This work was also supported by Susan G. Komen for the Cure grant KG100677 (to RSC).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2:127-137; PMID:11252954; http://dx.doi.org/10.1038/35052073
- Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014; 25:282-303; PMID:24651011; http://dx.doi.org/10.1016/j.ccr.2014.02.025
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA 2003; 100:8933-8938; PMID:12853564; http://dx.doi.org/10.1073/pnas.1537685100
- Jura N, Shan Y, Cao X, Shaw DE, Kuriyan J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc Natl Acad Sci USA 2009; 106:21608-21613; PMID:20007378; http://dx.doi.org/10.1073/pnas.0912101106
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987; 235:177-182; PMID:3798106; http://dx.doi.org/10.1126/science.3798106
- Johnston S, Trudeau M, Kaufman B, Boussen H, Blackwell K, LoRusso P, Lombardi DP, Ben Ahmed S, Citrin DL, DeSilvio ML, et al. Phase II study of predictive biomarker profiles for response targeting human epidermal growth factor receptor 2 (HER-2) in advanced inflammatory breast cancer with lapatinib monotherapy. J Clin Oncol 2008; 26:1066-1072; PMID:18212337; http://dx.doi.org/10.1200/JCO.2007.13.9949
- Kaufman B, Trudeau M, Awada A, Blackwell K, Bachelot T, Salazar V, DeSilvio M, Westlund R, Zaks T, Spector N, et al. Lapatinib monotherapy in patients with HER2-overexpressing relapsed or refractory inflammatory breast cancer: final results and survival of the expanded HER2+ cohort in EGF103009, a phase II study. Lancet Oncol 2009; 10:581-588; PMID:19394894; http://dx.doi.org/10.1016/S1470-2045(09)70087-7
- Singh JC, Jhaveri K, Esteva FJ. HER2-positive advanced breast cancer: optimizing patient outcomes and opportunities for drug development. Br J Cancer 2014; 111:1888-98; PMID:25025958; http://dx.doi.org/10.1038/bjc.2014.388
- Zhang S, Huang WC, Li P, Guo H, Poh SB, Brady SW, Xiong Y, Tseng LM, Li SH, Ding Z, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nature Medicine 2011; 17:461-469; PMID:21399647; http://dx.doi.org/10.1038/nm.2309
- Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, et al. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA 2011; 108:5021-5026; PMID:21385943; http://dx.doi.org/10.1073/pnas.1016140108
- Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sánchez V, Sutton CR, Cheng H, Perou CM, Zhao JJ, et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci USA 2013; 110:14372-14377; PMID:23940356; http://dx.doi.org/10.1073/pnas.1303204110
- Xia W, Petricoin EF, Zhao S, Liu L, Osada T, Cheng Q, Wulfkuhle JD, Gwin WR, Yang X, Gallagher RI, et al. An heregulin-EGFR-HER3 autocrine signaling axis can mediate acquired lapatinib resistance in HER2+ breast cancer models. Breast Cancer Res 2013; 15:R85; PMID:24044505; http://dx.doi.org/10.1186/bcr3480
- Kurokawa M, Kim J, Geradts J, Matsuura K, Liu L, Ran X, Xia W, Ribar TJ, Henao R, Dewhirst MW, et al. A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci Signal 2013; 6:ra32; PMID:23652204; http://dx.doi.org/10.1126/scisignal.2003741
- Jackson-Fisher AJ, Bellinger G, Shum E, Duong JK, Perkins AS, Gassmann M, Muller W, Kent Lloyd KC, Stern DF. Formation of Neu/ErbB2-induced mammary tumors is unaffected by loss of ErbB4. Oncogene 2006; 25:5664-5672; PMID:16652155; http://dx.doi.org/10.1038/sj.onc.1209574
- Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet 2009; 41:1127-1132; PMID:19718025; http://dx.doi.org/10.1038/ng.438
- Mendoza-Naranjo A, El-Naggar A, Wai DH, Mistry P, Lazic N, Ayala FR, da Cunha IW, Rodriguez-Viciana P, Cheng H, Tavares Guerreiro Fregnani JH et al. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO Mol Med 2013; 5:1019-1034; PMID:23681745; http://dx.doi.org/10.1002/emmm.201202343
- Rexer BN, Engelman JA, Arteaga CL. Overcoming resistance to tyrosine kinase inhibitors: lessons learned from cancer cells treated with EGFR antagonists. Cell Cycle 2009; 8:18-22; PMID:19106609; http://dx.doi.org/10.4161/cc.8.1.7324
- Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther 2001; 1:85-94; PMID:12467226
