
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Markers of cell cycle stage allow estimation of cell cycle dynamics in cell culture and during embryonic development. The Fucci system incorporates genetically encoded probes that highlight G1 and S/G2/M phases of the cell cycle allowing live imaging. However the available mouse models that incorporate Fucci are beset by problems with transgene inactivation, varying expression level, lack of conditional potential and/or the need to maintain separate transgenes—there is no transgenic mouse model that solves all these problems. To address these shortfalls we re-engineered the Fucci system to create 2 bicistronic Fucci variants incorporating both probes fused using the Thosea asigna virus 2A (T2A) self cleaving peptide. We characterize these variants in stable 3T3 cell lines. One of the variants (termed Fucci2a) faithfully recapitulated the nuclear localization and cell cycle stage specific florescence of the original Fucci system. We go on to develop a conditional mouse allele (R26Fucci2aR) carefully designed for high, inducible, ubiquitous expression allowing investigation of cell cycle status in single cell lineages within the developing embryo. We demonstrate the utility of R26Fucci2aR for live imaging by using high resolution confocal microscopy of ex vivo lung, kidney and neural crest development. Using our 3T3 system we describe and validate a method to estimate cell cycle times from relatively short time-lapse sequences that we then apply to our neural crest data. The Fucci2a system and the R26Fucci2aR mouse model are compelling new tools for the investigation of cell cycle dynamics in cell culture and during mouse embryonic development.
Abbreviations
| BrdU | = | 5-bromo-2′-deoxyuridine |
| DAPI | = | 4′, 6-diamidino-2-phenylindole |
| DMEM | = | Dulbeccos modified eagle medium |
| ECACC | = | European Collection of Cell Cultures |
| EMMA | = | European Mouse Mutant Archive |
| FACS | = | Fluorescence-activated cell sorting |
| Fucci | = | Fluorescent Ubiquitination-based Cell Cycle Indicator |
| GMEM | = | Glasgow minimum essential medium |
| hESC | = | Human embryonic stem cell |
| IRES | = | Internal ribosomal entry site |
| LIF | = | leukemia inhibitory factor |
| mAG | = | Monomeric Azami Green |
| mESC | = | Mouse embryonic stem cell |
| mKO2 | = | Monomeric Kusabira Orange |
| RBDB | = | Riken Bioresource Center DNA Bank |
| T2A | = | Thosea asigna virus 2A peptide |
Introduction
The cell cycle in the early embryo is tightly regulated but as development progresses control diversifies and increased asynchronous divisions lead to variation within and between tissues.Citation1 Differential proliferation within tissues has been implicated in branching morphogenesis of the developing lung and kidney and in limb bud formation.Citation2–4 Furthermore proliferation is thought to contribute to the active migration of the neural crest during embryogenesis.Citation5 The mechanisms underlying these processes are poorly understood and a lineage restricted cell cycle reporter system would be a powerful tool to help dissect them.
The E3 ligases APCCdh1 and SCFSkp2 ubiquitinate a number of proteins, targeting them for degradation during the cell cycle. SCFSkp2 is both a substrate and a direct inhibitor of APCCdh1 meaning that their levels (and the levels of the proteins they ubiquitinate) oscillate reciprocally. APCCdh1 is active in late M and G1 phases while SCFSkp2 is active in S and G2.Citation6–8 Geminin and Cdt1 play roles in the regulation of replication origins and are direct substrates of APCCdh1 and SCFSkpCitation2 respectively and therefore also oscillate.Citation9,10 The Fucci (Fluorescent Ubiquitination-based Cell Cycle Indicator) probe pair consists of a fusion of monomeric Kusabira Orange (mKO2) with a truncated hCdt1 containing amino acids 30-120 and a fusion of monomeric Azami Green and the 110 amino acid N-terminus of the hGeminin protein. The mKO2-hCdt1(30/120) probe accumulates during G1 phase and is degraded at the G1-S transition. The mAG-hGem(1/110) probe accumulates during S/G2/M phases and is rapidly degraded prior to cytokinesis.Citation11 Fucci2 replaces the fluorescent proteins mKO2 and mAG with mCherry and mVenus respectively.Citation12
A number of Fucci mouse lines exist. CAG-Fucci is not inducible and is composed of 2 lines; CAG-mKO2-hCdt1(30/120) and CAG-mAG-hGem(1/110) generated by random transgenesis.Citation11 Addition transgenics of this nature are prone to transgene inactivation causing variegated/low expression levels in some tissues.Citation13 This problem may be compounded by the independent integrations of each transgene; low expression has been reported for these lines in several tissues.Citation14 R26p-Fucci2 is a constitutive allele composed of a bidirectional transgene driving mCherry-hCdt1(30/120) and mVenus-hGem(1/110) using a fragment of the mouse Rosa26 promoter. It is also generated by random transgenesis and is homozygous lethal; only hemizygotes are used resulting in a waste of non-transgenic offspring. R26-mCherry-hCdt1(30/120) and R26-mVenus-hGem (1/110) are separate inducible lines recombined into the Rosa26 locus and driven by the endogenous promoter, the R26-mCherry-hCdt1(30/120) allele suffers from low expression levels.Citation14 Of these existing mouse models no Cre-recombinase inducible single construct has been developed. One approach to achieve this goal would be to produce a bicistronic construct driven by a strong ubiquitously expressed promoter targeted to a safe harbor such as the mouse Rosa26 or HPRT locus.Citation15,16
One method to achieve bicistronic gene expression might be to use a viral internal ribosomal entry site (IRES) utilizing a cap-dependent initiation of translation for the first open-reading-frame (ORF) and a cap-independent mechanism for translation of the second (for a review see Hellen and Sarnow, 2001).Citation17 However rarely are equimolar amounts of protein produced using an IRES sequence.Citation18 An attractive alternative to the IRES are the viral 2A peptides, these short peptide sequences can be inserted between genes to create a single ORF that yields separate proteins by ribosomal skipping during translation.Citation19 2A peptides share a highly conserved C-terminal region at which the cleavage event occurs between the penultimate glycine residue and the final proline, if the cleavage efficiency is high enough a near 1:1 stoichiometric relationship between the gene products can be achieved.Citation20 The CAG promoter is a strong synthetic promoter incorporating the cytomegalovirus early enhancer element; the promoter, first exon and first intron of the chick β-actin gene; and the β-globin splice acceptor sequence.Citation21 CAG has been used widely to drive transgene expression in mouse embryos but can be sensitive to position effects therefore careful choice of the integration site is required. The most widely used safe harbor for transgene insertion in mouse is the Rosa26 locus, identified as a site of ubiquitous gene expression.Citation22,23 Rosa26 has subsequently been used to target many reporters such as β-galactosidase, CFP and YFP to produce ubiquitously expressed inducible alleles both driven by the endogenous promoterCitation15,24 and by CAG for higher expression.Citation25,26 The orientation of such constructs within the Rose26 locus can directly impact on their expression level.Citation27,28
We describe here the design and validation of a bicistronic version of Fucci2 (Fucci2a) incorporating the Thosea asigna virus 2A peptide (T2A) and the production of transgenic mice by targeting an inducible version of Fucci2a driven by the CAG promoter to the mouse Rosa26 locus. We use these tools to investigate cell cycle dynamics in 3T3 cells, the developing lung and kidney and migrating mouse neural crest derived cells. Furthermore we describe and validate a generally applicable method to estimate cell cycle times from relatively short time lapse sequences and apply this method to our neural crest data. The Fucci2a cell line, constructs and the R26Fucci2aR mouse model we describe are compelling new tools for the investigation of cell cycle dynamics in cell culture and during mouse embryonic development.
