
Abstract
Enzastaurin is a selective inhibitor of protein kinase C β and a potent inhibitor of tumor angiogenesis. In addition, enzastaurin shows direct cytotoxic activity toward a subset of tumor cells including colorectal cancer cells (CRC). In spite of promising results in animal models, the clinical activity of enzastaurin in CRC patients has been disappointing although a subset of patients seems to derive benefit. In the present study we investigated the biological and cytotoxic activities of enzastaurin toward a panel of well-characterized CRC cell lines in order to clarify the mechanistic basis for the cytotoxic activity. Our results show that enzastaurin is significantly more cytotoxic toward CRC cells with chromosome instability (CIN) compared to cells with microsatellite instability (MSI). Since CIN is usually attributed to mitotic dysfunction, the influence of enzastaurin on cell cycle progression and mitotic transit was characterized for representative CIN and MSI cell lines. Enzastaurin exposure was accompanied by prolonged metaphase arrest in CIN cells followed by the appearance of tetraploid and micronuclei-containing cells as well as by increased apoptosis, whereas no detectable mitotic dysfunctions were observed in MSI cells exposed to isotoxic doses of enzastaurin. Our study identifies enzastaurin as a new, context dependent member of a heterogeneous group of anticancer compounds that induce “mitotic catastrophe," that is mitotic dysfunction accompanied by cell death. These data provide novel insight into the mechanism of action of enzastaurin and may allow the identification of biomarkers useful to identify CRC patients particularly likely, or not, to benefit from treatment with enzastaurin.
Abbreviations
| CRC | = | colorectal cancer |
| PKC | = | protein kinase C |
| CIN | = | chromosome instability |
| MSI | = | microsatellite instability |
| VEGF | = | vascular endothelial cell growth factor |
| VEGFR | = | vascular endothelial cell growth factor receptor |
| TP53 | = | tumor protein p53 |
| MEK | = | mitogen-activated protein kinase kinase |
| MAP | = | mitogen-activated protein |
| RACK | = | receptor of activated protein kinase C |
| MMC | = | mitomycin C |
| MN | = | micronuclei |
| DMSO | = | Dimethyl sulfoxide |
Introduction
The protein kinase C (PKC) family is a group of 8 closely related proteins that have been the focus of intense drug discovery efforts since their identification as the receptor for phorbol ester, a potent tumor promoter, more than 20 years ago.Citation1 However, until now, not a single PKC-selective anticancer agent has been approved for clinical use.Citation2 One reason is that different PKC isoforms may promote different, and even opposite, activities in a cell- and tissue-dependent manner, which has prompted the development of isoform-selective agents.Citation3
The PKC β isoform exists as 2 different splice variants, PKC βI and βII, with an important role for βII in angiogenesis as well as in the etiology of some tumors. The vascular activities of PKC βII were first described for diabetic mellitus, where retinal neovascularization can lead to blindness. PKC βII promotes angiogenesis as an element of the intracellular signaling pathway induced by vascular endothelial cell growth factor (VEGF) via a VEGF - VEGFR2/KDR/Flk1 - phospholipase C gamma - PKC β - Raf - MEK - MAP kinase pathwayCitation4 as well as by phosphorylation of the retinoblastoma (Rb) protein, as shown by overexpression of a dominant negative PKC βII isoform.Citation5 Endothelial cell proliferation and retinal vascular permeability can also be attenuated by administration of ruboxistaurin (LY333531), a PKC β selective inhibitor.Citation6 Subsequent studies have expanded the role of PKC β to other vascular pathologies including tumor angiogenesisCitation7,8 and the disruption of the blood-brain barrier (BBB) in multiple sclerosis.Citation9
Increased PKC β signaling is implicated in different tumor types including colorectal cancer (CRC). Overexpression of PKC βII is accompanied by colonic hyperproliferation and increased sensitivity to colon carcinogenesis in transgenic mice Citation10,11 suggesting that activation of PKC βII is an early promotive event in colon carcinogenesis. PKC βII drives tumorigenesis through different mechanisms including activation of Wnt/APC/β-catenin signaling, repression of transforming growth factor β (TGF-β) signalingCitation10 and activation of the p57 MAP kinase.Citation12
Enzastaurin (LY317615), a PKC βII-selective inhibitor that was developed for oncology,Citation13 showed antiproliferative activity in the low micromolar range toward a subset of tumor cells in the NCI60 tumor cell line panel, including CRC cell lines, as well as tumor growth inhibitory activity in CRC xenograft models.Citation14 Enzastaurin exposure was accompanied by apoptosis in sensitive cell lines and attenuation of the Akt-mTOR axis, as shown by decreased levels of phospho-Ser240/244 S6 and phospho-Thr308 Akt.Citation13 Subsequent studies showed that enzastaurin treatment is accompanied by hypophosphorylation of 4E-BP (the eukaryotic initiation factor 4E binding protein) thereby reducing the formation of translation initiation complexes.Citation15 It is currently not known if the cytotoxic activity of enzastaurin is a result of the attenuated survival signaling or if the drug needs to interfere with additional targets to induce cell death.
Enzastaurin has shown variable activity in patients with metastatic CRC (mCRC). Treatment of a small number of asymptomatic mCRC patients with enzastaurin in first line indicated that enzastaurin may have single-agent activity in some patients.Citation16 However, a later study where enzastaurin was used for maintenance therapy in combination with bevacizumab showed no improvement of the progression-free survival for the combination as compared to bevacizumab alone.Citation17
In the present study we investigated the biological and cytotoxic activities of enzastaurin toward a panel of well-characterized CRC cell lines in order to clarify the mechanistic basis for the cytotoxic activity. Our results show that enzastaurin has preferential cytotoxicity toward chromosome instable (CIN) CRC cells compared to cells with microsatellite instability (MSI). Enzastaurin exposure was accompanied by prolonged metaphase arrest in CIN cells followed by the appearance of tetraploid and micronuclei-containing cells as well as by increased apoptosis whereas no detectable mitotic dysfunctions were observed in MSI cells exposed to isotoxic doses of enzastaurin. These data provide novel insight into the mechanism of action of enzastaurin and may allow the development of biomarkers useful for identification of CRC patients particularly likely, or not, to benefit from treatment with enzastaurin.
