
Abstract
Melanoma has the highest mortality rate of all skin cancers and a major cause of treatment failure is drug resistance. Tumors heterogeneity requires novel therapeutic strategies and new drugs targeting multiple pathways. One of the new approaches is targeting the scaffolding function of tumor related proteins such as focal adhesion kinase (FAK). FAK is overexpressed in most solid tumors and is involved in multiple protein-protein interactions critical for tumor cell survival, tumor neovascularization, progression and metastasis. In this study, we investigated the anticancer activity of the FAK scaffold inhibitor C4, targeted to the FAK-VEGFR-3 complex, against melanomas. We compared C4 inhibitory effects in BRAF mutant vs BRAF wild type melanomas. C4 effectively caused melanoma tumor regression in vivo, when administered alone and sensitized tumors to chemotherapy. The most dramatic effect of C4 was related to reduction of vasculature of both BRAF wild type and V600E mutant xenograft tumors. The in vivo effects of C4 were assessed in xenograft models using non-invasive multimodality imaging in conjunction with histologic and molecular biology methods. C4 inhibited cell viability, adhesion and motility of melanoma and endothelial cells, specifically blocked phosphorylation of VEGFR-3 and FAK and disrupted their complexes. Specificity of in vivo effects for C4 were confirmed by a decrease in tumor FAK and VEGFR-3 phosphorylation, reduction of vasculogenesis and reduced blood flow. Our collective observations provide evidence that a small molecule inhibitor targeted to the FAK protein-protein interaction site successfully inhibits melanoma growth through dual targeting of tumor and endothelial cells and is effective against both BRAF wild type and mutant melanomas.
Introduction
Melanoma has become a major public health problem, having risen by 3% to 8% per year in the Caucasian population since the 1960s.Citation1 Insights into the genetic makeup of melanoma tumors revealed its heterogeneity with somatic mutations in BRAF, NRAS, and KRAS in 43%, 20%, and 2% of melanomas respectively (www.sanger.ac.uk/genetics/CGP/cosmic/), and have led to the development of promising targeted agents in case of BRAF mutant melanoma. Unfortunately, the progress is limited by the development of the resistance which occurred after 6 months on average using BRAF inhibitor monotherapy and after 9–10 months using BRAF–MEK inhibitor combination therapy.Citation2 In addition, 35% cases that do not possess BRAF and RAS mutations are hard to treat and combination treatment approach is very limited.Citation3,4 The prognosis of patients with distant metastases remains poor. Melanoma is highly vascularized tumor, which preferentially metastasizes via the lymphatic system, and the occurrence of lymph node metastases dramatically reduces the survival rate.Citation5 A key mediator of tumor lymphangiogenesis is the vascular endothelial growth factor receptor 3 (VEGFR-3). VEGFR-3 and its ligands, VEGF-C and VEGF-D, control tumor lymphangiogenesis by enhancing proliferation and survival of lymphatic endothelial cells.Citation6 The VEGFR-3 pathway is also active in vascular endothelial cells at the sprouting tips of tumor capillaries, and may play important role in tumor neovascularization.Citation7 Level of VEGFR-3 expression is higher in tumor blood and lymphatic vessels than in surrounding normal vessels.Citation8 Clinical studies in human cancers have highlighted the prognostic importance of VEGF-C, VEGF-D and VEGFR-3 expression levelsCitation9 and lymphatic capillary density for the risk for melanoma progression,Citation10 lymph node metastasisCitation11 and for patient survival.Citation12 In cutaneous melanoma VEGF-C, VEGF-D and VEGFR-3 are overexpressed when compared to normal melanocytes and it was shown that VEGFR-3 is overexpressed at early stages of melanoma progression.Citation13 Inhibition of VEGFR-3 signaling leads to both, regression of the lymphatic network and to suppression of tumor lymph node metastasis.Citation14 These data implicate VEGFR-3 in multiple aspects of tumorigenesis and suggest that therapeutic targeting of VEGFR-3 might selectively reduce tumor growth and metastasis in melanoma.
In preclinical studies, antibody and small molecule kinase inhibitors against VEGFR-3 have shown to inhibit lymphangiogenesis and metastasis.Citation15,16 However, the resistance of melanoma to conventional therapies as well as single targeted therapy, suggests that multiple signaling pathways may need to be targeted for maximal therapeutic efficacy.Citation17 Previously, we have shown that focal adhesion kinase (FAK) physically interacts with VEGFR-3 and provides important survival signals for breast cancer cells.Citation18 FAK is a critical tyrosine kinase serving as a signaling hub for integrins, growth factor receptors, G-protein coupled receptors, death receptors and mechanical stress forces. This makes FAK a significant player in cancer cell survival and proliferation, migration, invasion, and tumor vasculogenesis.Citation19–21 FAK deletion impaired vascular endothelial growth factor (VEGF)-induced neovascularization and in vitro deletion of FAK in endothelial cells resulted in reduced VEGF-stimulated Akt phosphorylation, reduced cellular proliferation, as well as increased cell death.Citation22 Studies in conditional FAK knockout mouse models have demonstrated a critical role of FAK in angiogenesis during embryonic development and cancer progression.Citation23
We have recently identified FAK scaffold inhibitor, small molecule C4 (Chloropyramine hydrochloride), targeted to the C terminus of FAK, that disrupts the FAK-VEGFR-3 interaction and inhibits signaling of these 2 proteins.Citation24 In the current study we analyzed the anti-tumorogenic and anti-angiogenic properties of the FAK-VEGFR-3 inhibitor C4 in melanoma BRAF wild type and mutant V600 models. We show that C4 inhibits the proliferation of melanoma and endothelial cells (HUVEC) in vitro and alters tumor growth and neovascularization in a xenotransplant models. Our results demonstrate, for the first time, that a small molecule inhibitor targeted to the FAK protein-protein interaction site successfully inhibits melanoma growth through dual targeting of tumor and endothelial cells.
Results
FAK scaffold inhibitor C4 selectively bound FAK and inhibited melanoma cell growth
To validate the specificity and in vitro efficacy of the FAK-VEGFR-3 inhibitor C4, it was tested in the fluorescence polarization competition binding assay with FAK FAT protein. The 12-mer VEGFR-3 derived peptide AV3Citation18 was TAMRA-conjugated and used as the fluorescent probe. In saturation experiment with AV3 we determined Kd = 21.63 μM () and unlabeled peptide AV3 competed this interaction with IC50 = 53 μM (). We found that C4 displaced labeled AV3 peptide with IC50 = 43.75 μM (). Compound C1, an analog of C4 without biological activity, was used as negative control and did not show binding (). These experimental results indicate that compound C4 specifically targets the FAK-VEGFR-3 protein-protein interaction. Next we measured compound efficacy in vitro in melanoma cells.
Figure 1. FAK scaffold inhibitor C4 selectively binds FAK and inhibits melanoma cell growth. A–D. Fluorescent polarization-based assay. (A) Saturation experiment, dose-response binding curve of TAMRA labeled peptide AV3 derived from the binding site of VEGFR-3 to FAK FAT protein. (B) Dose-response competitive binding curve: competition experiment with increased concentration of unlabeled peptide AV3. (C) Competition experiment with increased concentration of FAK scaffold inhibitor C4 and compound C4 structure. (D) Competition experiment with increased concentration of C4 analog, negative control compound C1 and its structure. (E) Western blot analysis of FAK and VEGFR-3 expression and activation in melanoma cell lines. (F). FAK scaffold inhibitor C4 IC50 in melanoma cell lines of different genetic background. Viability MTS assay.