Results
The Fucci2 probe mVenus-hGem(1/110) is sensitive to C-terminal modification
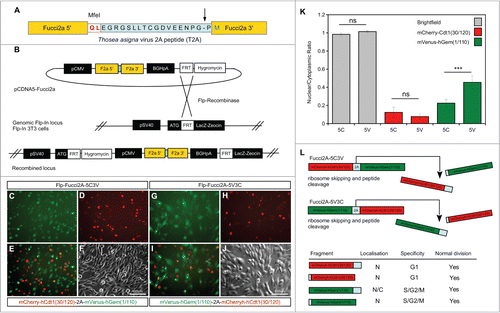
We built a bicistronic version of Fucci2 incorporating both probes into a single construct; mVenus-hGem(1/110) and mCherry-hCdt1(30/120) fused using the Thosea asigna virus 2A (T2A) self cleaving peptide sequence. The transcript from this construct should predictably produce equimolar quantities of both cell cycle probes. outlines the design and validation of the constructs. As 2A peptides cleave asymmetrically, and add the majority of amino acid residues to the C-terminus of the protein located at the N-terminal position (see ) 2 versions of the Fucci2a construct were designed to test the effects of this addition on each probe. The 2 fusions had the Fucci2 probes arranged so that either mCherry-hCdt1(30/120) or mVenus-hGem(1/110) were in the N-terminal position. We tested the constructs in 3T3 cells by generating stable lines containing a single integration of either fusion (). Analysis of these cell lines revealed that mCherry-hCdt1(30/120) localized normally to the nucleus despite the additional amino acids whereas the nuclear localization of mVenus-hGem(1/110) was partially disrupted. We confirmed this by quantification of the nuclear to cytoplasmic ratio of mVenus-hGem(1/110) in each cell line () and demonstrated an increase in this ratio (reflecting an increase in the cytoplasmic level of mVenus-hGem(1/110)). summarizes the results of this initial validation; we concluded that mVenus-hGem(1/110) was partially mis-localized on C-terminal modification. Consequently the construct with mCherry-hCdt1(30/120) in the N-terminal position was chosen for further validation and will subsequently be referred to as Fucci2a.
Figure 1. Design and validation of bicistronic Fucci2a expression constructs. (A) The Fucci2 probes mVenus-hGem(1/110) and mCherry-hCdt1(30/120) were fused using the Thosea asigna virus 2A peptide, the core T2A sequence is highlighted in blue and was inserted between the Fucci2 probes in both orientations. The T2A sequence comprises 18 amino acids, cleavage occurs between the final glycine and proline (arrow in A). In addition to these 17 amino acids 2 amino acids were added to create an MfeI restriction site for cloning; in total the 5′ Fucci probe has 19 amino acids added to its C-terminus while the 3′ Fucci probe incorporates one additional amino acid. (B) The resulting 2 versions of Fucci2a were termed Fucci2a-5C3V (5′ mCherry-hCdt1(30/120) 3′ mVenus-hGem(1/110)) and Fucci2a-5V3C (5′ mVenus-hGem(1/110) 3′ mCherry-hCdt1(30/120)) and were targeted to a single locus in NIH 3T3 cells using the Flp-In system to create 2 isogenic polyclonal Fucci2a cell lines. (C–J) Imaging of the resulting stable cell lines revealed that the addition of the 19 T2A amino acids resulted in mVenus-hGem(1/110) partially loosing its nuclear localization, mCherry-hCdt1(30/120) remained nuclear with the same addition (compare C to G and D to H). (K) Quantification of the nuclear to cytoplasmic ratio for both Fucci2 probes for the 2 Fucci2a cell lines revealed a statistically significant increase in the nuclear to cytoplasmic ratio of mVenus-hGem(1/110) in the Fucci2a-5V3C cell line (2-way ANOVA P = <0.0001, Tukey's HSD P < 0.0001). (L) Summary of the initial characterization showing that only mVenus-hGem(1/110) is sensitive to the additional 18 amino acids. Error bars in K = 95% Confidence interval.
3T3 cell cycle time is modulated by serum concentration and is concordant between daughter pairs
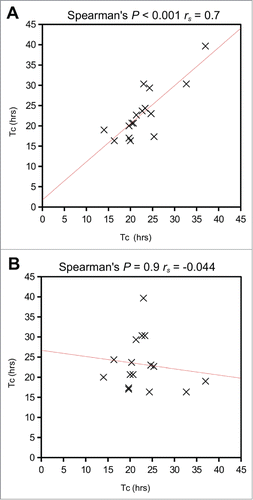
To validate the behavior of the stable Flp-Fucci2a 3T3 cells and demonstrate the utility of Fucci2a in quantitative cell cycle analyses, we performed live time-lapse imaging experiments and fluorescence activated cell sorting (FACS). summarizes the key results. Flp-Fucci2a 3T3 cells proceeded normally through the cell cycle; detectable levels of the mVenus and mCherry reporters appeared to be coordinated and cyclic; cells were either mCherry positive (red) in G1, mVenus positive (green) in S/G2/M or double positive (yellow) for a brief period in the transition between these phases (See Movie S1 and ). FACS analysis using DAPI to quantify DNA content () showed clearly that the mCherry and mVenus positive cell populations were associated with the G1 and S/G2/M fractions of the cell cycle in the same manner as the original Fucci constructs.Citation11 To obtain quantitative measurements of the lengths of the cell cycle phases highlighted by Fucci2a, we performed fluorescence live imaging of Flp-Fucci2a 3T3 cells in the presence of 10% or 15% fetal calf serum (FCS). The results were subjected to image analysis to measure the length of the mCherry (G1 – red), mVenus (S/G2/M – green) and double positive (G1/S – yellow) peaks. summarizes these results. The mean (± 95% CI) cell cycle time in the Flp-Fucci2a 3T3 cells grown in the presence of 10% FCS was 25.62 ± 2.69 hours (n = 20 mitoses). When the cells were grown in the presence of 15% FCS the cell cycle time was 20.23 ± 2.20 hours (n = 20 mitoses) representing a statistically significant reduction. The G1 (red) phase was also significantly reduced from 12.7 ± 2.57 hours to 8.90 ± 1.81 hours for 10% versus 15% serum (n = 20 mitoses in both cases). The other cell cycle phases (S/G2/M and G1/S) followed the same trend but showed smaller reductions that were not statistically significant. Interestingly there was a remarkable concordance between the cell cycle length of the 2 daughters of a mitosis over a roughly three-fold-range of cell cycle times from 15 up to 40 hours (). The positive correlation between cell cycle times of a given daughter pair was highly statistically significant (Spearman's rank order correlation P < 0.001, rs = 0.7) whereas there was no correlation between the same data if the pairs were assigned randomly (Spearman's rank order correlation P = 0.9, rs = −0.04).
Figure 2. Live imaging of Fucci2a stable 3T3 cell line. (A) A montage of a time-lapse sequence showing nuclear expression of Fucci2a throughout the progressing cell cycle. mCherry accumulates during G1 and is lost during the G1/S transition as mVenus reaches its peak. Both probes are lost at mitosis (asterisk in final panel). (B) Plot of the relative intensities of the Fucci2a probes during a single cell cycle showing the mCherry and mVenus peaks. (C) Confirmation by FACS that the Fucci2a probes accurately predict cell cycle phase defined by DAPI staining for DNA content. Cells positive for mCherry peak in the G1/2n population, while mVenus positive cells peak in the 4n population immediately prior to mitosis. (D) Quantification of the length of cell cycle phases by live cell imaging and image analysis. Increasing the serum concentration from 10–15% resulted in a statistically significant shortening of the cell cycle (students t-test with Bonferroni correction P < 0.001) and a reduction in the length of G1 ( students t-test with Bonferroni correction P < 0.05). Tc = cell cycle time; Tg1 = G1 length; Tsg2m = S/G2/M length; Ts = G1/S transition length. Error bars in D = 95% Confidence interval.
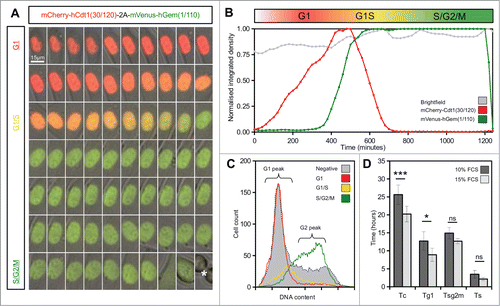
Figure 3. Comparison of mitosis times for daughter cell pairs. (A) Correlation of mitosis times for daughter cell pairs was highly statistically significant (Spearman's rank order correlation P < 0.001, rs = 0.7) (B) There was no correlation between the same data if the pairs were assigned randomly (Spearman's rank order correlation P = 0.9, rs = −0.04). Tc = cell cycle time.
