
Abstract
Upon DNA damage, cell cycle progression is temporally blocked to avoid propagation of mutations. While transformed cells largely maintain the competence to recover from a cell cycle arrest, untransformed cells past the G1/S transition lose mitotic inducers, and thus the ability to resume cell division. This permanent cell cycle exit depends on p21, p53, and APC/CCdh1. However, when and how permanent cell cycle exit occurs remains unclear. Here, we have investigated the cell cycle response to DNA damage in single cells that express Cyclin B1 fused to eYFP at the endogenous locus. We find that upon DNA damage Cyclin B1-eYFP continues to accumulate up to a threshold level, which is reached only in G2 phase. Above this threshold, a p21 and p53-dependent nuclear translocation required for APC/CCdh1-mediated Cyclin B1-eYFP degradation is initiated. Thus, cell cycle exit is decoupled from activation of the DNA damage response in a manner that correlates to Cyclin B1 levels, suggesting that G2 activities directly feed into the decision for cell cycle exit. Once Cyclin B1-eYFP nuclear translocation occurs, checkpoint inhibition can no longer promote mitotic entry or re-expression of mitotic inducers, suggesting that nuclear translocation of Cyclin B1 marks the restriction point for permanent cell cycle exit in G2 phase.
Abbreviations
| APC/C | = | anaphase-promoting complex/cyclosome |
| ATM | = | Ataxia telangiectasia mutated kinase |
| ATR | = | Ataxia telangiectasia and Rad3 related kinase |
| AU | = | arbitrary units |
| Cdk | = | cyclin-dependent kinase |
| Chk1/2 | = | checkpoint kinase 1/2 |
| DDR | = | DNA damage response |
| DNA-PK | = | DNA-dependent protein kinase |
| LMB | = | Leptomycin B |
| Mdm2 | = | mouse double minute 2 homolog |
| MK2 | = | MAPKAP kinase 2 |
| NCS | = | Neocarzinostatin |
| Plk1 | = | polo-like kinase 1 |
| γH2AX | = | histone variant |
| H2AX | = | phosphorylated on serine 139 |
Introduction
Preserving the integrity of the genome and preventing cell division in the presence of DNA damage are critical tasks for cell and organism survival. Consequently, eukaryotic cells possess complex networks for surveillance of DNA integrity, checkpoint activation, and DNA repair. Upon a DNA damage insult the DDR kinases ATM, ATR and DNA-PK are activated. Early on, the downstream activated effector kinases MK2, Chk1 and Chk2 mediate a checkpoint response that converges in inactivation of Cdc25s, which results in persistent inhibitory phosphorylation of Cdks.Citation1-3 In addition, the DDR kinases target p53 and Mdm2, resulting in the activation of a p53-dependent transcriptional program including the upregulation of the Cdk inhibitor p21 and the p53-dependent transcriptional repression of cell cycle-promoting factors.Citation4,5 Together these pathways enforce a checkpoint that allows time for repair and eventual reentry into the cell cycle. However, not all cells recover from a DNA damage arrest, but instead terminally exit the cell cycle.
Terminal cell cycle exit is presumably a mean to prevent propagation of mutations when damage cannot be repaired. In G2 phase it strictly depends on p53- and p21-dependent APC/CCdh1 activation and subsequent degradation of cell cycle proteins.Citation6-11 A key target for APC/CCdh1 is Cyclin B1, which in complex with Cdk1 acts as a master regulator of mitotic entry in an unperturbed cell cycle.Citation12,13 Indeed, we have previously found that upon DNA damage the levels of Cyclin B1 function as a marker for whether G2 cells are still competent to resume cell division.Citation14 Interestingly, in cancer cell lines with compromised p21 function APC/C-dependent degradation pathways are activated, but cells avoid degradation of Cyclin B1 as well as other mitotic proteins and do not terminally exit the cell cycle.Citation9,15
Traditionally, DNA damage checkpoint activation and DNA repair were seen as a molecular cascade directly activated downstream of the recognition of DNA damage. Recently however, a growing number of studies indicate that DNA-damage signaling and checkpoint activation have more integrated roles that rely not only on the type of damage, but also on the region in the genomeCitation16 and the cell cycle phaseCitation17 when the damage occurs. However, the timing and coordination of the checkpoint response and terminal cell cycle exit could not be conclusively addressed, as available studies depend largely on population averages and cell synchronization. How single cells decide between checkpoint recovery and terminal cell cycle exit remains currently unclear.