Results
Enzastaurin-sensitivity is associated with chromosomal instability (CIN), but not with PKC βII levels or TP53 status
A panel of 12 well-characterized CRC cell lines was used to establish the cytotoxic activity of enzastaurin. Enzastaurin was active in the low micromolar range (0.35 to 4 μM) toward all 12 cell lines as determined by the MTT viability assay after 120 hours continuous drug exposure (). Interestingly, the average IC50 value for the panel (2 μM) was within the same dose range as the clinically relevant plasma concentration.Citation18
Figure 1. Influence of PKC-βII expression, genetic stability and TP53 status on the sensitivity of CRC cells to enzastaurin. (A) The growth inhibitory effects of enzastaurin was determined by the MTT viability assay after 120 hours continuous drug exposure and is expressed as IC50 values (drug concentration inhibiting cell growth by 50% compared to untreated controls). The white columns correspond to cells with CIN whereas the dark columns represent cells with MSI. All values are averages of at least 3 independent experiments each done in duplicate. (B) Correlation between the IC50 values and the basal levels of PKC-βII protein as determined by Western blot analysis followed by quantitative analysis by Image J software. There was no statistical significant correlation between the 2 parameters (r2 = 2 × 10−5). (C) Average IC50 values of CRC cells with CIN (white columns, n = 6) or MSI (dark columns, n = 6). Bars, S.D; **, P < 0.01 as determined by Student's t-test. (D) Average IC50 values of CRC cells with wild-type TP53 (white columns representing LS513, LS174T, SW48, HCT-116 and LoVo, n = 5) or mutant TP53 (dark columns representing HT-29, SW1116, SW620, FET, SW480, HCT-15 and DLD-1, n = 7). Bars, SD.
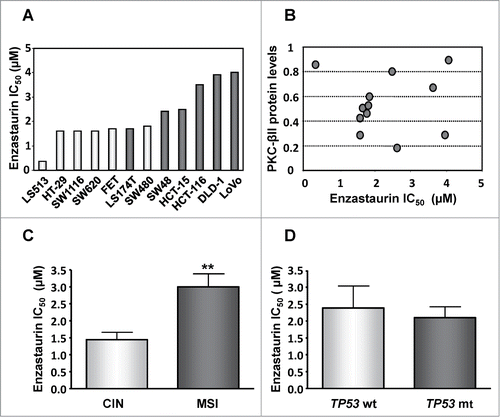
Enzastaurin is a selective inhibitor of the βII isoform of PKC. We therefore determined if PKC βII expression was correlated with drug response. The results showed no correlation between PKC βII protein levels and enzastaurin sensitivity () suggesting that PKC βII inhibition alone may not be sufficient for induction of enzastaurin-induced cell death.
Chromosomal instability (CIN) has been associated with poor prognosis and chemoresistance to a variety of agentsCitation19–22 whereas microsatellite instability (MSI) has been linked to chemosensitivity to at least some drugs.Citation23 We therefore characterized the correlation between enzastaurin sensitivity and the CIN or MSI phenotype in our CRC cell panel. Unexpectedly, CIN was significantly (p < 0.01) associated with enhanced sensitivity to enzastaurin (IC50 = 1.3 ± 0.54 μM, mean ± SD) whereas MSI cells were more resistant (IC50 = 3.0 ± 0.93 μM) ().
Since MSI cells often express wild-type p53 protein whereas many CIN cells have TP53 mutations,Citation24 we divided the cell line panel into 2 groups harboring either TP53 mutations or wt TP53. As shown in , TP53 status alone had no significant influence on the sensitivity to enzastaurin.
Enzastaurin induces mitotic arrest in CIN cells
Next, we characterized the influence of enzastaurin on the cell cycle progression. Continuous enzastaurin exposure (2 μM) of HT-29 cells (CIN) induced a transient cell cycle arrest in G2/M (). By 72 hours, the fraction of G2/M cells diminished whereas the fraction of cells with a sub-G1 DNA content increased, indicative of apoptotic cell death. Similar findings were observed for SW-620 cells (CIN) whereas the cell cycle progression of DLD-1 and LoVo cells, that display a MSI phenotype, was not affected at isotoxic doses (data not shown).
Figure 2. Enzastaurin exposure induces mitotic arrest in CIN cells. (A) HT-29 cells were exposed to 2 μM of enzastaurin for the indicated times and the cell cycle distribution was determined by flow cytometry analysis. The values indicate the average values for 2 experiments, each done in duplicate. (B) Bi-parametric flow analysis of HT-29 cells exposed to enzastaurin for 24 hours, reveals a dose-dependent accumulation of phospho-H3 positive mitotic cells. Positive control, cells treated with 10 μM nocodazole for 16 hours. Negative control, cells treated with the appropriate isotype-specific antibody. The values indicate the average of 2 experiments, each done in duplicate. (C) Fraction of phospho-H3 positive mitotic cells following exposure to enzastaurin for the indicated times. The columns indicate the average values for at least 2 experiments, each done in duplicate. Bars, SD; **, P < 0.01 and ***, P < 0.001 as determined by Student's t-test. (D) Enzastaurin exposure (24 hours) is accompanied by an increased fraction of phospho-H3 positive mitotic cells for CIN cells (HT-29 and SW-620) but not for MSI cells (DLD-1 and LoVo). The columns indicate the average values of at least 3 experiments, each done in duplicate.
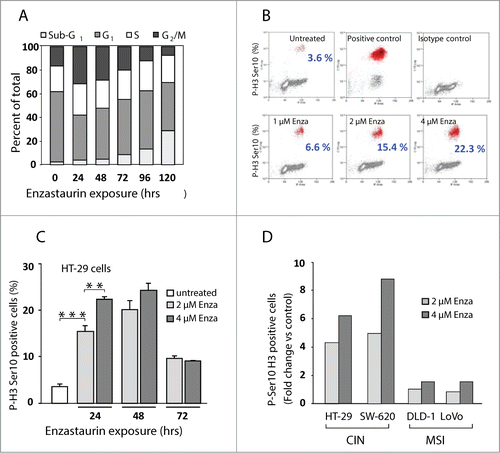
To establish if the increased G2/M fraction was due to G2 or mitotic arrest, flow cytometry analysis was carried out using an antibody that specifically recognizes the mitosis-specific phosphorylation of Ser10 on histone H3.Citation25,26 Bi-parametric flow cytometry analysis revealed that 24 hours exposure to enzastaurin induced a strong, dose-dependent increase in the fraction of phospho-H3-positive cells to reach 22.3% after exposure to 4 μM enzastaurin, compared to 3.6% for untreated control cells (). The mitotic arrest was transient, with a peak of phospho-H3-positive cells at 48 hours (). Similar results (data not shown) were obtained using the MPM-2 antibody that detects multiple mitotic phosphoepitopes.Citation27 Analysis of additional cell lines revealed that enzastaurin-treatment was accompanied by mitotic arrest in CIN cells, but not in MSI cells ().