Cell cycle time (Tc) can be estimated using the length of the mVenus positive phase
In an asynchronously cycling population, containing no non-cycling cells, the ratio of cells in a given cell cycle phase to the total number of cells is equal to the ratio of the length of that phase to the total cell cycle length. This relationship has been exploited using double labeling of cells with thymidine analogs to estimate cell cycle length (Tc).Citation29–31 By measuring the number of cells in S-phase (Scells) and the number that have left S-phase (Lcells) in the time between thymidine analog injections (Ti), the length of S-phase (Ts) can be derived in the following manner:
and Tc can be derived as:
Where Pcells is the total number of cells. Having accurately measured the mean cell cycle time in our 3T3 time-lapse experiments we decided to evaluate the accuracy of this theoretical approach. We reasoned that we could derive a cell cycle time from the length of S/G2/M (Tsg2m), the number of cells in S/G2/M (Nsg2m) and the total number of cells (Nall) as follows:
Using this approach we calculated cell cycle times (mean ± 95% CI) of 25.70 ± 2.50 hours and 18.40 ± 1.48 hours for the 10% and 15% serum treated populations respectively. The difference between these values was highly statistically significant (students t-test P < 0.001) and was in extremely close agreement with the values calculated from measuring complete mitoses described above.
R26Fucci2a embryonic stem cells do not accumulate mCherry-hCdt1(30/120) during G1 phase
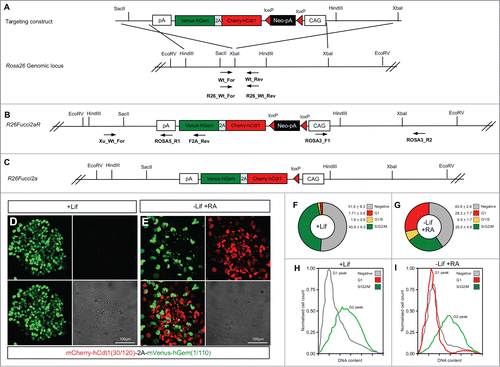
Fucci2a Cre recombinase activatable mouse embryonic stem cell (mESC) lines were made by targeting the Fucci2a construct to the Rosa26 locus creating the R26Fucci2aR allele. outlines the strategy used. Rather than rely on the endogenous Rosa26 promoter, Fucci2a expression was driven by the CAG promoter.Citation21 The construct was orientated in the opposite direction to the endogenous promoter to avoid transcriptional interference. Citation28 To test Fucci2a expression in the resulting mESC lines the construct was activated by transfection with a Cre expressing plasmid thus excising the neomycin resistance cassette to create the R26Fucci2a allele (). After transfection cells were plated at low density and individual clones picked and tested for G418 sensitivity. In R26Fucci2a mESCs clones, maintained under standard conditions in the presence of leukemia inhibitory factor (LIF), mVenus could be observed in a high proportion of nuclei () whereas mCherry positive nuclei could only be detected in a few differentiating cells at the periphery of some colonies. FACS analysis of single G418 sensitive R26Fucci2a mESC clones revealed a substantial population of cells that did not appear to be positive for either mCherry or mVenus (). Subsequent FACS analysis using DAPI to quantify DNA content revealed that the majority of this negative cell population were in G1 (). To investigate whether this was a problem with the expression construct or a mESC specific effect, R26Fucci2a ES cells were differentiated by LIF withdrawal and addition of retinoic acid (RA). After 4 days of culture mCherry became detectable in a large proportion of nuclei (compare ). DAPI staining and cell cycle analysis by FACS to quantify DNA content showed that the majority of the emerging mCherry positive population were in G1 (compare ).
Figure 4. Mouse embryonic stem cells expressing Fucci2a. A single copy of the Fucci2a transgene under the control of the CAG promoter was inserted into the Rosa26 locus by homologous recombination in mouse embryonic stem cells (mESCs). (A) Targeting construct used, a stop cassette containing a loxP flanked neomycin resistance gene and polyadenylation sequence was inserted between CAG and Fucci2a, this construct was inserted in the reverse orientation to the endogenous Rosa26 promoter to avoid transcriptional interference. (B) The targeted R26Fucci2aR inducible allele, screening for correct homologous recombination was done using PCR across the 5′ and 3′ homology arms of the targeting construct. To test the R26Fucci2aR ES cell lines they were transfected with a Cre-recombinase expressing plasmid (pPGK-Cre), plated at low density and screened for G418 sensitive Fucci2a expressing clones (R26Fucci2a). (C) The targeted R26Fucci2a allele after Cre-mediated excision of the floxed-Neo-pA stop cassette. (D) R26Fucci2a ES cells showed high levels of mVenus in the majority of cells but very few mCherry positive cells were evident. (E) However on withdrawal of Lif and culture in the presence of retinoic acid (RA) for 4 days high proportions of mCherry positive cells were evident. (F and G) FACS analysis showed that there were a large proportion of cells negative for both markers in R26Fucci2a ES cells and that on Lif removal and RA treatment the negative population was reduced and an mCherry positive population became apparent. (H and I) Quantification of DNA content by DAPI staining followed by FACS analysis showed clearly that the majority of G1 cells were negative for mCherry in the R26Fucci2a clone. After Lif withdrawal and culture in the presence of RA for 4 days, a G1 population of mCherry positive cells became apparent.
Regionalized variation in Fucci2a cell cycle status in R26Fucci2aR+/Tg/CAG-Cre+ve embryos
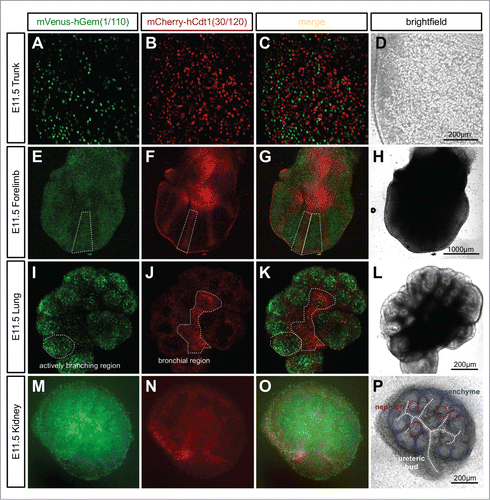
R26Fucci2aR ES cells were used to generate the global, bicistronic, Cre-activatable reporter mouse line R26Fucci2aR. No Fucci2a expression was observed in R26Fucci2aR animals due to the strong transcriptional stop sequences contained in the floxed-Neo-pA stop cassette (data not shown). Ubiquitous Fucci2a expression was examined in R26Fucci2aR+/Tg/CAG-Cre+ve embryos, summarizes some of the patterns observed. We observed strong Fucci2a expression at all time points examined including trunk, forelimbs, lung and kidney at E11.5 (). Further investigation of the developing limb bud at E11.5 showed there was a clear pattern in the distribution of cells in G1/G0 (red) and S/G2/M (green). In the condensing mesenchyme of the future digits there was a predominance of cells in G1/G0 (red, ) while in the interdigitary mesenchyme, cells continued to proliferate and were predominately in S/G2/M (green, ). Such regionalized differences in proliferation were also evident in the developing lung and kidney cultured from E11.5. In the lung the actively branching regions were predominantly in S/G2/M (green, ) while the future bronchial regions were beginning to exit the cell cycle and were predominantly populated by cells in G1/G0 (red, ). In the developing kidney, cells in S/G2/M (green) were primarily located within the ureteric bud as well as in clusters of cells immediately adjacent to the bud (). It was clear from inspection of the brightfield channel that these clusters were early nephron structures at either renal vesicle or comma-shaped body stage (). In contrast the cap mesenchyme, containing the nephron progenitor cells, was largely composed of cells in G1/G0 (red, ).
Figure 5. Ubiquitous expression of Fucci2a in R26Fucci2aR+/Tg/CAG-Cre+ve embryos. (A–D) Whole mount R26Fucci2aR+/Tg/CAG-Cre+ve embryonic trunk at E11.5. Fucci2a expression and localization is clearly apparent with red green and yellow cells evident. (E–H) Whole mount R26Fucci2aR+/Tg/CAG-Cre+ve embryonic limb bud at E11.5. Clear areas of mCherry-hCdt1(30/120) positive cells are evident in the condensing mesenchyme that will go on to form the future bone while the interdigitary areas are still highly proliferative. (I–L) A R26Fucci2aR+/Tg/CAG-Cre+ve E11.5 embryonic lung cultured for 24 hours, there is a clear bias in the distribution of cells in G1 and S/G2/M. The actively branching regions of the developing lung are highly proliferative while the future bronchial regions have begun to drop out of the cell cycle as demonstrated by the high proportion of G1 cells in these regions (dotted regions in I, J, K). (M–P) A dissected R26Fucci2aR+/Tg/CAG-Cre+ve E11.5 embryonic kidney cultured for 24hrs. Cells in S, G2, and M-phase were primarily detected within the ureteric bud and in clusters of cells immediately adjacent to the bud comprising early nephron structures. The cap mesenchyme was largely populated by cells in G1.