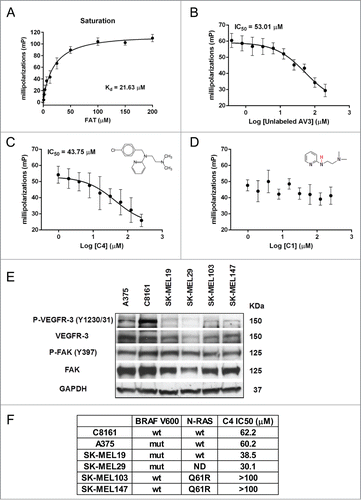
To examine for the presence of activated FAK and VEGFR-3 in adherent proliferating human melanoma cells we performed Western blot analysis. Six human melanoma cell lines were evaluated for the presence of total and phosphorylated FAK and VEGFR-3. We have identified the presence of FAK and VEGFR-3 in all cell lines (). These cell lines differ by the presence/absence of mutations in BRAF and RAS genes, and we compare their sensitivity to compound C4 in viability assay (). We found that cells with RAS mutation Q61R are the most resistant to FAK-VEGFR-3 inhibition and cells from primary tumors with BRAF V600 mutations are moderately sensitive to C4. Interestingly, highly aggressive, metastatic melanoma cell line C8161 is BRAF and RAS wild type, but has C4 sensitivity comparable with A375 cells. We selected C8161 and A375 cell lines for further analysis based on relatively high activation of both FAK and VEGFR-3 kinases and absence/presence of BRAF mutation respectively. We especially were interested in the response of C8161 melanoma cells because the treatment options for the patients with this type of tumors (BRAF and RAS wild type) are limited.Citation2
FAK scaffold inhibitor C4 reduced phosphorylation of FAK and VEGFR-3 and decreased their complex formation
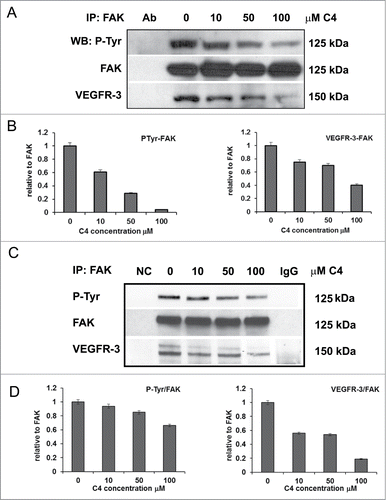
First, the effect of C4 on melanoma cells was analyzed in FAK and VEGFR-3 immunoprecipitates from C8161 cells. We analyzed precipitates with anti-phospho-tyrosine antibody and found that total tyrosine phosphorylation of FAK was reduced in the dose-dependent manner and at 100 μM C4 only 4.2% of FAK molcules are phosphorylated ( left panel). We probed the FAK precipitates for the presence of VEGFR-3 protein and found that in parallel with FAK deactivation, the amount of VEGFR-3 in co-precipitates was reduced and at 100 μM concentration of C4 60% of the VEGFR-3 molecules are lost from the complex with FAK ( right panel). Immunoprecipitation with VEGFR-3 antibody and analysis with anti-phospho-tyrosine antibody also confirmed increased dephosphorylation of VEGFR-3 in parallel with increased concentration of C4 (Supplementary Figs. S1A, B left panel). This was accompanied by a decrease of FAK in the complex with VEGFR-3 (Figs. S1A and B right panel). We found the same inhibitory effect of C4 on FAK phosphorylation and FAK-VEGFR-3 complexes formation in A375 melanoma cell line (). Thus, we confirmed that C4 disrupted FAK-VEGFR-3 complexes in the selected melanoma cells and this effect was dose-dependent.
Figure 2. FAK scaffold inhibitor C4 reduced phosphorylation of FAK and VEGFR-3 and decreased their complex formation. (A) C4 dose-dependent decrease of FAK-VEGFR-3 association in melanoma C8161 cells. Cells were treated with increased concentrations of C4 for 24 h. FAK protein complexes were immunoprecipitated (IP) with FAK antibody (Millipore, clone 4.47). FAK precipitates were analyzed in consequent protein gel blotting (WB) for FAK phosphorylation with pan-phospho-tyrosine antibody 4G10 (P-Tyr) and for the presence of VEGFR-3 protein with VEGFR-3 specific antibody (Millipore, clone 9D9). (B) Densitometry was performed for each IP experiment and data are presented at the bottom of the protein gel blot image. Left panel – decrease of P-Tyr phosphorylation of FAK; right panel – decrease of VEGFR-3 protein in FAK co-precipitates. (C) C4 dose-dependent decrease of FAK-VEGFR-3 association in melanoma A375 cells. Cells were treated with increased concentrations of C4 for 24 h and FAK protein complexes were immunoprecipitated (IP) with FAK antibody. FAK precipitates were analyzed in consequent protein gel blotting for FAK phosphorylation and for the presence of VEGFR-3 protein. Densitometry was performed for IP experiment and data are presented at the bottom of the protein gel blot image.
FAK scaffold inhibitor C4 affected FAK and VEGFR-3 signaling and sensitized melanoma cells to cytotoxic therapy in vitro
BRAFV600E-driven melanomas are characterized by elevated ERK activity. To elucidate the effects of FAK-VEGFR-3 complex disruption and dephosphorylation of both molecules on Erk1/2 signaling, melanoma BRAF wild type and mutant cells were treated with C4 for 24, 48 and 72 h and phosphorylation of Erk1/2 was measured. We found that Erk1/2 is highly phosphorylated in BRAF mutant A375 cells. Importantly, activation of Erk1/2 was inhibited by C4 in both cell lines and effect was detectable even 72 h post-treatment ().
Figure 3. FAK scaffold inhibitor C4 affected FAK/VEGFR-3 signaling and sensitized melanoma cells to cytotoxic therapy in vitro. (A–D). Comparison of inhibitory effect of C4 on FAK-VEGFR-3 downstream signaling in melanoma cell lines. Cells were treated 24 h, 48 h and 72 h with selected concentration of the small molecule C4 and analyzed by Western blot. Control – cells at 72 h time point, grown in media with 0.01% DMSO. (A) Western blot analysis of C8161 cells. (B) Densitometry analysis of the ERK dephosphorylation in C8161 cells. (C) Western blot analysis of A375 cells. P-Erk1/2 and Erk1/2 lines were cut and pasted from the same gel. (D) Densitometry analysis of the ERK dephosphorylation in A375 cells. (E) Viability MTS assay of C8161 cells treated for 48 h with C4 in combination with dacarbazine (DTIC). Data of MTS assay presented as ratio of OD treated to untreated cells, 1 corresponds to the 100% viability of the untreated cells. (F) Viability MTS assay of A375 cells treated for 48 h with C4 in combination with DTIC. All concentrations are μM. *P < 0.05 and **P < 0.01 relative to control, @ relative to C4 alone, # relative to chemotherapy alone. (G) Measurement of C4 and DTIC interaction in C8161 cells. (H) Measurement of C4 and DTIC interaction in A375 cells. The combination index (CI) indicates a quantitative measure of the degree of drug interaction in terms of synergistic (CI < 1), additive (CI = 1) or antagonistic effect (CI > 1).Citation49
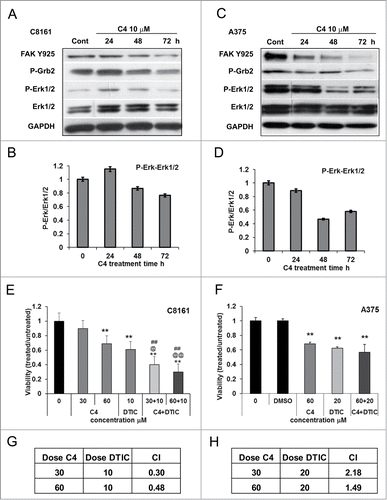
It is known that FAK and VEGFR-3 are involved in the regulation of cell motility through activation of Grb2/MEK/ERK pathways. We have shown that treatment with C4 inhibited melanoma cell motility (Figs. S2A and B) and biochemical analysis revealed that phosphorylation of Grb2 was decreased after treatment with C4 (). Importantly, this inhibitory effect correlated with a decrease in FAK Y925 phosphorylation in both cell lines.