Enzastaurin induces metaphase arrest in CIN cells
To establish which part of mitosis is affected by enzastaurin, HT-29 cells (CIN and enzastaurin-sensitive) and DLD-1 cells (MSI and enzastaurin refractory) were treated with enzastaurin for 24 hours and the mitotic figures were quantified by visual scoring after immunostaining for phospho-H3 (mitosis), lamin B (prophase to metaphase transition) and α/β tubulin (mitotic spindle formation) as illustrated in . The results show that enzastaurin induced a significant increase in the proportion of metaphase cells, with a corresponding decrease in the fraction of prophase and anaphase cells (). In contrast, enzastaurin had no detectable influence on mitosis in DLD-1 cells (data not shown).
Figure 3. Enzastaurin exposure is accompanied by accumulation of CIN cells in metaphase with an activated mitotic checkpoint. (A) HT-29 and DLD-1 cells were treated with the indicated doses of enzastaurin, and the mitotic figures were characterized following immunostaining for lamin B, α/β tubulin and phospho-H3. (B) The influence of enzastaurin on the proportion of prophase, metaphase and anaphase cells was characterized as described above after visual scoring of 100 mitotic cells. The data represent the average of 2 independent experiments. Bars, SD **, P < 0.01; ***, P < 0.001 as determined by Student's t-test. (C) The influence of enzastaurin on the accumulation of the active, hyperphosphorylated forms of the spindle assembly check-point proteins BubR1 and Chk1 in HT-29 and DLD-1 cells. The black arrows indicate the migration of the hyperphosphorylated forms of the 2 check-point proteins. The blot is representative of 2 independent experiments.
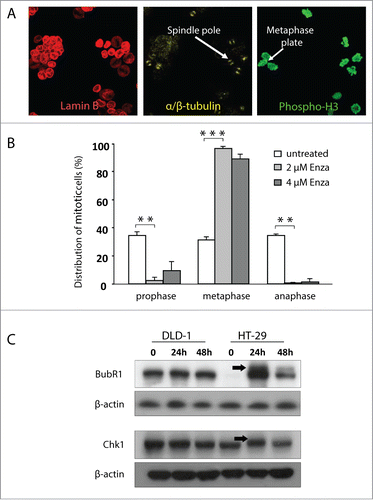
Metaphase arrest is characterized by the accumulation of cells with activated mitotic check-point proteins such as BubR1 and Chk1.Citation28–30 Western blot analysis showed that enzastaurin exposure of HT-29 cells was accompanied by a time-dependent accumulation of the active, phosphorylated forms of BubR1 and Chk1 as indicated by an electrophoretic mobility shift (, black arrows). In contrast, no accumulation of phosphorylated checkpoint proteins was observed for DLD-1 cells.
Enzastaurin induces chromosome misalignment and polyploidy
Prolonged mitotic arrest is indicative of mitotic dysfunction that may result in chromosome abnormalities. In agreement, 72 hours treatment of HT-29 cells with enzastaurin (2 μM) revealed the presence of tetraploid cells as indicated by morphological and biochemical criteria ( left, white arrows) and by flow cytometry analysis ( right, black arrow). Chromosome missegregation can result in the formation of chromosome fragments that can give rise to micronuclei (, black arrows). To establish if enzastaurin exposure leads to micronuclei formation, HT-29 and DLD-1 cells were exposed to enzastaurin for 36 hours in the presence of cytochalasin, an inhibitor of cytokinesis. Mitomycin C, a DNA cross-linking agent, was included as positive control. The results show that mitomycin C induced a strong increase in micronuclei formation in both cell lines (). In clear contrast, enzastaurin induced a significant, dose-dependent increase in the formation of micronuclei in HT-29 cells, but had no significant influence on micronuclei levels in DLD-1 cells.
Figure 4. Enzastaurin exposure is accompanied by chromosomal and centrosomal abnormalities in CIN cells. (A) Left, Immunocytochemical staining for lamin B, α/β- tubulin and phospho-H3 reveals the presence of polyploid HT-29 cells (white arrows) after 72 hours exposure to enzastaurin (2 μM). Right, HT-29 cells were incubated with enzastaurin (2 μM) for the indicated times followed by flow cytometry analysis. The black arrow indicates the presence of tetraploid cells. (B) Illustration of typical micronuclei formation in HT-29 cells (black arrows). (C) Enzastaurin induces a dose-dependent increase in the formation of micronuclei in HT-29 cells (left) but not in DLD-1 cells (right), in clear contrast to the positive control, mitomycin C (MMC) that is a strong micronuclei inducer in both cell types. Results represent the mean values of 1000 binucleated cells from 2 independent experiments. ***, P < 0.001 as determined by Student's t-test. (D) Enzastaurin induces numerical centrosome abnormalities in HT-29, but not in DLD-1 cells. Left, enzastaurin exposure is accompanied by the appearance of cells with an increased number of centrosomes (white arrows) as determined by immunocytochemistry with an antibody directed against the centrosomal marker, gamma-tubulin. Right, cells were incubated with enzastaurin (2 μM) for the indicated times and the formation of additional centrosomes was determined by immunocytochemistry with an antibody directed against gamma-tubulin. Results are expressed as mean values of 100 cells from 2 independent experiments. **, P < 0.01 as determined by Student's t-test.
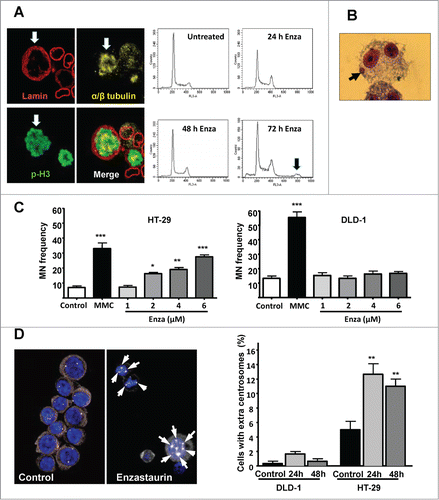
Numerical chromosomal aberrations such as chromosome misalignment and/or polyploidy may be due to centrosome amplification.Citation31 To determine the influence of enzastaurin on centrosome numbers, HT-29 or DLD-1 cells were exposed to isotoxic doses of enzastaurin (2 or 4 μM, respectively) for the indicated times and the number of centrosomes per cell was determined by immunohistochemistry with an antibody directed against gamma-tubulin, a centrosomal marker ( left. Multiple centrosomes are indicated with white arrows). The results show that enzastaurin exposure had minor effect on the centrosome number in DLD-1 cells. In contrast, prolonged enzastaurin exposure was accompanied by a significant increase in the fraction of HT-29 cells with numerical centrosome abnormalities ( right). It should be noted that the background levels of cells with numerical centrosome abnormalities was higher for HT-29 (CIN) than for DLD-1 (MSI) cells, in agreement with their respective phenotype.