Rapid proliferation at the budding tip drives branching morphogenesis in the embryonic lung and kidney
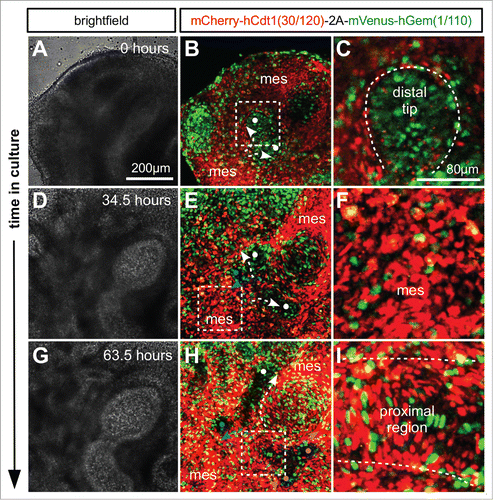
We used time lapse imaging of tissue from R26Fucci2a+/Tg/CAG-Cre+ve mice to investigate cell cycle progression during lung and kidney development. Whole lung and kidney samples were dissected from E11.5 embryos and cultured. In the lung it was possible to observe the process of branching morphogenesis of the respiratory epithelium (; Movie S2). Branching was observed via both bifurcation of the tip of an epithelial branch () and by domain branching from the side of an existing branch (). In both cases cells at the distal tip of an epithelial branch were observed to be highly proliferative, demonstrated by the high proportion of S/G2/M phase cells () labeled with mVenus (green). This localized proliferation appears to drive elongation of an epithelial branch into the surrounding mesenchyme which was composed of cells predominantly positive for mCherry and therefore in G1/G0 () suggesting a much lower rate of proliferation. As an epithelial branch elongated, cells of the proximal region began to slow their cell cycles ending in a stable G1/G0 state and perdurance of the mCherry label ().
Figure 6. Live confocal imaging of branching morphogenesis during lung development. Lungs were cultured ex vivo from E11.5. (A and B) An elongating branch of lung epithelium begins to bifurcate at right angles to the plane of imaging (white dots and arrows in B). (C) Cells in the distal tip of the extending branch of lung epithelium are proliferating rapidly shown by the high proportion of S/G2/M (green) nuclei. (D and E) The left and right branches elongate driven by proliferation at the growing tip. By 34.5 hours a daughter branch can be seen to emerge from the left parent branch by domain branching (blue dot and arrow in E). (F) In contrast to the epithelial branch the surrounding mesenchyme is composed of cells predominantly in G1/G0 (red). (G and H) Subsequently the right parent branch bulges and then bifurcates into 2 sister branches (magenta arrows and dots in H). (I) In the proximal regions of the branching lung epithelium cells begin to exit the cell cycle and enter G1/G0 (red). Abbreviations: mes = mesenchyme.
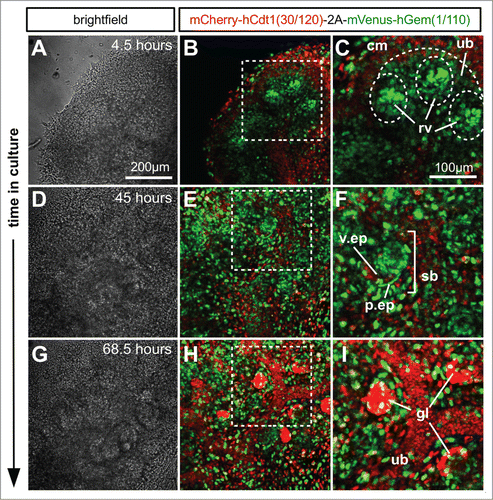
In the kidney, time lapse images were captured of the developing cap-mesenchyme, ureteric bud tips, ureteric bud stalk, as well as developing nephrons (Fig. 6; Movie S3). The cap-mesenchyme population consisted of a mixture of cells positive for mVenus (green) and mCherry (red) and therefore in both S/G2/M and G1/G0. When nephrons progressed through development and formed the more mature structure of S-shaped bodies () a large proportion of cells remained in S/G2/M (green). However, presumptive podocytes in the visceral epithelium were beginning to enter G1/G0 (red). Cells within the ureteric bud successively entered G1/G0 (red) as the ureteric bud tips grew further away. In more mature glomerular structures almost all podocytes were in G1/G0 (red, ; Movie S3). The nephron progenitor niche, i.e. the cap mesenchyme, contained cells in S/G2/M (green), and G1/G0 (red) that kept cycling throughout the culture period. The ureteric bud tips primarily contained cells in S/G2/M (green) but stalk regions became progressively populated by cells in G1/G0 (red). Renal vesicles (early nephrons) forming adjacent to the ureteric bud tips contained a high proportion of cells in S/G2/M (green). As the nephrons developed further through S-shaped body stage, the distal and medial segments (identified by their morphology) were in S/G2/M (green) but the podocytes in the presumptive glomeruli were in G1/G0 (red).
Figure 7. Ex vivo time-lapse imaging of mouse embryonic kidney development. (A–C) Part of an E12.5 kidney after 4.5 hours in culture. The cap mesenchyme contains a larger proportion of cells in G1 (red) than either ureteric bud or renal vesicles. The renal vesicles in particular contain tight condensations of cells predominately in S/G2/M (green). (D–F) After 45 hours in culture the S-shaped bodies have formed and while the majority of cells were still cycling it was clear that the presumptive podocytes of the visceral epithelium were beginning to exit the cell cycle and enter G1/G0 (red). (G–I) By 68.5 hours in culture the majority of podocytes in the mature glomerular structures appeared to have exited the cell cycle. Abbreviations: cm = cap mesenchyme, ub = ureteric bud; rv = renal vesicle; v.ep = visceral epithelium; p.ep = parietal epithelium; sb = S-shaped body.
Melanoblast specific expression of Fucci2a and estimation of cell cycle time
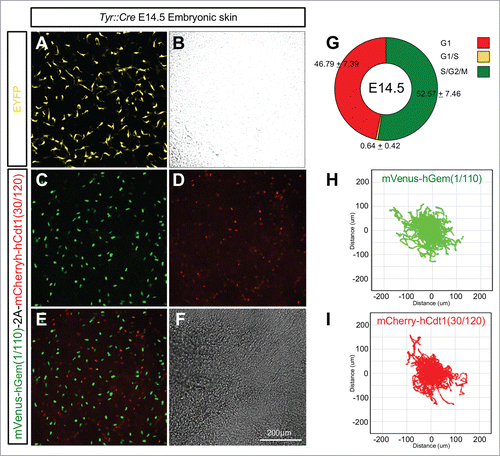
To test the tissue specificity of R26Fucci2aR mice they were crossed with Tyr::CreB animals expressing Cre-recombinase in the neural crest derived melanocyte lineage.Citation32 Embryonic mouse skin from E14.5 Tyr::CreBTg+ve/R26Fucci2aRTg/+ animals was cultured as previously describedCitation33 and analyzed by live confocal microscopy for Fucci2a expression. As expected Fucci2a expression was observed in melanoblasts, a highly migratory population of melanocyte precursors in the developing epidermis (; Movie S4). It was possible to calculate the relative proportions of Fucci2 labeled cells and also track the cells as they migrated (). Roughly equal proportions of mCherry positive and mVenus positive cells were observed at the beginning of the time lapse sequence in all the samples analyzed (52.57 ± 7.46% vs 46.79 ± 7.39% respectively n = 10 embryos) while only a very low percentage were observed in the G1/S transition (0.64 ± 0.42%). Strikingly both the mCherry positive and mVenus positive populations were able to migrate freely with very similar mean (±95% CI) velocities of 0.58 ± 0.07 um/min (n = 1562 tracks from 10 embryos) and 0.52 ± 0.05 um/min (n = 1080 tracks from 10 embryos) respectively. Because cells could not be distinguished from the surrounding keratinocytes after cytokinesis (see the green cells disappearing in Movie S4) and because our time lapse sequences were relatively short (∼18 hours) it was not possible to follow individual cells through an entire cell cycle. However it was clear from the observation of time-lapse sequences that the randomly migrating melanoblast population was asynchronously cycling. Furthermore it was previously shown that 2 BrdU doses at 20 min intervals followed by a 2 hour chase labeled around 60% of the entire epidermal melanoblast population at E14.5.Citation34 This strongly suggests that the vast majority if not all melanoblasts are cycling. We therefore reasoned we could estimate the cell cycle time of the epidermal melanoblast population using the method described above for our Flp-Fucci2a 3T3 cells. We calculated the length of S/G2/M (mean ± 95% CI) in the migrating melanoblast population to be 7.73 ± 0.41 hours (n = 26 cells, 7 independent movies/embryos). Using this data we were able to derive a cell cycle time (mean ± 95% CI) of 17.79 ± 4.6 hours for the epidermal melanoblast population at E14.5.