We have set up a system that allows us to study Cyclin B1 dynamics in single untransformed cells and to assess the resulting cell fate decisions during a DNA damage checkpoint response. We find that terminal cell cycle exit is temporally decoupled from the DNA damage insult in a manner that depends on the cell cycle position. Moreover, we find that nuclear translocation of Cyclin B1 marks a restriction point for terminal cell cycle exit.
Results
Gene-targeted Cyclin B1 as a novel setup to study the DNA-damage checkpoint response
We wanted to set up a system to study permanent cell cycle exit in individual untransformed cells. We first analyzed an ongoing DNA-damage checkpoint response in hTERT-immortalized RPE cells and compared it to transformed U2OS cells. When continuously exposed to low concentration of the topoisomerase II-inhibitor Etoposide, U2OS cells retain expression of G2 specific markers, with the exception of Plk1.Citation15 In contrast, RPE cells lose the expression of Cyclin A2, Cyclin B1, Plk1, Cdk1, and Cdk2, all key regulators of S and G2 phase, indicating a cell cycle exit ().Citation6 We next established a U2OSCitation18 and an RPE (Fig. S1A) cell line encoding a Cyclin B1-eYFP fusion protein at the endogenous CCNB1 locus, which allowed us to directly monitor Cyclin B1 protein dynamics in single live cells. We have previously shown that expression levels of Cyclin B1, a key regulator of mitotic entry, correlate closely with the competence to recover from a DNA damage checkpoint.Citation14 Furthermore, the decrease in Cyclin B1 levels after DNA damage correlates with cellular senescence.Citation19,20
Figure 1. Gene-targeted Cyclin B1 as a novel setup to study checkpoint recovery competence. (A) U2OS or RPE cells were treated with 1 or 5 μM Etoposide for the indicated time periods and subjected to immunoblotting with the indicated antibodies. (C) Untreated control. (B) Representative images of U2OS Cyclin B1-eYFP and RPE Cyclin B1-eYFP cell populations during ongoing Etoposide treatment. Scale bar: 50 μm. (C–F) Equal amounts of U2OS Cyclin B1-eYFP cells (C and E) and RPE Cyclin B1-eYFP cells (D and F) were treated with Etoposide (C and D) or NCS (E and F) at time point 0, and followed by time-lapse microscopy. Average Cyclin B1-eYFP signal was quantified and passage through mitosis was determined. (G) U2OS Cyclin B1-eYFP and RPE Cyclin B1-eYFP cells were treated with 1 μM Etoposide for 5, 10 or 15 h, and subsequently cellular DNA content and eYFP positivity were assessed by flow cytometry.
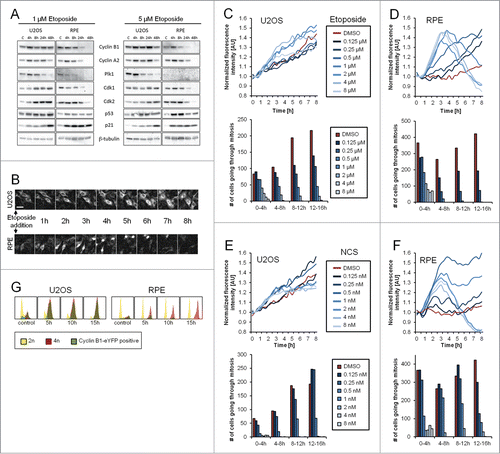
We monitored Cyclin B1-eYFP levels using time-lapse microscopy () and quantified the total fluorescence of multiple positions after careful subtraction of background fluorescence (materials and methods), allowing a time-resolved readout of Cyclin B1-eYFP levels in a population. At the same time, we monitored a checkpoint arrest by scoring whether cells could enter mitosis. We find that Cyclin B1-eYFP levels increase over a range of Etoposide and NCS concentrations in U2OS cells ( and ). In fact, the increase in Cyclin B1-eYFP levels is more pronounced at Etoposide and NCS concentrations that block mitotic entry. In accordance, FACS analysis shows an accumulation of 4n U2OS cells containing high levels of Cyclin B1 (). Thus, U2OS cells block in G2 phase without impairing the ability to retain Cyclin B1.