Discussion
In this study we investigated the biological and cytotoxic activities of enzastaurin toward a panel of well-characterized colorectal cancer cells. The results show that enzastaurin was significantly more cytotoxic toward cells with chromosome instability (CIN) than to cells that display microsatellite instability (MSI). CIN cells are characterized by an aneuploid/polyploid karyotype whereas MSI cells are typically diploid or near-diploid due to important differences in mitotic fidelity between the 2 cell types. In concordance, the cytotoxic activity of enzastaurin toward CIN cells was characterized by prolonged metaphase arrest followed by the appearance of tetraploid and micronuclei-containing cells as well as by increased apoptosis. In comparison, no detectable mitotic dysfunction was observed for MSI cells exposed to isotoxic doses of enzastaurin. Therefore, our study identifies enzastaurin as a new, context dependent, member of a heterogeneous group of anticancer compounds that induce “mitotic catastrophe," that is mitotic dysfunction accompanied by cell death.Citation32
Most cancers are characterized by some degree of aneuploidy, which is usually attributed to defective chromosome cohesion, mitotic checkpoint alterations or the presence of multiple centrosomes.Citation33 A meta-analysis of 63 studies representing more than 10000 CRC patients indicates that CIN tumors are associated with a significantly worse prognosis compared to MSI tumors.Citation20 A subsequent study provided a more complex picture since the outcome may not be dependent on chromosome instability as such, but rather on the identity of the implicated gene loci since specific chromosome gains or deletions could be associated with either survival or disease progression.Citation21
Interestingly, mitotic dysfunction is not restricted to tumor cells, but has also been described for other pathologies and cell types. Cell division fidelity is altered during the vascular response to injury where the rapidly proliferating smooth muscle cells show increased division defects as indicated by the presence of binucleate cells and cells with micronuclei or multiple centrosomes, which likely contribute to the formation of atherosclerotic plaques.Citation34 Aneuploidy and other chromosomal abnormalities are also present in tumor-associated endothelial cells, in particular in the microenvironment of highly metastatic tumors.Citation35,36
The preferential cytotoxicity of enzastaurin toward CIN cells was unexpected. A recent study that determined the activity of 160 small molecule protein kinase inhibitors targeting different protein kinase families toward a panel of CRC cells reported that CIN cells were consistently more resistant than cells that are chromosome stable.Citation22 The CIN phenotype was also associated with taxane resistance in cellular models as well as in the OV01 ovarian cancer clinical trialCitation37 whereas the same signature was linked to carboplatin sensitivity. In vitro studies using a large panel of CRC cell lines suggested that MSI is associated with resistance to 5-fluorouracil.Citation38 In agreement, in adjuvant trials, MSI status was associated with 5-fluorouracil resistance for at least some subgroups of CRC patients.Citation39 Together, these findings suggest that although the CIN phenotype is frequently accompanied by increased resistance to anticancer agents, some drugs may be preferentially toxic to CIN cells, at least within certain cellular contexts.
The mitotic abnormalities observed in CIN cells after enzastaurin exposure could be due to defects in chromosome cohesion, defective mitotic check-point function and/or centrosome dysfunction. Enzastaurin did not show any obvious influence on chromosome cohesion in CIN cells based on the morphology of mitotic chromosomes. Furthermore, the prolonged metaphase arrest is indicative of a functional spindle assembly checkpoint. In contrast, enzastaurin-exposure of CIN cells, but not of cells with MSI, was accompanied by a significant increase in centrosome abnormalities.
Centrosomes are small organelles that are needed for microtubule nucleation and organization. Anchoring of the microtubules and their associated gamma-tubulin complexes to the centrosomes is mediated by pericentrin during mitosis, whereas pericentrin has no detectable influence on gamma-tubulin localization or microtubule organization in interphase cells.Citation40 Orderly mitotic progression in at least some tumor cells requires the docking of catalytically active PKC βII to pericentrin.Citation41,42 Disruption of the PKC βII-pericentrin association with blocking peptidesCitation41 was accompanied by microtubule disorganization and chromosome missegregation, in full agreement with the mitotic abnormalities observed herein following treatment with the PKC βII inhibitor enzastaurin. Interestingly, overexpression of the disrupting peptide fragment had no effect in cells lacking PKC βII, indicating a specific regulatory role of this isoform in centrosome function that is essential for some, but not all, cells. These findings are consistent with our observation that catalytically active PKC βII was required for orderly mitotic progression of CRC cells with CIN, but had no detectable effect on MSI cells at isotoxic doses.
The role of PKC βII may not be restricted to CRC. Expression and organization of pericentrin is often perturbed in prostate cancer, not only for the tumor cells but also for the associated endothelial cells and is characterized by an abnormal organization of pericentrin into elongated filaments rather than as focalized dots. Treatment of prostate cancer xenografts with a small peptide that disrupted the binding of PKC βII to its membrane-associated RACK1 receptor, thereby markedly enhancing the levels of pericentrin-associated PKC βII, was accompanied by normalization of pericentrin structure and function in the tumor cells as well as in the surrounding tumor-associated endothelial cells.Citation42
Cell death in enzastaurin-treated CIN cells was preceded by prolonged metaphase arrest coherent with the presence of activated BubR1 and Chk1. There is conflicting data concerning the influence of the spindle assembly checkpoint on mitotic catastrophe, with some reports suggesting that prolonged metaphase arrest promotes cell deathCitation43,44 while other results suggest that it might be cytoprotective.Citation45 It is possible, that the influence of the spindle assembly checkpoint on cellular survival may depend on the type of mitotic abnormalities that triggered it. Interestingly, prolonged metaphase arrest seems to be essential for induction of mitotic catastrophe and cell death linked to centrosomal dysfunction after targeted depletion of centrosomal proteins.Citation44 One possibility why prolonged mitotic arrest would be accompanied by cell death is that condensed chromatin is associated with transcriptional inhibition thereby preventing the replacement of the short-lived mRNAs that encode survival proteins.Citation46
Prolonged metaphase arrest might also influence signaling pathways needed for cellular survival during mitosis such as the phosphatidylinositol 3-kinase (PI3K)-Akt pathway.Citation47,48 As recently shown for the VEGFR pathway, that promotes CRC cell survival under hypoxia, different cell lines have different signaling intensity.Citation49 As a result, some cells may be able to maintain strong survival signaling throughout even a prolonged metaphase arrest, whereas other cells would only be able to activate the pathway temporarily.