Figure 8. Lineage specific Fucci2a expression in developing melanoblasts. (A and B) Embryonic skin in culture from a Tyr::Cre+ve/R26EYFPRTg/+ embryo at E14.5. EYFP positive melanoblasts show their characteristically dendritic morphology. (C–F) Embryonic skin in culture from a Tyr::Cre+ve/R26Fucci2aRTg/+ positive embryo at E14.5, melanoblasts from all stages of the cell cycle are visible. (G) Quantification of the proportions of melanoblast in the 3 cell cycle phases in E14.5 embryonic skin samples (n = 10 embryos). (H) Automated cell tracking of mVenus-hGem(1/110) labeled melanoblasts (in S/G2/M) over an 18 hour time-lapse sequence showing the population spread. (I) Automated cell tracking of mCherry-hCdt1(30/120) labeled melanoblasts (in G1) over an 18 hour time-lapse sequence showing the population spread. Error values in G = 95% confidence intervals.
Discussion
We describe the design and validation of Fucci2a, a bicistronic reporter of cell cycle progression. Fucci2a performs identically to the original Fucci and Fucci2 probes,Citation11,14 but offers the key advantage of being composed of a single genetic construct. highlights the resources developed during this study and the repositories in which they have been housed. In particular the R26Fucci2aR transgenic mouse line will prove an invaluable tool especially where live imaging and ex vivo culture are required. A clear advantage of the R26Fucci2aR allele over the other published mouse models is that because both probes are expressed from a single genomic locus only one reporter strain is required, simplifying maintenance and experimental crosses. Furthermore the R26Fucci2aR animals can be maintained as homozygotes. This is not possible with the recently published R26p-Fucci2 constitutive allele (because it is homozygous lethal) or the R26-mCherry-hCdt1(30/120) and R26-mVenus-hGem (1/110) alleles because they are integrated at the same genomic locus meaning a mouse can only be heterozygous for each probe.Citation14 Furthermore, because the Fucci2a construct is bicistronic, it is likely that roughly equimolar amounts of each fluorescent probe are produced which is advantageous for imaging purposes. Previous reports have suggested that in some contexts the T2A peptide used in this study can have a cleavage efficiency of >99%.Citation35 In the present study the tight correlation between Fucci2a fluorescence and cell cycle stage, demonstrated by live imaging and FACS analysis, suggests that either the cleavage efficiency of the T2A peptide used is extremely efficient or that any uncleaved peptide is rapidly degraded because it contains the degradation signals of both hGeminin and hCdt1.
Table 1. Available Fucci2a resources and repository information
The present study shows that 3T3 cells modulate the length of their G1 phase in response to increased serum concentration and demonstrate the utility of the Fucci2a system to detect a change in G1 length in response to a relatively subtle increase from 10% to 15% serum. However the observed concordance between cell cycle length in sister pairs following a mitosis suggests that as well as extrinsic factors such as confluence and composition of the culture media there are heritable cell autonomous factors that can influence cell cycle length even in transformed cells. This is perhaps not surprising given that a polyclonal 3T3 cell line was used and that there is known genetic heterogeneity between clonally selected populations.Citation36 Using our Flp-Fucci2a 3T3 cell line we were able to validate a method to very accurately estimate cell cycle time from relatively short time lapse sequences by measuring only the length of S/G2/M. This method shroud be generally applicable and useful in situations where long time-lapse sequences are difficult to capture or where it is difficult to track cells though late M-phase when the Fucci2a probes are both degraded.
In R26Fucci2a mESCs very little accumulation of mCherry-hCdt1(30/120) in G1 was observed. Similarly 2 previous reports using mESCs infected with Fucci expressing lentivirus vectors have described only between 6 and 9% mKO2-hCdt1(30/120) positive cells with substantial negative populations.Citation37 In both cases the authors concluded that the lack of mKO2-hCdt1(30/120) fluorescence in early G1 was because the short G1 phase in mESCs did not allow enough time for the mKO2-hCdt1(30/120) probe to accumulate. In the present study it was estimated that in 3T3 cells it only takes ∼100 minutes (see ) to accumulate detectable levels of mCherry-hCdt1(30/120). G1 length in mESCs has previously been estimated at around 180 minutesCitation38 which should be enough time for mKO2-hCdt1(30/120) to accumulate. Therefore if the previously published explanation holds true it seems likely the relative expression level of the Fucci probes must be affecting how soon during G1 the hCdt1(30/120)-tagged probe becomes detectable. The expression level of the R26Fucci2a mESC line described in the present study is likely to be lower than the lentiviral systems used in the above papers and this may be the reason we see fewer mCherry-hCdt1(30/120) labeled cells. In support of this theory in human embryonic stem cells (hESCs) both Fucci probes are readily detectableCitation39,40 consistent with the longer cell cycle time reported for hESCs compared to mESCs.Citation41,42
The present study demonstrates the feasibility of using Fucci2a in ex vivo embryonic organ culture to investigate the role of proliferation during development. Both probes were readily detectable in live tissue using only modest confocal laser power and tissues remained viable throughout the culture period. We observed Fucci2a expression in all tissues examined between E10.5 and E15.5. In the embryo, areas that appeared predominantly red (mCherry-hCdt1 – G1/G0) generally corresponded with areas of differentiation such as; the developing digits of the limb bud, the proximal regions of lung epithelial branches and the stalk of the ureteric bud in the developing kidney. Using time-lapse confocal microscopy we captured the growth of lungs, and kidneys. Our data clearly show active, rapid proliferation driving branching morphogenesis in the lung and kidney. In the kidney the actively branching regions of the ureteric bud tips were seen to be highly proliferative while proximal regions of the ureteric bud began to drop out of the cell cycle. This is consistent with BrdU based studies of proliferation in a similar organ culture based system, showing increased BrdU incorporation at the ureteric bud tips compared to the stalks proximal to these regions.Citation3 Similarly in the present study, regions of high proliferation were observed in the tips of lung epithelial branches with lower rates of proliferation in regions proximal to the tips. Indeed budding tips in the lung epithelium were often so active that few or no G1 cells could be observed in this region at all, suggesting cells were moving through the G1 phase in less than 100 minutes. These results are consistent with a previous BrdU based proliferation study that showed that while bud initiation does not require differential proliferation bud outgrowth does.Citation43 However using a BrdU pulse only serves to highlight cells in S phase during the pulse period, it is a snapshot that does not provide information about the cell cycle status of the non-labeled cells. The R26Fucci2aR reporter mouse presented here allows one to map cell cycle fate in all the cells of a developing branch in real-time providing unprecedented detail of the fate and cell cycle status of every cell both spatially and temporally.