Our data suggest that down-regulation of FAK compromised pro-survival pathways and may augment tumor sensitivity to cytotoxic therapy. Previous studies in our laboratory have shown that down regulation of FAK with antisense oligonucleotides resulted in increased sensitivity to 5-fluorouracil (5-FU) in human melanoma cell lines.Citation25 This supports the concept that FAK inhibition may aid in sensitizing melanoma cells to chemotherapeutics. We validated this by combining C4 with Dacarbazine (DTIC). C8161 and A375 cells were treated at drug concentrations corresponding to IC50 of both drugs and viability was measured after 48 h of treatment with a single agent or a combination of the drugs. The combination of 60 μM C4 with 10 μM DTIC reduced viability of C8161 cells by 70 ± 2% (). The analysis of the dose-effect curves using the Calcusyn Software confirmed the synergistic effect of the drugs with the combination index CI = 0.48 (). However, the combination of C4 with DTIC in A375 cells lead only to moderate additive inhibition effect (). We have shown that C4 treatment caused dose- and time-dependent apoptosis in both melanoma cell lines and combining C4 with DTIC lead to the activation of the apoptotic pathways (Figs. S2 and S3). Still, A375 BRAF mutant cell line was more resistant to the treatment. This finding prompt us to study the possible different response to compound C4 in BRAF wild type and mutant melanomas in vivo.
FAK scaffold inhibitor C4 inhibited melanoma growth in vivo and sensitized tumors to chemotherapy
We examined the activity of C4 as a single agent and in combination with chemotherapy against melanoma growth using C8161 and A375 xenograft models. Our preliminary analysis of the C4 toxicity in the SCID mice have shown that 62 d of intraperitoneal injections at 50 mg/kg does not affect mouse behavior and growth (Figs. S5A–C). Intraperitoneal injections of C4 were started when tumors reached on average a size of 100 mm3. After 17 d of C4 treatment, tumor volumes of established C8161 tumors in C4 treated groups were significantly smaller than those in control group (fast growing and ulcerating) (). The effect was dose dependent with approximate tumor growth reduction of 77% (P = 0.006) in 50 mg/kg group and 61% in 25 mg/kg group. These data demonstrated that the small molecule C4 was able to reduce tumor growth in vivo as a single agent. Concomitant administration of C4 (25 mg/kg IP once a day) with DTIC (8 mg/kg 1 × 4d) reduced C8161 tumor growth by 83% ± 3% (P = 0.003) (; Fig. S5D) in comparison with control and by 40% (C4) and 38% ± 2.1% (DTIC) in comparison with each drug alone. Dual treatment in a second xenograft model with implanted A375 cells reduced tumor growth by 67 ± 3% in comparison with control. However, the effect was not synergistic and tumor burden in the dual treatment group was not significantly different from C4 alone (P > 0.05) (; Fig. S5E). This additive effect of 2 drugs on BRAF mutant melanoma reproduced our in vitro data. Nevertheless, the immunohistochemistry analysis of the tumors from these experiments with Caspase 3 antibody confirmed the increase in apoptotic cell death after treatment with C4 and significant apoptosis after dual treatment (not shown).
Figure 4. FAK scaffold inhibitor C4 reduced tumor growth in vivo and sensitized tumors to chemotherapy. (A) C4 dose-dependent effect in C8161 melanoma xenograft model. Female nude mice were subcutaneously inoculated with C8161 cancer cells (5 mice/ group). Intraperitoneal injections (IP) with compound C4 at 25 mg/kg and 50 mg/kg daily were started when tumor reached 100 mm3. *P < 0.05 and **P < 0.01 relative to control at day 17, # relative to C4 50 mg/kg at day 23. (B) C4 sensitized C8161 tumors to dacarbazine (DTIC) chemotherapy. Female nude mice were subcutaneously inoculated with C8161 cancer cells (5 mice/ group). IP with compound C4 (25 mg/kg, daily), DTIC (8 mg/kg, Q4D) and combination were started when tumor reach 100 mm3 and continued for 23 d. TR – percent tumor growth reduction relative to controls: *P < 0.05 and **P < 0.01 relative to untreated control, @ relative to C4 alone, # relative to chemotherapy alone. (B) left panel, C8161 xenograft dual treatment C4 and DTIC. (B) right panel, representative C8161 tumors from each corresponding treatment group. Tumors from the groups treated with C4 are avascular. (C) left panel, A375 xenograft model, dual treatment C4 and DTIC. Female nude mice were subcutaneously inoculated with melanoma A375 cells. IP with compound C4 (25 mg/kg, daily), DTIC (8 mg/kg, Q4D) and combination were started when tumor reach 100 mm3 and continued for 31 d. (C) Right panel, representative A375 tumors from each corresponding treatment group. Tumors from the groups treated with C4 are avascular. (D) Western blot analysis of representative C8161 tumors from the control PBS and C4 treated groups showed reduced phosphorylation of VEGFR-3 on Tyr 1230/1231 in C4 treated group. (E) Immunohistochemistry analysis of C8161 xenograft tumors performed for total vessels (CD31) (left panel), and lymphatic (LYVE1) (right panel) vessels density.
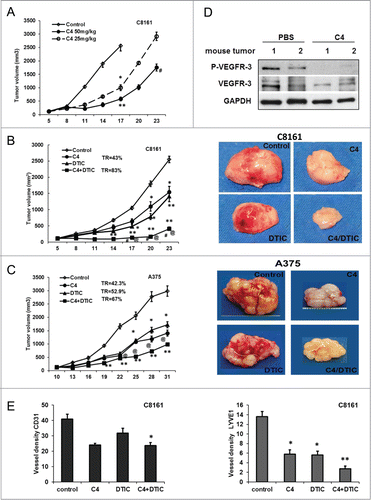
Analyzes of the excised tumors showed dramatic differences in tumor appearance: C4 treated tumors were less vascularized and lost peritumoral blood vessels (, right panels). Comparison of tumors of equal size from chemotherapy alone and C4 alone treated groups suggest that observed effects are related to treatment, not to the size of the tumors. Western blot analysis of treated tumors reveals reduced levels of phosphorylated and total VEGFR-3 (). Tumor specimens were immunostained with CD31 antibody (marker for endothelial cells), and LYVE1 antibody (specific to lymphatic vessels) to provide information on blood and lymphatic vessels present. The analysis revealed a decrease of CD31 vessel density in the C4 treated group and vessel density was significantly decreased in the C4 plus DTIC treatment group (, left panel). Importantly, we found significant reductions of lymphatic, LYVE1 positive, vasculature in C8161 xenografts, treated with C4 (, right panel). That analysis led us to conclude that FAK scaffold inhibitor C4 affects tumor vasculogenesis in vivo.