- Schaefer G, Shao L, Totpal K, Akita RW. Erlotinib directly inhibits HER2 kinase activation and downstream signaling events in intact cells lacking epidermal growth factor receptor expression. Cancer Res 2007; 67:1228-1238; PMID:17283159; http://dx.doi.org/10.1158/0008-5472.CAN-06-3493
- Piechocki MP, Yoo GH, Dibbley SK, Lonardo F. Breast cancer expressing the activated HER2/neu is sensitive to gefitinib in vitro and in vivo and acquires resistance through a novel point mutation in the HER2/neu. Cancer Res 2007; 67:6825-6843; PMID:17638894; http://dx.doi.org/10.1158/0008-5472.CAN-07-0765
- Azuma K, Tsurutani J, Sakai K, Kaneda H, Fujisaka Y, Takeda M, Watatani M, Arao T, Satoh T, Okamoto I, et al. Switching addictions between HER2 and FGFR2 in HER2-positive breast tumor cells: FGFR2 as a potential target for salvage after lapatinib failure. Biochem Biophys Res Commun 2011; 407:219-224; PMID:21377448; http://dx.doi.org/10.1016/j.bbrc.2011.03.002
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009; 15:429-440; PMID:19411071; http://dx.doi.org/10.1016/j.ccr.2009.03.020
- O'Brien NA, McDonald K, Tong L, von Euw E, Kalous O, Conklin D, Hurvitz SA, di Tomaso E, Schnell C, Linnartz R et al. Targeting PI3K/mTOR overcomes resistance to HER2-targeted therapy independent of feedback activation of AKT. Clin Cancer Res 2014; 20:3507-3520; PMID:24879796; http://dx.doi.org/10.1158/1078-0432.CCR-13-2769
- Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007; 67:11924-11932; PMID:18089823; http://dx.doi.org/10.1158/0008-5472.CAN-07-1885
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011; 29:1046-1051; PMID:22037378; http://dx.doi.org/10.1038/nbt.1990
- Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1998; 54:105-115; PMID:2898299; http://dx.doi.org/10.1016/0092-8674(88)90184-5
- Liby K, Black CC, Royce DB, Williams CR, Risingsong R, Yore MM, Liu X, Honda T, Gribble GW, Lamph WW, et al. The rexinoid LG100268 and the synthetic triterpenoid CDDO-methyl amide are more potent than erlotinib for prevention of mouse lung carcinogenesis. Mol Cancer Ther 2008; 7:1251-1257; PMID:18483313; http://dx.doi.org/10.1158/1535-7163.MCT-08-0023
- To C, Kim EH, Royce DB, Williams CR, Collins RM, Risingsong R, Sporn MB, Liby KT. The PARP inhibitors, veliparib and olaparib, are effective chemopreventive agents for delaying mammary tumor development in BRCA1-deficient mice. Cancer Prev Res 2014; 7:698-707; PMID:24817481; http://dx.doi.org/10.1158/1940-6207.CAPR-14-0047
- Ni CY, Murphy MP, Golde TE, Carpenter G. Gamma-secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 2001; 294:2179-2181; PMID:11679632; http://dx.doi.org/10.1126/science.1065412
- Kalous O, Conklin D, Desai AJ, O'Brien NA, Ginther C, Anderson L, Cohen DJ, Britten CD, Taylor I, Christensen JG, et al. Dacomitinib (PF-00299804), an irreversible Pan-HER inhibitor, inhibits proliferation of HER2-amplified breast cancer cell lines resistant to trastuzumab and lapatinib. Mol Can Ther 2012; 11:1978-1987; PMID:22761403; http://dx.doi.org/10.1158/1535-7163.MCT-11-0730
- Canonici A, Gijsen M, Mullooly M, Bennett R, Bouguern N, Pedersen K, O'Brien NA, Roxanis I, Li JL, Bridge E, et al. Neratinib overcomes trastuzumab resistance in HER2 amplified breast cancer. Oncotarget 2013; 4:1592-1605; PMID:24009064
- Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, Wang YC, Shou J, Bissell MJ, Osborne CK, Schiff R. β1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res 2011; 13:R84; PMID:21884573; http://dx.doi.org/10.1186/bcr2936
- Muraoka-Cook RS, Feng SM, Strunk KE, Earp HS. ErbB4/HER4: role in mammary gland development, differentiation and growth inhibition. Journal of Mammary Gland Biology and Neoplasia 2008; 13:235-246; PMID:18437540; http://dx.doi.org/10.1007/s10911-008-9080-x
- Portier BP, Minca EC, Wang Z, Lanigan C, Gruver AM, Downs-Kelly E, Budd GT, Tubbs RR. HER4 expression status correlates with improved outcome in both neoadjuvant and adjuvant Trastuzumab treated invasive breast carcinoma. Oncotarget 2013; 10:1662-72; PMID:24091566
- Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL. H1047R phosphatidylinositol 3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene 2010; 29:5193-203; PMID:20581867; http://dx.doi.org/10.1038/onc.2010.257