The findings in this report highlight some of the complexities of modern anticancer therapy. The activity of anticancer agents is based on their capacity to preferentially kill tumor cells rather than their normal counterparts, which is usually denoted as the therapeutic index. Although it was originally believed that the therapeutic index was principally a question of differences in proliferation rates, increasing evidence suggest that the most crucial component is the molecular and functional differences between normal and malignant cells. Thus, the use of classical DNA-targeted agents is dependent on the almost universal loss of cell cycle checkpoints and DNA repair proteins in malignant cells Citation50 as well as cancer-associated changes in chromatin organization or, alternatively, on the cancer-associated upregulation of target enzymes such as topoisomerase I and II.Citation51 Exposure to treatment with DNA damaging agents is typically accompanied by up-regulation of survival signaling, like the EGFR pathwayCitation52 that would need to be attenuated by molecular targeted agents in parallel.Citation53 For targeted agents, the therapeutic response may be explained by oncogene addiction Citation54 as for c-Kit and ErbB2/HER2 as well as the capacity of many oncogenic signaling pathways to promote tumor cell survival within the hostile tumor environment.
However, neoplastic cells may also possess specific features that can limit the efficiency of anticancer agents such as karyotypic complexity and chromosomal instabilityCitation55 or expression of mutidrug transportersCitation56 which would required either selective agents as being the case for enzastaurin in the current study or a combination of restrictive combinations designed to narrow the variety of targeted cells.Citation56,57
Recent results have enlarged the number of mechanisms that may contribute to tumor cell resistance including expression of specific microRNAsCitation58 or the secretion of exosomes with altered cargoCitation59 that influence cellular communication. Communication between different cell types in the tumor microenvironment may also influence tumor cell survival. A particularly interesting example is adipose tissue in obese patients, that can modify tumor cell behavior directly or indirectly, in part via activation of mTOR signaling.Citation60
Taken together, we here show that enzastaurin, a selective PKC βII inhibitor shows preferential cytotoxicity toward CRC cells with CIN compared to cells with MSI. Specifically, enzastaurin exposure was accompanied by prolonged metaphase arrest in CIN cells followed by the appearance of tetraploid and micronuclei-containing cells as well as by increased cell death. In comparison, no detectable mitotic dysfunctions were observed in MSI cells exposed to isotoxic doses of enzastaurin. These data provide novel insight into the mechanism of action of enzastaurin and may allow the identification of biomarkers useful to identify CRC patients particularly likely, or not, to benefit from treatment with enzastaurin.
Materials and Methods
Reagents and antibodies
Enzastaurin (LY317615.HCl) was obtained from Eli Lilly and Co (Indianapolis, IN) and was prepared as a 10 mM stock solution in DMSO. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide), propidium iodide, 4′,6-diamidino-2-phenylindole (DAPI), RNase A, cytochalasin B, mitomycin C and nocodazole were purchased from Sigma. The antibodies used were mouse anti-phospho-Ser10 Histone H3 (Cell Signaling, Clone 6G3, #9706), mouse anti-phospho-Ser/Thr-Pro-MPM-2 (Upstate, #05–368), goat anti-lamin B (Santa-Cruz Biotechnology, sc-6216), mouse anti-γ-tubulin (Sigma clone GTU-88, T6557), rabbit anti-α/β-tubulin (Cell Signaling, #2148), mouse anti-BubR1 (BD Bioscience, #612502), mouse anti-Chk1 (Cell Signaling, #2360) and mouse anti-β-actin (Sigma, clone AC-15, #A5441). Horseradish peroxidase-conjugated anti-mouse, CyTM5-conjugated anti-mouse, CyTM5-conjugated anti-rabbit, CyTM3-conjugated anti-goat and FITC-conjugated anti-mouse antibodies were from Jackson ImmunoResearch.
Cell culture
LS413, SW1116, FET, LS174T, SW48, HCT-15, DLD-1 and LoVo cells were kindly provided by Richard Hamelin (Paris, France). SW-480 colon carcinoma cells were purchased from American Type Culture Collection (Rockville, MD). HT-29 and SW620 cells were kindly provided by Richard Camalier, (National Cancer Institute, Bethesda, MD) while HCT-116 cells were a kind gift from Bert Vogelstein (Baltimore, MD),
The cells were maintained in, McCoy's A (HCT-116), DMEM (HT-29, LS513, SW1116, FET, LS174T, SW48, LoVo, SW480) or RPMI (HCT-15, DLD-1, SW620) supplemented with 5% fetal calf serum and 1% penicillin/streptomycin (PAA).
Cytotoxicity assay
The growth inhibitory effects of enzastaurin was determined by the MTT viability test as previously described Citation61 with minor modifications. Cells (5,000 to 2,0000 per well) were seeded in 24-well plates in culture media containing 1% FCS, incubated in drug-free media for 24 hours followed by enzastaurin exposure for 120 hours. Cellular viability was determined by exposing cells to the MTT tetrazolium salt for 4 hours at 37°C, and the formation of formazan was measured at 560 nm by a microplate reader. The IC50 value is defined as the drug concentration causing 50% reduction of viable cells compared to the untreated control cells. All values are averages of at least 3 independent experiments each done in duplicate.
Cell cycle analysis and flow cytometry
Cell-cycle analysis was carried out as described previouslyCitation62 with minor modifications. The percentage of mitotic cells was determined using antibodies directed against phospho-histone H3 or MPM-2 as follows. Cells were fixed in 70% ethanol at −20°C, rehydrated in PBS and incubated with primary antibody (1:100 overnight at 4°C). After 2 washes with PBS, cells were incubated with the appropriate CyTM5-conjugated antibody (1:100 for 1 hour at room temperature). Cells were washed twice with PBS and stained in PBS containing RNase A and propidium iodide (100 and 20 μg/ml, respectively, for 30 min at 37°C), followed by flow cytometry analysis.