FAK scaffold inhibitor C4 inhibited viability of endothelial cells and prevented vessel formation and sprouting
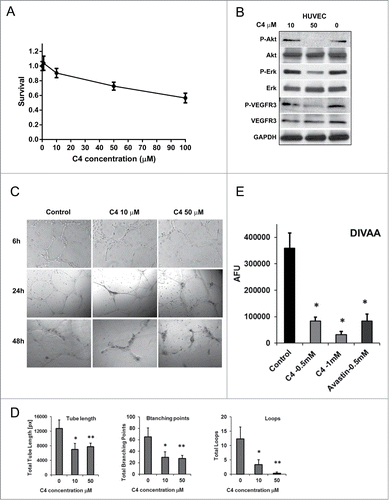
Neoangiogenesis plays an important role in tumor growth and metastasis. FAK is expressed not only in tumor cells, but plays an indispensable role in endothelial cells. In addition, VEGFR-3 expression was found in sprouting endothelial cells in tumors. We next evaluated the effects of C4 on endothelial cells and used HUVEC (Human umbilical vein endothelial cells) cells as a model system. We found that C4 decreased viability of these cells in a dose-dependent manner with very moderate activity (IC50 > 100 μM) and 50 μM of C4 decreased HUVEC viability by 29% ± 3% (P = 0.005) (). However, 50 μM of C4 dramatically reduced phosphorylation of Akt and decreased significantly phosphorylation of Erk1/2 and VEGFR-3 (). Tube formation assay (), mimicking neovascularization, demonstrated that even a 10 μM concentration of C4 inhibited proper vessel formation and reduced total tube length by 45% (P = 0.016), decreasing the number of branching points and loops (). We also performed a quantitative in vivo assessment of an angiogenic response to C4 by the Directed in Vivo Angiogenesis Assay (DIVAA)Citation26 to measure cellular inhibition. We found that the inhibitory effect of C4 is dose dependent and comparable with the effect of the anti-angiogenic drug bevacizumab (Avastin) (). Thus, 0.5 mM of C4 caused a decrease in blood vessels outgrowth up to 4.3 times in comparison with FGF2 stimulated control (P = 0.022) and a twice higher C4 concentration of 1 mM caused an 11.5 times outgrowth reduction (P = 0.016) (). These data demonstrate for the first time the importance of the FAK-VEGFR-3 complex formation in vasculogenesis.
Figure 5. FAK scaffold inhibitor C4 inhibited FAK/VEGFR-3 signaling in endothelial cells and prevented vessel formation and sprouting. (A) C4 inhibited viability of endothelial cells. Viability MTS assay. HUVEC cells were plated on 96 well plates, grown 24 h and treated 72 h with selected concentrations of small molecule C4. Data of MTS assay presented as ratio of OD treated to untreated cells, 1 corresponds to the 100% viability of the untreated cells. (B) HUVEC cells were treated with selected concentration of C4 for 24 h and protein gel blotting was performed to analyze effect of C4 on FAK and VEGFR-3 signaling. Antibody for P-Akt (Ser473), P-Erk1/2 (Tre202/Tyr204), P-VEGFR-3 (Tyr1230/1231) were used. (C) Inhibition of HUVEC tube formation by C4. The HUVEC cells were treated with increased concentration of C4 and the formation of the tubes was analyzed at 6, 24, 48 h time points (10× magnification). (D) Analysis of the tube formation assay at 24 h time point with WimTube software from Wimasis by examining different tube structures: total tube length (pixels, px), branching points and number of loops. Results represent the mean ± standard error of the mean (SEM) of 3 different experiments. *P < 0.05 and **P < 0.01 compared with controls (treatment with DMSO only). (E) Dose-dependent inhibition of in vivo vessels growth and sprouting in Directed in vivo angiogenesis assay (DIVAA). Angioreactors were implanted subcutaneously into the SCID mice for 12 d, and vessels allowed to infiltrate. C4 was mixed at concentrations of 0.5 and 1 mM with Matrigel (Trevigen), and bevacizumab (Avastin) was used at 0.5 mM concentration as positive control. DIVAA was quantitated by recovery of the angioreactors, dispase digestion, and fluorescence spectroscopy. Results are expressed in fluorescence units as the mean ±SEM of 3 experiments.
FAK scaffold inhibitor C4 decreased tumor blood flow in vivo in melanoma mouse model
Next, we examined the vascular response of C8161 xenografts to C4 treatment using contrast-enhanced MRI. SCID mice bearing subcutaneous C8161 human melanoma xenografts were randomized to control (PBS) or treatment groups (C4) based on comparable sizes of the tumor. T1-weighted contrast-enhanced MRI was performed on days 7 and 14 post therapy and compared to volume-matched controls (n = 3/group) (). Quantitative estimates of contrast agent concentration (ΔR1) in tumors and in blood were obtained to compute changes in blood volume (Y-intercept). At 1 week () and at 2 weeks () post treatment, a significant reduction in blood volume (p < 0.05) in tumors treated with C4 compared to PBS controls was found. Relative blood volume maps of an animal in the control and treatment groups are shown in . C4 treatment resulted in reduced perfusion as evidenced by decreased contrast agent uptake in the tumor.
Figure 6. Vascular response of C8161 melanoma xenografts to C4 treatment. (A) MRI analysis of blood flow. Plots show the change in R1 (ΔR1) post contrast in PBS and C4 treated tumors at 1 week (A) and at 2 weeks (B) post treatment. Values were normalized to blood to account for potential differences in contrast agent injection between the animals. A significant reduction (P < 0.05) in relative blood volume (y-intercept) was seen following C4 treatment at 1 week and 2 weeks post treatment. (C) Relative blood volume maps of an animal in the PBS control group and C4 treatment group. Three contiguous slices of the tumor are shown. (D) Optical imaging of PBS and C4 treated tumors at 1 week and at 2 weeks post treatment showed decrease in blood flow in comparison to unchanged blood flow in muscles of the treated mice.