Mouse embryonic skin culture has been widely used to study skin development and more recently in conjunction with live-imaging to understand the behavior of melanoblasts during embryonic development.Citation33,44,45 Here for the first time the feasibility of using live cell cycle probes to investigate this lineage is demonstrated. The labeled populations migrated at speeds that were consistent with previous reports that did not consider cell cycle phase.Citation33 This is the first report that demonstrates that melanoblasts do not modulate their migratory mechanisms during cell cycle progression except immediately prior to mitosis when they pause to divide. This is consistent with a recent report on the mitotic behavior of chick neural crest cells, showing that they do not lose their position in the migrating population as they undergo mitosis.Citation5 The present study includes the first direct measurement of the cell cycle characteristics in the migrating melanoblast population. Our estimate of an E14.5 cell cycle time of ∼17 hours is in close agreement with the work of Luciani et alCitation34 who estimated the population doubling time by BrdU labeling and mathematical modeling to be between 16 and 18 hours. This system, will be very useful to investigate the large number of mouse mutants with pigment phenotypes especially those in genes implicated in migration and proliferation such as Kit, Kitl, Mitf and Pax3.Citation46–49
In conclusion we describe the design and validation of Fucci2a a novel bicistronic cell cycle reporter system appropriate for live imaging and FACS analysis. We demonstrate the utility of Fucci2a for quantitative assessment of cell cycle progression in a stable 3T3 cell line and in mESCs and describe a method to estimate cell cycle times from short time lapse sequences without having to track complete mitoses. Furthermore we describe the design, development and characterization of the R26Fucci2aR mouse model which will prove a valuable tool for the study of proliferation during mouse embryonic development when combined with a tissue-specific Cre-recombinase.
Materials and Methods
Construct design
In order to concatenate mVenus-hGeminin(1/110) and mCherry-hCdt1(30/120) in both orientations the second gene was amplified first by PCR using a generic forward primer incorporating the T2A sequence preceded by MluI and MfeI restriction sites (Fucci2t2a_For) and a reverse primer specific to either hGem(1/110) (hGemKpnI_Rev) or hCdt1(30/120) (hCdt1KpnI_Rev) flanked by a KpnI restriction site. The resulting amplicon was restriction cloned as an MluI-KpnI fragment into pCAGiP (a kind gift from Dr Thomas Pratt). The first gene was then amplified with a generic forward primer containing an MluI restriction site (Fucci2_For) and a reverse primer specific to either hGeminin (hGemMfeI_Rev) or hCdt1(hCdt1MfeI_Rev) flanked by an MfeI site. The second amplicon was then restriction cloned as an MfeI/MluI fragment to give pCAG-5C3V and pCAG-5V3C. All primer sequences are outlined in , the restriction sites used are underlined. To make the Flp-targeting plasmids pCDNA5–5C3V and pCDNA5–5V3C the plasmid pCDNA5/Frt (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/V601020) was modified with a polylinker (see for oligonucleotide sequences—pCDNAPoly_For and pCDNAPoly_Rev) cloned between the NheI and ApaI restriction sites and introducing EcoRI and AscI restriction sites. Subsequently Fucci–5V3C and Fucci2–5C3V were cloned between the EcoRI and AscI restriction sites. To generate pRosa26-CAG-floxNeo-Fucci2a, pCAGiP was modified with a polylinker (pCAGPoly_For and pCAGPoly_Rev) allowing the subcloning of CAG-Fucci2-pA as a SalI/HindIII fragment. A floxed Neo cassette was then restriction cloned between CAG and Fucci2 as an EcoRI fragment from the plasmid pCAGfloxNEOpA (a kind gift from Prof Ian Chambers). The pRosa26-PA plasmid (Addgene, https://www.addgene.org/21271) was modified with a polylinker (pRosa26-pA_For and pRosa26_pA_Rev) so that the AscI and PacI sites were inverted. Subsequently the entire CAG-floxNeopA-Fucci2–5C3V-pA construct was transferred as a PacI/AscI fragment so that it was aligned in the opposite direction to the endogenous Rosa26 promoter to make pRosa26-CAG-floxNeo-Fucci2a.
Table 2. Oligonucleotide sequences used for vector construction
Generation of stable Flp-Fucci2a cell lines
Flp-in 3T3 cells (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/R76107) were grown to confluence in a T75 tissue culture flask under standard conditions. The Neon electroporation system (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/MPK5000S) was used to co-transfect the cells with the modified pCDNA5/FRT plasmids described above and the pOG44 Flp recombinase expressing plasmid (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/V600520). Briefly, the cells were trypsinised, washed in PBS and resuspended in Buffer R. 18ug of pOG44 and 3ug of the targeting construct were added to each tube. Each tube was then split into 2 × 100 ul electroporations (2 pulses: 1,350V, 20ms) and pooled into a single T75 flask containing pre-warmed OPTIMEM (Life Technolgies, https://www.lifetechnologies.com/order/catalog/product/31985070) and incubated overnight. On the second day cells were transferred into dulbeccos modified eagle medium (DMEM) containing, 10% fetal calf serum, 1% Penicillin/Streptomycin and 100μg/ml Hygromycin B. After 14 days of Hygromycin B selection the polyclonal cell lines were passaged as normal and used for subsequent analyses.
Fluorescence microscopy and live imaging of Flp-Fucci2a cell lines
For the initial screening of the Flp-Fucci2a-5V3C and Flp-Fucci2a-5V3C cell lines, single images were captured on a widefield Nikon Ti fluorescence microscope with a 20x dry lens. To compare the nuclear to cytoplasmic ratio of the Fucci2a probes and to measure the length of mitosis, cells were seeded in glass bottomed dishes in DMEM (10% or 15% FCS, 1% Pen/Strep). The dish was enclosed in an environmental chamber maintained at 37°C with 5% CO2 in air. Images were captured using a Nikon C1 confocal system at 20 minute time intervals. For Movie S1 and the montage in cells were seeded at low densities in glass dishes and cultured in DMEM (10% FCS, 1% Pen/Strep) on the stage of Nikon TiE microscope equipped with a Nikon perfect focus system. The dish was enclosed in an environmental chamber maintained at 37°C with 5% CO2 in air. Images were captured with a 40x dry objective every 20 minutes using a Photometrics Evolve 512 EMCCD camera and a Lumen Dynamics Xcite 120Q light source (120W mercury vapor).
ES Cell targeting and generation of transgenic mice
E14 ES cells were maintained in Glasgow Minimum Essential Medium (GMEM) supplemented with 10% fetal calf serum, 0.1mM non-essential amino acids, 2mM L-Glutamine, 1mM sodium pyruvate, 0.1mM β-mercaptoethanol and 106 units/L LIF. They were electroporated with linearized pRosa26-CAG-floxNeo-Fucci2a plasmid using standard procedures. Clones were picked after 14 days of G418 selection. Colonies were screened by PCR across the Rosa26 5′ homology arm using the primers Xu_Wt_ForCitation50 and Rosa5_R1 (See and ) to generate a 1.4kb targeted band. A second control PCR was conducted to demonstrate DNA integrity using the primers Wt_For and Wt_RevCitation15 to generate a 450bp wildtype band from the Rosa26 locus. Correct targeting was confirmed on the positive clones by PCR amplification of a 4kb targeted band across the Rosa26 3′ homology arm using the primers Rosa3_F1 and Rosa3_R2, all primer sequences are outlined in . All PCR reactions were carried out using 50 ng genomic DNA using Phusion Hotstart II DNA polymerase (Thermo Fisher Scientific,
Table 3. Oligonucleotide sequences used for PCR reactions
Table 4. PCR conditions for all reactions
Maintenance and genotyping of mice
All animal work was approved by a University of Edinburgh internal ethics committee and was performed in accordance with institutional guidelines under license by the UK Home Office. Mice were maintained in the animal facilities of the University of Edinburgh. R26Fucci2aR mice were genotyped using a duplex PCR reaction with the primers R26_Wt_For, R26_Wt_Rev and F2A_Rev () the R26_Wt_Rev primer was used at 50μM rather than 100μM; all other conditions were the same as described above, and the cycling conditions are detailed in .
FACS analysis
Flow Cytometry sorting of Flp-Fucci2a cells was performed using a FACS Aria2 SORP cell sorter (Becton Dickinson) equipped with a 405nm violet laser, a 488nm blue laser and a 560nm yellow-green laser. mVenus was detected using the 488nm laser and a 525/50nm bandpass filter. mCherry was detected using the 560nm laser and a 610/20nm bandpass filter. Flp-Fucci2a cells were sorted into mCherry positive, mVenus positive, double positive and negative fractions. The cells were subsequently fixed in 70% Ethanol at −20°C overnight. The following day they were briefly stained with DAPI (5μg/ml in PBS) and analyzed for DNA content using the 405nm laser and a 450/50nm bandpass filter. FACSDiVa Version 6.1 (BD) was used to operate the instrument and to analyze the data.