Fluorescent immunocytochemistry analysis
Cells were grown on coverslips in the absence or presence of enzastaurin for the indicated times. After fixation and permeabilization, cells were incubated with the indicated antibodies overnight at 4°C (anti-phospho-H3 at 1:200, anti-lamin B at 1:100, anti-γ-tubulin at 1:1000 and anti-α/β-tubulin antibodies at 1:50 dilution). After washing, cells were exposed to the appropriate fluorescence-coupled secondary antibody (1:100 dilution), counterstained with DAPI and mounted with vectashield (Vector) followed by confocal microscopy (Leica).
Western blot analysis
Western blot analysis was carried out as described previouslyCitation63 with minor modifications. Proteins (50000 cells per well) were resolved on SDS-PAGE gels (5–20% acrylamide) in a denaturing buffer followed by transfer to nitrocellulose membranes. Membranes were incubated with anti-BubR1 (1:500) or anti-Chk1 antibodies (1:500) followed by incubation with the appropriate secondary horseradish peroxydase-conjugated antibodies (1:20000 dilution). Protein expression was revealed with enhanced chemiluminescence reagents (ECL Amersham).
Micronucleus test
The micronucleus test was performed as previously describedCitation64,65 with minor modifications. HT-29 and DLD-1 cells were plated in 6-well plates at 30,0000 cells/well and incubated in drug-free media for 24 hours followed by enzastaurin exposure for 18 hours. Cells were collected and incubated with cytochalasin B (3 μg/ml) for an additional 36 hours. Then, cells were fixed, added to glass slides and stained with Diff-quick staining kit (Dabe-Behring) according to the manufacturer's protocol. Positive (Mitomycin C, 3 μg/ml) and negative (culture media) controls were included in all experiments. Micronuclei (MN) formation was determined from 1000 binucleated cells in at least 2 independent experiments.
Statistical Analysis
The statistical analysis of experimental data was performed using a Student's paired t-test, and results are presented as mean ± SD.
Disclosure of Potential Conflicts of Interest
This work was financed in part by research funding to Annette K Larsen from Eli Lilly. The sponsors had no role in the study design, data collection and analysis, interpretation of the results, the preparation of the manuscript, or the decision to submit the manuscript for publication.
Supplemental File
Download Zip (1.5 MB)Acknowledgments
We thank Alexandre Escargueil and Paul Mésange for careful reading of the manuscript. We like to acknowledge the contribution of Philippe Fontanges and Romain Morichon from the confocal microscopy platform of IFR65 (Institut Fédératif de Recherche 65) and Anne-Marie Faussat from the flow cytometry platform of IFR65.
References
- Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem 1982; 257:7847-51; PMID: 7085651
- Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov 2012; 11:937-57; PMID: 23197040; http://dx.doi.org/10.1038/nrd3871
- Reyland ME. Protein kinase C isoforms: Multi-functional regulators of cell life and death. Front Biosci 2009; 14:2386-99; http://dx.doi.org/10.2741/3385
- Takahashi T, Ueno H, Shibuya M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 1999; 18:2221-30; PMID: 10327068; http://dx.doi.org/10.1038/sj.onc.1202527
- Suzuma K, Takahara N, Suzuma I, Isshiki K, Ueki K, Leitges M, Aiello LP, King GL. Characterization of protein kinase C beta isoform's action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci U S A 2002; 99:721-6; PMID: 11805327; http://dx.doi.org/10.1073/pnas.022644499
- Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, Mori F, Ciulla TA, Ways K, Jirousek M, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes 1997; 46:1473-80; http://dx.doi.org/10.2337/diab.46.9.1473
- Yoshiji H, Kuriyama S, Ways DK, Yoshii J, Miyamoto Y, Kawata M, Ikenaka Y, Tsujinoue H, Nakatani T, Shibuya M, et al. Protein kinase C lies on the signaling pathway for vascular endothelial growth factor-mediated tumor development and angiogenesis. Cancer Res 1999; 59:4413-8; PMID: 10485491
- Teicher BA, Alvarez E, Menon K, Esterman MA, Considine E, Shih C, Faul MM. Antiangiogenic effects of a protein kinase Cbeta-selective small molecule. Cancer Chemother Pharmacol 2002; 49:69-77; PMID: 11855754; http://dx.doi.org/10.1007/s00280-001-0386-2
- Lanz TV, Becker S, Osswald M, Bittner S, Schuhmann MK, Opitz CA, Gaikwad S, Wiestler B, Litzenburger UM, Sahm F, et al. Protein kinase Cβ as a therapeutic target stabilizing blood-brain barrier disruption in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2013; 110:14735-40; PMID: 23959874; http://dx.doi.org/10.1073/pnas.1302569110
- Murray NR, Davidson LA, Chapkin RS, Clay Gustafson W, Schattenberg DG, Fields AP. Overexpression of protein kinase C betaII induces colonic hyperproliferation and increased sensitivity to colon carcinogenesis. J Cell Biol 1999; 145:699-711; PMID: 10330400; http://dx.doi.org/10.1083/jcb.145.4.699
- Murray NR, Weems J, Braun U, Leitges M, Fields AP. Protein kinase C betaII and PKCiota/lambda: collaborating partners in colon cancer promotion and progression. Cancer Res 2009; 69:656-62; PMID: 19147581; http://dx.doi.org/10.1158/0008-5472.CAN-08-3001
- Sauma S, Yan Z, Ohno S, Friedman E. Protein kinase C beta 1 and protein kinase C beta 2 activate p57 mitogen-activated protein kinase and block differentiation in colon carcinoma cells. Cell Growth Differ 1996; 7:587-94.