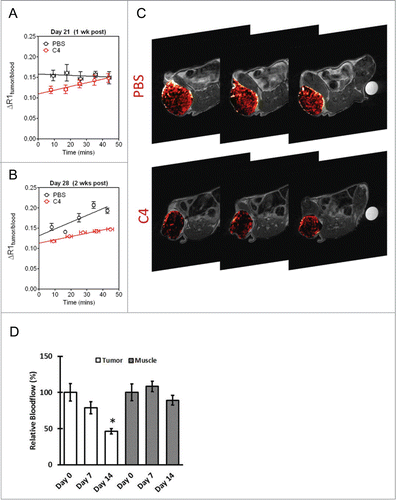
In addition to MRI, blood flow measurements were also obtained from tumors in mice from the C4 treated group at baseline and 2 weeks post C4 treatment using diffuse correlation spectroscopy. Three mice were treated with C4 and followed for 2 weeks. As shown in , the skeletal muscle did not show significant changes (p > 0.05, changes were less than 10%) in blood flow during treatment. However, a ∼22% decrease (p > 0.05) and a 54% decrease in blood flow (p = 0.002) were observed at day 7 and day 14, respectively. Thus, the significant changes in tumor vascular function were detected by both MRI and optical imaging in response to FAK scaffold inhibitor C4 treatment in vivo.
Discussion
Multiple studies confirmed importance of FAK for tumor development, invasion and metastasis, its involvement in cancer cells drug resistance and support of neovascularization and cancer stem cells.Citation20,27 A few FAK kinase inhibitors were developedCitation28,29 and some of them show promising results in early clinical trials.Citation30,31 However, it is well established now that in addition to kinase function FAK serves as a scaffold for multiple protein signaling complexes and its scaffolding function is very important for tumor progression.Citation32 Targeting such protein complexes brings the possibility to affect, modulate, or shut down multiple signaling pathways simultaneously. We have shown for the first time that FAK scaffold inhibitor, a small molecule targeted to the FAK-VEGFR-3 protein-protein interaction, can be used successfully to inhibit tumor growth through dual inhibition of tumor and endothelial cells. We have shown that FAK scaffold inhibitor C4 inhibited proliferation of melanoma and endothelial cell (HUVEC) in vitro and inhibited tumor growth and neovascularization in a xenotransplant melanoma models.
FAK plays an important role in melanoma disease progression. Correlation between expression of FAK and increased melanoma cell migration and metastasis has been describedCitation33 and it was shown that down regulation of FAK with FAK-CD inhibited melanoma cell growth and led to cell deathCitation34 and reduction in the lung metastases.Citation35 FAK activation is enhanced in aggressive melanoma cells as a result of increased phosphorylation on tyrosines 397 and 576.Citation36 This is associated with increased invasive behavior and increased tumor cell plasticity and vascular mimicry.Citation37 FAK tumor signaling requires both FAK kinase activity and its ability to form multiple protein complexes.Citation38 Thus, the scaffolding function of FAK plays a pivotal role in cancer and endothelial cell signaling.Citation32, Citation39
In the present study, we show that in melanoma cells FAK scaffold inhibitor C4 disrupted the FAK-VEGFR-3 complex formation and led to dephosphorylation of both proteins and decrease of cell proliferation. We found that total phosphorylation of FAK is diminished and phosphorylation of Tyr925 is decreased in a time- and C4 dose-dependent manner. This is consistent with the previous observation of Kaneda et al.Citation40 that mutation Y925F of FAK suppresses melanoma cell proliferation. More importantly, they found that Y925F mutation in B16F10 melanoma cells suppressed metastasis in an experimental model. Their results provide clear evidence that Tyr925 of FAK is critical for melanoma metastasis and we plan to study the possibility that compound C4 has anti-metastatic activity in a few different metastatic cancer models.
In parallel with the FAK dephosphorylation we saw increased dephosphorylation of extracellular signal–related kinase 1/2 (Erk1/2). This indicated the deactivation of important signaling pathway, which supports melanoma cell proliferation and survival in as many as 90% of cases.Citation41 Elevated ERK activity is a characteristic of BRAFV600E melanomas and can be inhibited by RAF inhibitors such as vemurafenib that selectively target mutant BRAF monomers; however, acquired resistance to these drugs often develops. We have shown that treatment with C4 decrease Erk phosphorylation/activation in both wild type and mutant BRAF cells and we hypothesize that treatment with C4 can overcome resistance to BRAF inhibitor and we are now evaluating this hypothesis.
Disruption of the FAK-VEGFR-3 interaction is also accompanied by dephosphorylation of VEGFR-3, which we saw in vitro in melanoma and endothelial cells and in vivo in the C4 treated tumors. VEGFR-3 phosphorylation Y1230/Y1231 contributes to proliferation, migration, and survival of endothelial cells. Phospho-Y1230/Y1231 directly recruits growth factor receptor–bonus protein (Grb2) to the receptor, inducing the activation of both AKT and Erk1/2 signaling.Citation42 Our experiments confirmed deactivation of Grb2 and Erk1/2 and motility inhibition of melanoma and endothelial cells. Our data are also in nice concordance with the data of Mitra et al.Citation43 that loss of a FAK-Grb2-MAPK linkage reduced vascular endothelial growth factor (VEGF) expression and resulted in small avascular tumors in mice.
The VEGF/VEGFR signaling pathway is integral to endothelial migration and proliferation in melanoma-associated angiogenesis.Citation44 The VEGF ligand and receptor inhibitors bevacizumab, sorafenib and axitinib have anti-tumor activity in preclinical studies.Citation45 However axitinib, a tyrosine kinase inhibitor of VEGFR family, has demonstrated single-agent activity, but no benefits for combination therapy,Citation46 and bevacizumab has demonstrated a failure as a single-agent cancer treatment.Citation47 This data suggest that VEGF signaling targeted therapies will not have a stand-alone role and effective second-line or co-therapeutic agents that mediate anti-tumor effects via non-overlapping mechanisms are needed. FAK plays an indispensable role in endothelial cells. Increased expression of FAK in tumor angiogenic vessels has been reportedCitation48 and deletion of Fak in endothelial cells impaired VEGF-mediated tumor angiogenesis in vivo and in vitro.Citation22 We have shown in 2 melanoma models, BRAF wild-type and mutant, that the FAK scaffold inhibitor C4 reduced melanoma growth as a single agent. We demonstrated that treatment with the FAK-VEGFR-3 protein–protein interaction inhibitor C4 reduced tumor blood and lymphatic vessel density and affected tumor blood flow and permeability. Our data on inhibitory effect of C4 on BRAF wild-type C8161 melanoma growth are especially important as prognoses are still very bad for patients harboring BRAF wild-type. Combination of FAK scaffold inhibitors with other targeted therapy can be promising new approach in treatment of metastatic melanoma.
Materials and Methods
Cell lines
C8161, A375, and HUVEC cells were purchased from American Type Culture Collection. Cells were maintained in RPMI-1640 with 10% fetal bovine serum. The human metastatic melanoma cell lines SK-MEL19, SK-MEL29, SK-MEL103, SK-MEL147 were kindly provided by Memorial Sloan-Kettering Cancer Center and were cultured in Dulbecco's modified Eagle's medium (Life Technologies, #11965118) supplemented with 10% fetal bovine serum (Invitrogen, # 10437028). All cell lines were incubated at 37°C in 5% CO2.