Embryonic skin culture
Embryonic mouse skin was dissected from E14.5 embryos and mounted on a custom made imaging clip filled with 1% agarose to support the tissue. The skin was then clamped with the epidermal side against a lumox gas-permeable membrane (Greiner Bio-One; http://www.greinerbioone.com/en/row/articles/catalog/articles/423_11/) in a custom made culture chamber filled with DMEM (10% FCS, 1% Glutamax, 1% Penicillin/Streptomycin). The chamber was placed on the stage of an inverted Nikon C1 confocal microscope surrounded by an environmental chamber providing 5% CO2 in air and maintained at a constant stage top temperature of 37°C. Confocal Z-stacks were captured every 2 minutes for 18 hours.
Lung and kidney culture
To capture kidneys at low magnification an inverted epifluorescent microscope equipped with a 4x objective was used. For high resolution confocal time lapse of lungs and kidneys the tissue was immobilized with reduced growth factor matrigel (BD biosciences) on lumox gas-permeable membranes mounted in a custom chamber and submerged in DMEM (10% FCS, 1% Glutamax, 1% Penicillin/Streptomycin). The chamber was placed on the stage of an inverted Nikon A1R confocal microscope surrounded by an environmental chamber providing 5% CO2 in air and maintained at a constant stage top temperature of 37°C. Images were captured with a 20x objective every 30 minutes through roughly 60 μm z-stacks.
Image analysis
All image analysis tasks were performed using custom written macros for the FijiCitation51 distribution of ImageJ an open source image anlaysis package based on NIH Image.Citation52 To track Flp-Fucci2a 3T3 cells and analyze the length of cell cycle phases a semi automated tracking method was used. Intensity was measured as the integrated density of a 10 pixel circular ROI that was centered on the local maxima within a cell's nucleus. Pairs of cells were tracked from the first frame after cytoplasmic cleavage until the frame before cleavage at the next mitosis. The intensity values for each track were normalized (between 0-1) and a 3-point rolling average was applied to smooth the data. Peaks were called as positive for mCherry or mVenus if the normalized integrated density was greater than 0.15. To automatically track mCherry and mVenus labeled melanoblasts a modified version of the wrMTrck plugin (http://www.phage.dk/plugins/wrmtrck.html) was used on segmented TIFF stacks of each channel, the script used relied on Gabriel Landini's morphology collection (http://www.mecourse.com/landinig/software/software.html).
Statistics
All statistical tests were performed using the ‘R’ statistics package, an open source software package based on the ‘S’ programming language (http://www.R-project.org). The length of cell cycle phases after culture in 10% or 15% serum were compared with students’ t-tests using Bonferroni correction for multiple testing. The nuclear to cytoplasmic ratios of mCherry and mVenus in Flp-3T3-Fucci2a cells were compared using a one-way analysis of variance (ANOVA) followed by pairwise post hoc tests using Tukey's honest significant difference (HSD) test. The correlation between mitosis times for daughter cells and randomly matched pairs were compared using a Spearman's rank order test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Movie S4: Melanoblast specific Fucci2a expression in mouse embryonic skin culture.
Download MP4 Video (9.9 MB)Movie S3: Fucci2a expression in mouse embryonic kidney culture.
Download MP4 Video (5.4 MB)Movie S2: Fucci2a expression in mouse embryonic lung culture.
Download MP4 Video (5.8 MB)Movie S1: Stable Flp-Fucci2a 3T3 cells progressing through the cell cycle.
Download MP4 Video (1 MB)Acknowledgments
The authors are grateful to: Joe Mee and the Scottish Center for Regenerative Medicine Transgenic Service for ES cell targeting; Fiona Kilanowski and Julia Dorin for ES cell karyotyping; Matthew Pearson and Paul Perry for imaging support; Elisabeth Freyer for help and advice on FACS analysis; Olivia Harrison for providing plasmids.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Additional information
Funding
References
- Edgar B. Diversification of cell cycle controls in developing embryos. Curr Opin Cell Biol 1995; 7:815-24; http://dx.doi.org/10.1016/0955-0674(95)80065-4
- Nogawa H, Morita K, Cardoso W V. Bud formation precedes the appearance of differential cell proliferation during branching morphogenesis of mouse lung epithelium in vitro. Dev Dyn 1998; 213:228-35; PMID:9786423; http://dx.doi.org/10.1002/(SICI)1097-0177(199810)213:2<228::AID-AJA8>3.0.CO;2-I
- Michael L, Davies JA. Pattern and regulation of cell proliferation during murine ureteric bud development. J Anat 2004; 204:241-55; PMID:15061751; http://dx.doi.org/10.1111/j.0021-8782.2004.00285.x
- Boehm B, Westerberg H, Lesnicar-Pucko G, Raja S, Rautschka M, Cotterell J, Swoger J, Sharpe J. The role of spatially controlled cell proliferation in limb bud morphogenesis. PLoS Biol 2010; 8:e1000420; PMID:20644711; http://dx.doi.org/10.1371/journal.pbio.1000420
- Ridenour DA, McLennan R, Teddy JM, Semerad CL, Haug JS, Kulesa PM. The neural crest cell cycle is related to phases of migration in the head. Development 2014; 141:1095-103; PMID:24550117; http://dx.doi.org/10.1242/dev.098855
- Vodermaier HC. APC/C and SCF: controlling each other and the cell cycle. Curr Biol 2004; 14:R787-96; PMID:15380093; http://dx.doi.org/10.1016/j.cub.2004.09.020
- Wei W, Ayad NG, Wan Y, Zhang G-J, Kirschner MW, Kaelin WG. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004; 428:194-8; PMID:15014503; http://dx.doi.org/10.1038/nature02381
- Benmaamar R, Pagano M. Involvement of the SCF complex in the control of Cdh1 degradation in S-phase. Cell Cycle 2005; 4:1230-2; PMID:16123585; http://dx.doi.org/10.4161/cc.4.9.2048
- Nishitani H, Lygerou Z, Nishimoto T. Proteolysis of DNA replication licensing factor Cdt1 in S-phase is performed independently of geminin through its N-terminal region. J Biol Chem 2004; 279:30807-16; PMID:15138268; http://dx.doi.org/10.1074/jbc.M312644200
- Ang XL, Harper JW. Interwoven ubiquitination oscillators and control of cell cycle transitions. Sci STKE 2004; 2004:pe31; PMID:15266102
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008; 132:487-98; PMID:18267078; http://dx.doi.org/10.1016/j.cell.2007.12.033
- Sakaue-Sawano A, Kobayashi T, Ohtawa K, Miyawaki A. Drug-induced cell cycle modulation leading to cell-cycle arrest, nuclear mis-segregation, or endoreplication. BMC Cell Biol 2011; 12:2; PMID:21226962; http://dx.doi.org/10.1186/1471-2121-12-2
- McClenaghan M, Mehtali M, Dobie K, Lathe R. Variegated gene expression in mice. Trends Genet 1997; 13:127-30; PMID:9097721; http://dx.doi.org/10.1016/S0168-9525(97)01097-4
- Abe T, Sakaue-Sawano A, Kiyonari H, Shioi G, Inoue K, Horiuchi T, Nakao K, Miyawaki A, Aizawa S, Fujimori T. Visualization of cell cycle in mouse embryos with Fucci2 reporter directed by Rosa26 promoter. Development 2013; 140:237-46; PMID:23175634; http://dx.doi.org/10.1242/dev.084111
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 1999; 21:70-1; PMID:9916792; http://dx.doi.org/10.1038/5007
- Evans V, Hatzopoulos A, Aird WC, Rayburn HB, Rosenberg RD, Kuivenhoven JA. Targeting the Hprt locus in mice reveals differential regulation of Tie2 gene expression in the endothelium. Physiol Genomics 2000; 2:67-75; PMID:11015584
- Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 2001; 15:1593-612; PMID:11445534; http://dx.doi.org/10.1101/gad.891101