- Faul MM, Gillig JR, Jirousek MR, Ballas LM, Schotten T, Kahl A, Mohr M. Acyclic N-(azacycloalkyl)bisindolylmaleimides: isozyme selective inhibitors of PKCbeta. Bioorg Med Chem Lett 2003; 13:1857-9; PMID: 12749884 http://dx.doi.org/10.1016/S0960-894X(03)00286-5
- Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, Banks C, Capen A, Goode R, Lewis JE, et al. The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HCl), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses growth of human colon cancer and glioblastoma xenografts. Cancer Res 2005; 65:7462-9; PMID: 16103100; http://dx.doi.org/10.1158/0008-5472.CAN-05-0071
- Dumstorf CA, Konicek BW, McNulty AM, Parsons SH, Furic L, Sonenberg N, Graff JR. Modulation of 4E-BP1 function as a critical determinant of enzastaurin-induced apoptosis. Mol Cancer Ther 2010; 9:3158-63; PMID: 20971826; http://dx.doi.org/10.1158/1535-7163.MCT-10-0413
- Glimelius B, Lahn M, Gawande S, Cleverly A, Darstein C, Musib L, Liu Y, Spindler KL, Frödin JE, Berglund A, et al. A window of opportunity phase II study of enzastaurin in chemonaive pat-ients with asymptomatic metastatic colorectal cancer. Ann Oncol 2010; 21:1020-6; PMID: 19901015; http://dx.doi.org/10.1093/annonc/mdp521
- Wolff RA, Fuchs M, Di Bartolomeo M, Hossain AM, Stoffregen C, Nicol S, Heinemann V. A double-blind, randomized, placebo-controlled, phase 2 study of maintenance enzastaurin with 5-fluorouracil/leucovorin plus bevacizumab after first-line therapy for metastatic colorectal cancer. Cancer 2012; 118:4132-8; PMID: 22213153; http://dx.doi.org/10.1002/cncr.26692
- Carducci MA, Musib L, Kies MS, Pili R, Truong M, Brahmer JR, Cole P, Sullivan R, Riddle J, Schmidt J, et al. Phase I dose escalation and pharmacokinetic study of enzastaurin, an oral protein kinase C beta inhibitor, in patients with advanced cancer. J Clin Oncol 2006; 24:4092-9; PMID: 16943527; http://dx.doi.org/10.1200/JCO.2005.05.3447
- Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet 2006; 38:1043-8; PMID: 16921376; http://dx.doi.org/10.1038/ng1861
- Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut 2008; 57:941-50; PMID: 18364437; http://dx.doi.org/10.1136/gut.2007.135004
- Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, Domany E. Association of survival and disease progression with chromosomal instability: a genomic exploration of colorectal cancer. Proc Natl Acad Sci U S A 2009; 106:7131-6
- Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res 2011; 71:1858-70; PMID: 21363922; http://dx.doi.org/10.1158/0008-5472.CAN-10-3604
- Fallik D, Borrini F, Boige V, Viguier J, Jacob S, Miquel C, Sabourin JC, Ducreux M, Praz F. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res 2003; 63:5738-44; PMID: 14522894
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7; PMID: 22810696; http://dx.doi.org/10.1038/nature11252
- Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 1999; 97:99-109; PMID: 10199406; http://dx.doi.org/10.1016/S0092-8674(00)80718-7
- Hans F, Dimitrov S. Histone H3 phosphorylation and cell division. Oncogene 2001; 20:3021-7; PMID: 11420717; http://dx.doi.org/10.1038/sj.onc.1204326
- Escargueil AE, Larsen AK. Mitosis-specific MPM-2 phosphorylation of DNA topoisomerase IIalpha is regulated directly by protein phosphatase 2A. Biochem J 2007; 403:235-42; PMID: 17212588; http://dx.doi.org/10.1042/BJ20061460
- Li W, Lan Z, Wu H, Wu S, Meadows J, Chen J, Zhu V, Dai W. BUBR1 phosphorylation is regulated during mitotic checkpoint activation. Cell Growth Differ 1999; 10:769-75; PMID: 10593653
- Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP-E functions at kinetochores and binds the cyclosome/APC. J Cell Biol 1999; 146:941-54; PMID: 10477750; http://dx.doi.org/10.1083/jcb.146.5.941
- Enomoto M, Goto H, Tomono Y, Kasahara K, Tsujimura K, Kiyono T, Inagaki M. Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. J Biol Chem 2009; 284:34223-30; PMID: 19837665; http://dx.doi.org/10.1074/jbc.C109.051540
- Ghadimi BM, Sackett DL, Difilippantonio MJ, Schröck E, Neumann T, Jauho A, Auer G, Ried T. Centrosome amplification and instability occurs exclusively in aneuploid, but not in diploid colorectal cancer cell lines, and correlates with numerical chromosomal aberrations. Genes Chromosomes Cancer 2000; 27:183-90; PMID: 10612807; http://dx.doi.org/10.1002/(SICI)1098-2264(200002)27:2%3c183::AID-GCC10%3e3.0.CO;2-P
- Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 2011; 12:385-92; PMID: 21527953; http://dx.doi.org/10.1038/nrm3115
- Colombo R, Moll J. Targeting aneuploid cancer cells. Expert Opin Ther Targets 2011; 15:595-608; PMID: 21314491
- Silverman-Gavrila R, Silverman-Gavrila L, Bendeck MP. Cell division fidelity is altered during the vascular response to injury: its novel role in atherosclerosis progression. Am J Pathol 2013; 182:628-39; PMID: 23260773; http://dx.doi.org/10.1016/j.ajpath.2012.11.007
- Akino T, Hida K, Hida Y, Tsuchiya K, Freedman D, Muraki C, Ohga N, Matsuda K, Akiyama K, Harabayashi T, et al. Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am J Pathol 2009; 175:2657-67; PMID: 19875502; http://dx.doi.org/10.2353/ajpath.2009.090202
- Ohga N, Ishikawa S, Maishi N, Akiyama K, Hida Y, Kawamoto T, Sadamoto Y, Osawa T, Yamamoto K, Kondoh M, et al. Heterogeneity of tumor endothelial cells: comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am J Pathol 2012; 180:1294-307; PMID: 22245217; http://dx.doi.org/10.1016/j.ajpath.2011.11.035
- Swanton C, Nicke B, Schuett M, Eklund AC, Ng C, Li Q, Hardcastle T, Lee A, Roy R, East P, et al. Chromosomal instability determines taxane response. Proc Natl Acad Sci U S A 2009; 106:8671-6
- Bracht K, Nicholls AM, Liu Y, Bodmer WF. 5-Fluorouracil response in a large panel of colorectal cancer cell lines is associated with mismatch repair deficiency. Br J Cancer 2010; 103:340-6; PMID: 20606684; http://dx.doi.org/10.1038/sj.bjc.6605780