Antibodies and reagents
VEGFR-3 and p-VEGFR-3 antibody from Cell Aplications, Inc. (CB5792, CB5793), Santa Cruz Biotechnology, Inc. (SC-321) and Millipore (MAB3757). Pro-caspase-8, p-FAK, Erk 1/2, p-Erk, Akt, p-Akt, PARP from Cell Signaling Technology (# 9746S, 3284S, 9102S, 4376S, 9272S, 9271S, 9542). FAK (clone 4.47, #16–173), Paxillin (#05–417), phospho-tyrosin 4G10 (#05–321) antibody from Millipore. Compound C4 – Chloropyramine hydrochloride, C1915 and Dacarbazine – D2390; were dissolved in DMSO and stored at −20°C, (Sigma-Aldrich). Compound C4 – Chloropyramine hydrochloride solution for injections (Suprastin, 20 mg/ml), (Egis Pharmaceuticals). Antibody for IHC: anti-murine CD31 (BD-PharMingen, BD Biosciences, 550274) and LYVE1 (AbCam, ab14917).
Protein purification
His-tagged recombinant FAK C-terminal FAT domain protein (residues 892–1052) was expressed from the vector pET15b (Novagen) in E. coli BL21 (DE3) cells. Cells were grown at 37°C in LB containing antibiotics to an OD600 density of 0.6. Protein expression was induced by 0.2 mM IPTG at 37°C for 6 h. Cells were lysed in 1X PBS, 2% Triton-X and EDTA-free complete ULTRA solution (Roche Diagnostics) on ice followed by ultrasonication (Sonicator 3000 with a microprobe, Misonix) at 4°C. The protein was purified using HisPur Ni-NTA spin columns (Thermo Scientific) according to the manufacturer's protocol. The purified protein was concentrated using Amicon Ultra-4 Centrifugal filter devices (5 kDa molecular weight cutoff; Millipore). The protein was stored in 1× EDTA-free complete ULTRA solution at −80°C.
Fluorescence polarization (FP)–based binding assays
The 12-mer VEGFR-3 derived peptide AV3Citation18 was 5-carboxytetramethylrhodamine (TAMRA)-conjugated (Biomatik) and used as the fluorescent probe in the FP-based binding assays (TAMRA- Acp-WHWRPWTPCKMF-NH2). The corresponding unlabeled peptide AV3 (Biomatik) was used in the competition assay. The Kd and IC50 values were determined using the Perkin Elmer EnVision 2103 Multilabel Reader, software Wallac Envision Manager 1.12 and Graphpad Prism 6.0 software. Detailed method description provided in the Supplementary Data.
Assays of cell viability
Cell survival was assayed by measuring mitochondrial dehydrogenase activity with CellTiter 96® Aqueous One Solution Cell Proliferation Assay (Promega, #G3581) according to the manufacturer's protocol. The dose-effect curves were analyzed by the median-effect method of Chou and Talalay using the Calcusyn Software as a non-constant ratio combination. The combination index (CI) indicates a quantitative measure of the degree of drug interaction in terms of synergistic (CI < 1), additive (CI = 1) or antagonistic effect (CI > 1).Citation49
Western blot and immunoprecipitation analysis
Appropriately treated or non-treated cells were allowed to grow until they are 80–85% confluent or until treatment was completed. Cells were twice washed with ice-cold phosphate-buffered saline (PBS), then incubated on ice for 30 minutes with 1% NP-40 lysis buffer with inhibitors and proceeded as previously published.Citation24 Densitometry analysis was done with program Scion Image (Scion Corporation).
Angiogenesis assays
Capillary-like tube formation assay. HUVECs were seeded onto the Matrigel layer in 24-well plates at a density of (4 to 7) × 104 cells. ECGM was with or without various dilutions of C4. After 6 h and 24 h tubular structure of endothelial cells was photographed using an inverted microscope (Olympus; magnification ×5). Three independent experiments were performed.
Directed in vivo Angiogenesis Assay (DIVAA) (Trevigen Inc., 3450-048-DA) Implant grade silicone cylinders (angioreactors) closed at one end are filled with 20 μl of basement membrane extract (BME) premixed with or without angiogenic-modulating factors were implanted subcutaneously in the dorsal flank of nude mice. Vascular endothelial cells proceed to grow into the BME and form vessels in the angioreactor. After 12 d post-implantation angioreactors were extracted and cells counted with FITC-lectin to determine an effective dose response to angiogenic modulating factors.
Animal models
In accordance with the RPCI IACUC approved protocol (1 or 5) × 106 C8161 cells or 7 × 106 A375 cells (100 μl 50% Matrigel) were subcutaneously injected into the right flank of the 6-week old female Nude (Hsd:Athymic Nude-Foxn1nu, Harlan) or SCID/CB17 (RPCI bred colony) mice, 5–8 in each group. Treatment with compound C4 was started when tumor reached approximately 100 mm3. Tumor size was measured twice weekly and volume was calculated using the formula length X width2 × 0.5. Animals were sacrificed after 21 d of treatment or when tumor size reached protocol end point. Tumors were excised, measured and preserved for protein and RNA preparation and cytochemistry. Mice were treated with compound C4 at 50 mg/kg or 25 mg/kg intraperitoneally (IP) once a day for 5 d a week. Dacarbazine was administered at 8 mg/kg or 13 mg/kg once a day IP for 4 d at first week with 3 weeks interval.
Magnetic resonance imaging
MRI examinations were performed in mice bearing C8161 melanoma xenografts with matched tumor volumes (n = 3 per group) on a 4.7T MR scanner according to previously published methods.Citation50 Raw image data sets were transferred to a workstation for post processing using AnalyzeTM (AnalyzeDirect, Overland Park, KS) and MATLAB (Version 7.0, Mathworks Inc., Natick, MA). Relative blood volume maps were calculated on a pixel-by-pixel basis using MATLAB (Version 7.0, Mathworks Inc., Natick, MA) and pseudo-colorized in Analyze™. Detailed method description provided in the Supplementary Data.
Diffuse correlation spectroscopy for blood flow quantification
Tissue blood flow was measured using a previously described and validated diffuse correlation spectroscopy (DCS) instrument.Citation51 For each measurement, the 4 detector signals were averaged to obtain average blood flow. Skeletal muscle away from the tumor was measured for comparison. Blood flow was measured before treatment to obtain baseline levels and subsequent measurements were performed 7 d and 14 d after treatment. Measurements were normalized to baseline values to quantify changes in relative blood flow due to C4 treatment. Detailed method description provided in the Supplementary Data.
Immunohistochemistry and scoring
All staining procedures were done as previously described.Citation52 For detection, we used Vectastain Elite ABC kit (Vector Laboratories, PK-6200). Diaminobenzidine (DAB) was used as the chromogen, and the slides were counterstained with hematoxylin. Negative and positive controls were included in each staining. IHC-stained tissue slides were scanned in an Aperio ScanScope CS, viewed using ImageScope software, and quantified using Aperio Image Analysis algorithms (Aperio Technologies, Inc., Vista, CA). A full time research pathologist (W.B.) analyzed tissue sections. To obtain relative staining levels, 5 fields/sample containing a minimum of 100 cells each, were analyzed for each stain. Staining analysis for CD31 and LYVE 1 expression was done using the microvessel algorithms (v9). Both algorithms were modified and the original input parameters were adjusted to enhance the performance and accuracy of the analysis for tissue and stains.
Statistical analysis
For all experiments comparison between groups were made using a 2-tailed 2-sample Student's t test. Differences for which P value was less than 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
W.G.C. and E.K. are stockholders and founders of CureFAKtor Pharmaceuticals, LLC. University of Florida and Roswell Park Cancer Institute have patents pending and awarded that are based on their work developing FAK inhibitors.