- Bochkov YA, Palmenberg AC. Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. Biotechniques 2006; 41:283-4, 286, 288 passim; PMID:16989088; http://dx.doi.org/10.2144/000112243
- Donnelly ML, Gani D, Flint M, Monaghan S, Ryan MD. The cleavage activities of aphthovirus and cardiovirus 2A proteins. J Gen Virol 1997; 78(Pt 1):13-21; PMID:9010280
- Szymczak-workman AL, Vignali KM, Vignali DAA. Design and Construction of 2A Peptide-Linked Multicistronic Vectors Design and Construction of 2A Peptide-Linked Multicistronic Vectors. Cold Spring Harb Protoc 2012; (2):199-204; PMID:22301656; http://dx.doi.org/10.1101/pdb.ip067876
- Miyazaki J, Takaki S, Araki K, Tashiro F, Tominaga A, Takatsu K, Yamamura K. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene 1989; 79:269-77; PMID:2551778; http://dx.doi.org/10.1016/0378-1119(89)90209-6
- Fiering S, Zambrowicz BP, Soriano P, Imamoto A, Herzenberg LA, Kerr WG. Disruption of overlapping transcripts in the ROSA βgeo 26 gene trap strain leads to widespread expression of β-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci U S A 1997; 94; PMID:9108056
- Friedrich G, Soriano P. Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev 1991; 5:1513-23; PMID:1653172; http://dx.doi.org/10.1101/gad.5.9.1513
- Srinivas S, Watanabe T, Lin C-SS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 2001; 1:4; PMID:11299042; http://dx.doi.org/10.1186/1471-213X-1-4
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 2007; 45:593-605; PMID:17868096; http://dx.doi.org/10.1002/dvg.20335
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic analysis with double markers in mice. Cell 2005; 121:479-92; PMID:15882628; http://dx.doi.org/10.1016/j.cell.2005.02.012
- Chen C, Krohn J, Bhattacharya S, Davies B. A comparison of exogenous promoter activity at the ROSA26 locus using a ΦiC31 integrase mediated cassette exchange approach in mouse ES cells. PLoS One 2011; 6:e23376; PMID:21853122; http://dx.doi.org/10.1371/journal.pone.0023376
- Grant SGN, Strathdee D, Ibbotson H. Expression of transgenes targeted to the Gt(ROSA)26Sor locus is orientation dependent. PLoS One 2006; 1:1-9; http://dx.doi.org/10.1371/journal.pone.0000001
- Martynoga B, Morrison H, Price DJ, Mason JO. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev Biol 2005; 283:113-27; PMID:15893304; http://dx.doi.org/10.1016/j.ydbio.2005.04.005
- Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol 1989; 18:311-8; PMID:2746304; http://dx.doi.org/10.1007/BF01190834
- Quinn JC, Molinek M, Martynoga BS, Zaki PA, Faedo A, Bulfone A, Hevner RF, West JD, Price DJ. Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol 2007; 302:50-65; PMID:16979618; http://dx.doi.org/10.1016/j.ydbio.2006.08.035
- Delmas V, Martinozzi S, Bourgeois Y, Holzenberger M, Larue L. Cre-mediated recombination in the skin melanocyte lineage. Genesis 2003; 36:73-80; PMID:12820167
- Mort RL, Hay L, Jackson IJ. Ex vivo live imaging of melanoblast migration in embryonic mouse skin. Pigment Cell Melanoma Res 2010; 23:299-301; http://dx.doi.org/10.1111/j.1755-148X.2010.00669.x
- Luciani F, Champeval D, Herbette A, Denat L, Aylaj B, Martinozzi S, Ballotti R, Kemler R, Goding CR, De Vuyst F, et al. Biological and mathematical modeling of melanocyte development. Development 2011; 138:3943-54; PMID:21862558; http://dx.doi.org/10.1242/dev.067447
- Donnelly MLL, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. The “Cleavage” activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring “A-like” sequences. J Gen Virol 2001; 82:1027-41; PMID:11297677
- Oh M-K, Scoles DR, Haipek C, Strand AD, Gutmann DH, Olson JM, Pulst S-M. Genetic heterogeneity of stably transfected cell lines revealed by expression profiling with oligonucleotide microarrays. J Cell Biochem 2003; 90:1068-78; PMID:14624465; http://dx.doi.org/10.1002/jcb.10712
- Coronado D, Godet M, Bourillot P-Y, Tapponnier Y, Bernat A, Petit M, Afanassieff M, Markossian S, Malashicheva A, Iacone R, et al. A short G1 phase is an intrinsic determinant of naïve embryonic stem cell pluripotency. Stem Cell Res 2013; 10:118-31; PMID:23178806; http://dx.doi.org/10.1016/j.scr.2012.10.004
- Koledova Z, Kafkova LR, Calabkova L, Krystof V, Dolezel P, Divoky V. Cdk2 inhibition prolongs G1 phase progression in mouse embryonic stem cells. Stem Cells Dev 2010; 19:181-94; PMID:19737069; http://dx.doi.org/10.1089/scd.2009.0065
- Singh AM, Chappell J, Trost R, Lin L, Wang T, Tang J, Wu H, Zhao S, Jin P, Dalton S. Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. Stem Cell Reports 2013; 1:532-44; PMID:24371808; http://dx.doi.org/10.1016/j.stemcr.2013.10.009
- Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell 2013; 155:135-47; PMID:24074866; http://dx.doi.org/10.1016/j.cell.2013.08.031
- Dalton S. Exposing hidden dimensions of embryonic stem cell cycle control. Cell Stem Cell 2009; 4:9-10; PMID:19128789; http://dx.doi.org/10.1016/j.stem.2008.12.003
- Ohtsuka S, Dalton S. Molecular and biological properties of pluripotent embryonic stem cells. Gene Ther 2008; 15:74-81; PMID:17989701; http://dx.doi.org/10.1038/sj.gt.3303065
- Nogawa H, Morita K, Cardoso WV. Bud formation precedes the appearance of differential cell proliferation during branching morphogenesis of mouse lung epithelium in vitro. Dev Dyn 1998; 213:228-35; PMID:9786423; http://dx.doi.org/10.1002/(SICI)1097-0177(199810)213:2<228::AID-AJA8>3.0.CO;2-I
- Li A, Ma Y, Jin M, Mason S, Mort RL, Blyth K, Larue L, Sansom OJ, Machesky LM. Activated mutant NRas(Q61K) drives aberrant melanocyte signaling, survival, and invasiveness via a Rac1-dependent mechanism. J Invest Dermatol 2012; 132(11):2610-21; PMID:22718121; http://dx.doi.org/10.1038/jid.2012.186
- Li A, Ma Y, Yu X, Mort RL, Lindsay CR, Stevenson D, Strathdee D, Insall RH, Chernoff J, Snapper SB, et al. Rac1 drives melanoblast organization during mouse development by orchestrating pseudopod- driven motility and cell-cycle progression. Dev Cell 2011; 21:722-34; PMID:21924960; http://dx.doi.org/10.1016/j.devcel.2011.07.008
- Wehrle-Haller B, Meller M, Weston JA. Analysis of melanocyte precursors in Nf1 mutants reveals that MGF/KIT signaling promotes directed cell migration independent of its function in cell survival. Dev Biol 2001; 232:471-83; PMID:11401406; http://dx.doi.org/10.1006/dbio.2001.0167
- Brannan CI, Lyman SD, Williams DE, Eisenman J, Anderson DM, Cosman D, Bedell MA, Jenkins NA, Copeland NG. Steel-Dickie mutation encodes a c-kit ligand lacking transmembrane and cytoplasmic domains. Proc Natl Acad Sci U S A 1991; 88:4671-4; PMID:1711207; http://dx.doi.org/10.1073/pnas.88.11.4671
- Nakayama A, Nguyen MT, Chen CC, Opdecamp K, Hodgkinson CA, Arnheiter H. Mutations in microphthalmia, the mouse homolog of the human deafness gene MITF, affect neuroepithelial and neural crest-derived melanocytes differently. Mech Dev 1998; 70:155-66; PMID:9510032; http://dx.doi.org/10.1016/S0925-4773(97)00188-3
- Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell 1991; 67:767-74; PMID:1682057; http://dx.doi.org/10.1016/0092-8674(91)90071-6
- Hohenstein P, Slight J, Ozdemir DD, Burn SF, Berry R, Hastie ND. High-efficiency Rosa26 knock-in vector construction for Cre-regulated overexpression and RNAi. Pathogenetics 2008; 1:3; PMID:19014667; http://dx.doi.org/10.1186/1755-8417-1-3
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676-82; PMID:22743772; http://dx.doi.org/10.1038/nmeth.2019
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9:671-5; PMID:22930834; http://dx.doi.org/10.1038/nmeth.2089