- Sinicrope FA, Foster NR, Thibodeau SN, Marsoni S, Monges G, Labianca R, Kim GP, Yothers G, Allegra C, Moore MJ, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst 2011; 103:863-75; PMID: 21597022; http://dx.doi.org/10.1093/jnci/djr153
- Zimmerman WC, Sillibourne J, Rosa J, Doxsey SJ. Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and -mitotic entry. Mol Biol Cell 2004; 15:3642-57; PMID: 15146056; http://dx.doi.org/10.1091/mbc.E03-11-0796
- Chen D, Purohit A, Halilovic E, Doxsey SJ, Newton AC. Centrosomal anchoring of protein kinase C βII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J Biol Chem 2004; 279:4829-39; PMID: 14594954; http://dx.doi.org/10.1074/jbc.M311196200
- Kim J, Choi YL, Vallentin A, Hunrichs BS, Hellerstein MK, Peehl DM, Mochly-Rosen D. Centrosomal PKCbetaII and pericentrin are critical for human prostate cancer growth and angiogenesis. Cancer Res 2008; 68:6831-9; PMID: 18701509; http://dx.doi.org/10.1158/0008-5472.CAN-07-6195
- Bekier ME, Fischbach R, Lee J, Taylor WR. Length of mitotic arrest induced by microtubule-stabilizing drugs determines cell death after mitotic exit. Mol Cancer Ther 2009; 8:1646-54; PMID: 19509263; http://dx.doi.org/10.1158/1535-7163.MCT-08-1084
- Kimura M, Yoshioka T, Saio M, Banno Y, Nagaoka H, Okano Y. Mitotic catastrophe and cell death induced by depletion of centrosomal proteins. Cell Death Dis 2013; 4:e603; PMID: AMBIGUOUS
- Giovinazzi S, Bellapu D, Morozov VM, Ishov AM. Targeting mitotic exit with hyperthermia or APC/C inhibition to increase paclitaxel efficacy. Cell Cycle 2013; 12:2598-607; PMID: 23907120; http://dx.doi.org/10.4161/cc.25591
- Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007; 6:70-4; PMID: 17245109; http://dx.doi.org/10.4161/cc.6.1.3682
- Hemström TH, Sandström M, Zhivotovsky B. Inhibitors of the PI3-kinase/Akt pathway induce mitotic catastrophe in non-small cell lung cancer cells. Int J Cancer 2006; 119:1028-38; PMID: 16570272; http://dx.doi.org/10.1002/ijc.21927
- Skladanowski A, Bozko P, Sabisz M, Larsen AK. Dual inhibition of PI3K/Akt signaling and the DNA damage checkpoint in p53-deficient cells with strong survival signaling: implications for cancer therapy. Cell Cycle 2007; 6:2268-75; PMID: 17890906; http://dx.doi.org/10.4161/cc.6.18.4705
- Mésange P, Poindessous V, Sabbah M, Escargueil AE, de Gramont A, Larsen AK. Intrinsic bevacizumab resistance is associated with prolonged activation of autocrine VEGF signaling and hypoxia tolerance in colorectal cancer cells and can be overcome by nintedanib, a small molecule angiokinase inhibitor. Oncotarget 2014; 5:4709-21; PMID: 25015210
- Bartek J, Lukas J, Bartkova J. DNA damage response as an anti-cancer barrier: damage threshold and the concept of 'conditional haploinsufficiency'. Cell Cycle 2007; 6:2344-7; PMID: 17700066; http://dx.doi.org/10.4161/cc.6.19.4754
- Larsen AK, Escargueil AE, Skladanowski A. From DNA damage to G2 arrest: the many roles of topoisomerase II. Prog Cell Cycle Res 2003; 5:295-300; PMID: 14593724
- Larsen AK, Ouaret D, El Ouadrani K, Petitprez A. Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther 2011; 131:80-90; PMID: 21439312; http://dx.doi.org/10.1016/j.pharmthera.2011.03.012
- Petitprez A, Larsen AK. Irinotecan resistance is accompanied by upregulation of EGFR and Src signaling in human cancer models. Curr Pharm Des 2013; 19:958-64; PMID: 22973964; http://dx.doi.org/10.2174/138161213804547204
- Blagosklonny MV. NCI's provocative questions on cancer: some answers to ignite discussion. Oncotarget 2011; 2:1352-67; PMID: 22267462
- Roschke AV, Kirsch IR. Targeting cancer cells by exploiting karyotypic complexity and chromosomal instability. Cell Cycle 2005; 4:679-82; PMID: 15846096; http://dx.doi.org/10.4161/cc.4.5.1687
- Blagosklonny MV. Targeting cancer cells by exploiting their resistance. Trends Mol Med 2003; 9:307-12; PMID: 12900218; http://dx.doi.org/10.1016/S1471-4914(03)00111-4
- Blagosklonny MV. "Targeting the absence" and therapeutic engineering for cancer therapy. Cell Cycle 2008; 7:1307-12; PMID: 18487952; http://dx.doi.org/10.4161/cc.7.10.6250
- Li H, Yang BB. MicroRNA in drug resistance. Oncoscience 2014; 1(1):3-4.
- Ragusa M, Statello L, Maugeri M, Barbagallo C, Passanisi R, Alhamdani MS, Li Destri G, Cappellani A, Barbagallo D, Scalia M, et al. Highly skewed distribution of microRNA. Oncoscience 2014; 1(2):132-57.
- Blagosklonny MV. Common drugs and treatments for cancer and age-related diseases: revitalizing answers to NCI's provocative questions. Oncotarget 2012; 3:1711-24; PMID: 23565531
- Poindessous V, Koeppel F, Raymond E, Comisso M, Waters SJ, Larsen AK. Marked activity of irofulven toward human carcinoma cells: comparison with cisplatin and ecteinascidin. Clin Cancer Res 2003; 9:2817-25; PMID: 12855662
- Skladanowski A, Côme MG, Sabisz M, Escargueil AE, Larsen AK. Down-regulation of DNA topoisomerase IIalpha leads to prolonged cell cycle transit in G2 and early M phases and increased survival to microtubule-interacting agents. Mol Pharmacol 2005; 68:625-34; PMID: 15942022
- Escargueil AE, Poindessous V, Soares DG, Sarasin A, Cook PR, Larsen AK. Influence of irofulven, a transcription-coupled repair-specific antitumor agent, on RNA polymerase activity, stability and dynamics in living mammalian cells. J Cel l Sci 2008; 121:1275-83; http://dx.doi.org/10.1242/jcs.023259
- Fenech M. Cytokinesis-block micronucleus cytome assay. Nat Protoc 2007; 2:1084-104; PMID: 17546000; http://dx.doi.org/10.1038/nprot.2007.77
- Hovhannisyan G, Aroutiounian R, Glei M. Butyrate reduces the frequency of micronuclei in human colon carcinoma cells in vitro. Toxicol In Vitro 2009; 23:1028-33; PMID: 19539745; http://dx.doi.org/10.1016/j.tiv.2009.06.011