941760_Supplemental_File.pdf
Download PDF (465.7 KB)Acknowledgments
The authors would like to thank the staff of the shared resources at RPCI for their technical assistance in performing these studies: Small Molecule Screening Core Facility, Laboratory Animal Resource, Small Animal Bio-Imaging Resource, The Pathology Core Facility, Pathology Resource Network, Data Bank and BioRepository.
Additional information
Funding
References
- Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet 2005; 365:687–701; PMID: 15721476; http://dx.doi.org/10.1016/S0140-6736(05)17951-3
- Held MA, Langdon CG, Platt JT, Graham-Steed T, Liu Z, Chakraborty A, Bacchiocchi A, Koo A, Haskins JW, Bosenberg MW, et al. Genotype-selective combination therapies for melanoma identified by high-throughput drug screening. CancerDiscov 2013; 3:52–67; PMID: 23239741; http://dx.doi.org/10.1158/2159-8290.CD-12-0408
- Al-Lazikani B, Workman P. Unpicking the combination lock for mutant BRAF and RAS melanomas. CancerDiscov 2013; 3:14–9; PMID: 23319765; http://dx.doi.org/10.1158/2159-8290.CD-12-0520
- Alex J. Eustace SK, Anne-Marie Larkin, Thamir Mahgoub, Dimitrios Tryfonopoulos LOD, Martin Clynes, John Crown, O'Donovan N. Predictive biomarkers for dasatinib treatment in melanoma. Oncoscience 2014; 1:158–66
- Zbytek B, Carlson JA, Granese J, Ross J, Mihm MC, Jr., Slominski A. Current concepts of metastasis in melanoma. Expert RevDermatol 2008; 3:569–85; PMID: 19649148; http://dx.doi.org/10.1586/17469872.3.5.569
- Alitalo K, Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. CancerCell 2002; 1:219–27; PMID: 12086857; http://dx.doi.org/10.1016/S1535-6108(02)00051-X
- Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008; 454:656–60; PMID: 18594512; http://dx.doi.org/10.1038/nature07083
- Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, Sproat G, Swann R, Gray N, Ryan A, et al. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin CancerRes 2010; 16:3548–61; PMID: 20606037; http://dx.doi.org/10.1158/1078-0432.CCR-09-2797
- Clarijs R, Schalkwijk L, Hofmann UB, Ruiter DJ, de Waal RM. Induction of vascular endothelial growth factor receptor-3 expression on tumor microvasculature as a new progression marker in human cutaneous melanoma. CancerRes 2002; 62:7059–65; PMID: 12460927
- Emmett MS, Symonds KE, Rigby H, Cook MG, Price R, Metcalfe C, Orlando A, Bates DO. Prediction of melanoma metastasis by the Shields index based on lymphatic vessel density. BMCCancer 2010; 10:208; PMID: 20478045; http://dx.doi.org/10.1186/1471-2407-10-208
- Hoshida T, Isaka N, Hagendoorn J, di Tomaso E, Chen YL, Pytowski B, Fukumura D, Padera TP, Jain RK. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. CancerRes 2006; 66:8065–75; PMID: 16912183; http://dx.doi.org/10.1158/0008-5472.CAN-06-1392
- Bogos K, Renyi-Vamos F, Dobos J, Kenessey I, Tovari J, Timar J, Strausz J, Ostoros G, Klepetko W, Ankersmit HJ, et al. High VEGFR-3-positive circulating lymphatic/vascular endothelial progenitor cell level is associated with poor prognosis in human small cell lung cancer. Clin CancerRes 2009; 15:1741–6; PMID: 19240177; http://dx.doi.org/10.1158/1078-0432.CCR-08-1372
- Mehnert JM, McCarthy MM, Jilaveanu L, Flaherty KT, Aziz S, Camp RL, Rimm DL, Kluger HM. Quantitative expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 in melanoma tissue microarrays. HumPathol 2010; 41:375–84; PMID: 20004943; http://dx.doi.org/10.1016/j.humpath.2009.08.016
- Roberts N, Kloos B, Cassella M, Podgrabinska S, Persaud K, Wu Y, Pytowski B, Skobe M. Inhibition of VEGFR-3 activation with the antagonistic antibody more potently suppresses lymph node and distant metastases than inactivation of VEGFR-2. CancerRes 2006; 66:2650–7; PMID: 16510584; http://dx.doi.org/10.1158/0008-5472.CAN-05-1843
- Alam A, Blanc I, Gueguen-Dorbes G, Duclos O, Bonnin J, Barron P, Laplace MC, Morin G, Gaujarengues F, Dol F, et al. SAR131675, a potent and selective VEGFR-3-TK inhibitor with antilymphangiogenic, antitumoral, and antimetastatic activities. Mol CancerTher 2012; 11:1637–49; PMID: 22584122; http://dx.doi.org/10.1158/1535-7163.MCT-11-0866-T
- Norrmen C, Tammela T, Petrova TV, Alitalo K. Biological basis of therapeutic lymphangiogenesis. Circulation 2011; 123:1335–51; PMID: 21444892; http://dx.doi.org/10.1161/CIRCULATIONAHA.107.704098
- Smalley KSM, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol CancerTherap 2006; 5:1136–44; PMID: 16731745; http://dx.doi.org/10.1158/1535-7163.MCT-06-0084
- Garces CA, Kurenova EV, Golubovskaya VM, Cance WG. Vascular endothelial growth factor receptor-3 and focal adhesion kinase bind and suppress apoptosis in breast cancer cells. CancerRes 2006; 66:1446–54; PMID: 16452200; http://dx.doi.org/10.1158/0008-5472.CAN-05-1661
- Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J CellBiochem 2006; 99:35–52; PMID: 16823799; http://dx.doi.org/10.1002/jcb.20956
- Lechertier T, Hodivala-Dilke K. Focal adhesion kinase and tumour angiogenesis. JPathol 2012; 226:404–12; PMID: 21984450; http://dx.doi.org/10.1002/path.3018
- Zhao X, Guan J-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug DeliverRev 2011; 63:610–5; PMID: 21118706; http://dx.doi.org/10.1016/j.addr.2010.11.001
- Tavora B, Batista S, Reynolds LE, Jadeja S, Robinson S, Kostourou V, Hart I, Fruttiger M, Parsons M, Hodivala-Dilke KM. Endothelial FAK is required for tumour angiogenesis. EMBO MolMed 2010; 2:516–28; PMID: 21154724; http://dx.doi.org/10.1002/emmm.201000106
- Wary KK, Kohler EE, Chatterjee I. Focal adhesion kinase regulation of neovascularization. MicrovascRes 2012; 83:64–70; PMID: 21616084; http://dx.doi.org/10.1016/j.mvr.2011.05.002
- Kurenova EV, Hunt DL, He D, Magis AT, Ostrov DA, Cance WG. Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo. J MedChem 2009; 52:4716–24; PMID: 19610651; http://dx.doi.org/10.1021/jm900159g
- Smith CS, Golubovskaya VM, Peck E, Xu LH, Monia BP, Yang X, Cance WG. Effect of focal adhesion kinase (FAK) downregulation with FAK antisense oligonucleotides and 5-fluorouracil on the viability of melanoma cell lines. MelanomaRes 2005; 15:357–62; PMID: 16179862; http://dx.doi.org/10.1097/00008390-200510000-00003
- Guedez L, Rivera AM, Salloum R, Miller ML, Diegmueller JJ, Bungay PM, Stetler-Stevenson WG. Quantitative assessment of angiogenic responses by the directed in vivo angiogenesis assay. Am JPathol 2003; 162:1431–9; PMID: 12707026; http://dx.doi.org/10.1016/S0002-9440(10)64276-9
- Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial–mesenchymal transition to suppression of anoikis. J CellSci 2013; 126:21–9; PMID: 23516327; http://dx.doi.org/10.1242/jcs.120907
- Schultze A, Fiedler W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin InvestDrugs 2010; 19:777–88; PMID: 20465362; http://dx.doi.org/10.1517/13543784.2010.489548
- Ward KK, Tancioni I, Lawson C, Miller NL, Jean C, Chen XL, Uryu S, Kim J, Tarin D, Stupack DG, et al. Inhibition of focal adhesion kinase (FAK) activity prevents anchorage-independent ovarian carcinoma cell growth and tumor progression. Clin ExpMetastasis 2013; 30:579–94; PMID: 23275034; http://dx.doi.org/10.1007/s10585-012-9562-5
- Infante JR, Camidge DR, Mileshkin LR, Chen EX, Hicks RJ, Rischin D, Fingert H, Pierce KJ, Xu H, Roberts WG, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J ClinOncol 2012; 30:1527–33; PMID: 22454420; http://dx.doi.org/10.1200/JCO.2011.38.9346
- Schultze A, Fiedler W. Clinical importance and potential use of small molecule inhibitors of focal adhesion kinase. Anticancer Agents MedChem 2011; 11:593–9; PMID: 21787277; http://dx.doi.org/10.2174/187152011796817727
- Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase-targeted cancer therapeutics. SciSignal 2013; 6:10–15; http://dx.doi.org/10.1126/scisignal.2004021
- Akasaka T, van Leeuwen RL, Yoshinaga IG, Mihm MC, Jr., Byers HR. Focal adhesion kinase (p125FAK) expression correlates with motility of human melanoma cell lines. J InvestDermatol 1995; 105:104–8; PMID: 7615962; http://dx.doi.org/10.1111/1523-1747.ep12313396
- Xu LH, Yang X, Craven RJ, Cance WG. The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell GrowthDiffer 1998; 9:999–1005; PMID: 9869300
- Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. Focal adhesion kinase activated by beta(4) integrin ligation to mCLCA1 mediates early metastatic growth J BiolChem 2002; 277:34391–400; PMID: 12110680; http://dx.doi.org/10.1074/jbc.M205307200
- Hess AR, Postovit LM, Margaryan NV, Seftor EA, Schneider GB, Seftor RE, Nickoloff BJ, Hendrix MJ. Focal adhesion kinase promotes the aggressive melanoma phenotype. CancerRes 2005; 65:9851–60; PMID: 16267008; http://dx.doi.org/10.1158/0008-5472.CAN-05-2172
- Hess AR, Hendrix MJ. Focal adhesion kinase signaling and the aggressive melanoma phenotype. CellCycle 2006; 5:478–80; PMID: 16552181; http://dx.doi.org/10.4161/cc.5.5.2518
- Abbi S, Guan JL. Focal adhesion kinase: protein interactions and cellular functions. HistolHistopathol 2002; 17:1163–71; PMID: 12371144
- Fan H, Zhao X, Sun S, Luo M, Guan J-L. Focal adhesion kinase scaffolding function to mediate endophilin A2 phosphorylation promotes epithelial-mesenchymal transition and mammary cancer stem cell activities in vivo. J BiolChem 2013; 288: 3322-33; PMID: 23255596; http://dx.doi.org/10.1074/jbc.M112.420497
- Kaneda T, Sonoda Y, Ando K, Suzuki T, Sasaki Y, Oshio T, Tago M, Kasahara T. Mutation of Y925F in focal adhesion kinase (FAK) suppresses melanoma cell proliferation and metastasis. CancerLett 2008; 270:354–61; PMID: 18606490; http://dx.doi.org/10.1016/j.canlet.2008.05.042
- Cohen C, Zavala-Pompa A, Sequeira JH, Shoji M, Sexton DG, Cotsonis G, Cerimele F, Govindarajan B, Macaron N, Arbiser JL. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin CancerRes 2002; 8:3728–33; PMID: 12473582
- Salameh A, Galvagni F, Bardelli M, Bussolino F, Oliviero S. Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood 2005; 106:3423–31; PMID: 16076871; http://dx.doi.org/10.1182/blood-2005-04-1388
- Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, et al. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene 2006; 25: 5969–84; PMID: 16682956
- Streit M, Detmar M. Angiogenesis, lymphangiogenesis, and melanoma metastasis. Oncogene 2003; 22:3172–9; PMID: 12789293; http://dx.doi.org/10.1038/sj.onc.1206457
- Schicher N, Paulitschke V, Swoboda A, Kunstfeld R, Loewe R, Pilarski P, Pehamberger H, Hoeller C. Erlotinib and Bevacizumab Have Synergistic Activity against Melanoma. Clin CancerRes 2009; 15:3495–502; PMID: 19447871; http://dx.doi.org/10.1158/1078-0432.CCR-08-2407
- Zhang XH, Qiao EQ, Gao Z, Yuan HQ, Cai PF, Li XM, Gu YH. Efficacy of combined axitinib with dacarbazine in a B16F1 melanoma xenograft model. OncolLett 2013; 6:69–74; PMID: 23946779
- Helfrich I, Scheffrahn I, Bartling S, Weis J, von Felbert V, Middleton M, Kato M, Ergün S, Augustin HG, Schadendorf D. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J ExpMed 2010; 207:491–503; PMID: 20194633; http://dx.doi.org/10.1084/jem.20091846
- Haskell H, Natarajan M, Hecker TP, Ding Q, Stewart J, Grammer JR, Gladson CL. Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin CancerRes 2003; 9:2157–65; PMID: 12796381
- Chou T-C. Drug combination studies and their synergy quantification using the chou-talalay method. CancerRes 2010; 70:440–6; PMID: 20068163; http://dx.doi.org/10.1158/0008-5472.CAN-09-1947
- Seshadri M, Sacadura N, Coulthard T. Monitoring antivascular therapy in head and neck cancer xenografts using contrast-enhanced MR and US imaging. Angiogenesis 2011; 14:491–501; PMID: 21901534; http://dx.doi.org/10.1007/s10456-011-9233-1
- Sunar U, Rohrbach D, Rigual N, Tracy E, Keymel K, Cooper MT, Baumann H, Henderson BH. Monitoring photobleaching and hemodynamic responses to HPPH-mediated photodynamic therapy of head and neck cancer: a case report. OptExpress 2010; 18:14969–78; PMID: 20639983; http://dx.doi.org/10.1364/OE.18.014969
- Ucar DA, Kurenova E, Garrett TJ, Cance WG, Nyberg C, Cox A, Massoll N, Ostrov DA, Lawrence N, Sebti SM, et al. Disruption of the protein interaction between FAK and IGF-1R inhibits melanoma tumor growth. CellCycle 2012; 11:3250–9; PMID: 22894899; http://dx.doi.org/10.4161/cc.21611