In contrast, Cyclin B1-eYFP levels start decreasing in RPE cells 3 to 4 hours after addition of Etoposide or NCS at concentrations where there is an apparent cell cycle arrest ( and ). The loss of Cyclin B1 does not depend on checkpoint slippage or an enforced G1/S checkpoint, as a large proportion of the Cyclin B1 eYFP-negative cells contain 4n DNA content (). This shows that there is a correlation between a cell cycle block and the loss of Cyclin B1 in RPE cells.
Cyclin B1 is degraded in a p21-, p53- and APC/CCdh1-dependent manner
It has been reported that Cyclin B1 is actively degraded in an APC/CCdh1-dependent manner in untransformed cells after DNA damage.Citation6,21 While Cyclin B1 and other APC/CCdh1 targets are also regulated at the mRNA level late after DNA damage, timely destruction relies on APC/CCdh1-dependent degradation.Citation6,19 In line, we find that addition of the proteasome inhibitor MG-132 leads to sustained Cyclin B1-eYFP presence in RPE cells, whereas it has no effect on U2OS cells (). Moreover, siRNA-mediated depletion of Cdh1, but not of Cdc20, NIPA, or β-TrCP, stabilized Cyclin B1-eYFP after Etoposide addition in a similar way as the APC/C inhibitor proTAME ( and ). This indicates that in RPE cells APC/CCdh1 targets Cyclin B1-eYFP for degradation after DNA damage.
Figure 2. Degradation of Cyclin B1 during ongoing DNA damage is p53-, p21- and APC/CCdh1-dependent. (A) Time-lapse microscopy quantification of populations of RPE and U2OS Cyclin B1-eYFP cells. Cells were treated with 1 μM Etoposide from time point ‘−1 h’. At 0 h cells were treated with MG-132 (inhibitor of the proteasome) or mock treated. (B–D) Time-lapse microscopy quantifications of RPE Cyclin B1-eYFP cells in different conditions. Cells were transfected with the indicated siRNA 48 h and 24 h prior to Etoposide addition. Etoposide was added at a concentration of 1 μM at the ‘−1 h’ time point. At 0 h cells were treated with MG-132, proTAME, or DMSO as indicated. The error bars represent standard error of the mean signal of at least 8 positions.
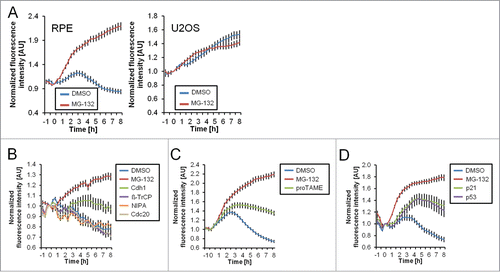
APC/CCdh1 activation during DNA damage was suggested to depend on p53 and p21.Citation6,9,11 Indeed, siRNA-mediated depletion of p53 or p21 led to sustained Cyclin B1-eYFP levels (). Thus, our live-cell setup recapitulates that Cyclin B1-eYFP is degraded in a p53-, p21-, and APC/CCdh1-dependent manner in RPE cells apparent 3 to 4 hours after induction of DNA damage.
Cyclin B1 degradation requires nuclear translocation
Cyclin B1 is predominantly cytoplasmic in interphase, but can translocate to the nucleus upon p21 induction, although the relevance of this translocation remains unclear.Citation7 When we followed the levels of Cyclin B1-eYFP in U2OS and RPE cells we observed obvious differences in the intracellular localization. Whereas Cyclin B1-eYFP stays almost exclusively cytoplasmic in U2OS cells, the majority of RPE cells translocate Cyclin B1-eYFP to the cell nucleus 1 to 2 hours before degradation commences ( and ). In fact, we found that nuclear translocation of Cyclin B1-eYFP in RPE cells appeared to be highly predictive for the initiation of Cyclin B1-eYFP degradation (). Nuclear translocation of Cyclin B1-eYFP is not dependent on active APC/CCdh1, as addition of MG-132, proTAME, or depletion of Cdh1 rather increased the amount of cells with nuclear Cyclin B1-eYFP. However, siRNA-mediated knockdown of p53 or p21 abolished both nuclear translocation and degradation of Cyclin B1-eYFP (). This suggests that although controlled by the same pathways, nuclear translocation of Cyclin B1 is independent of its degradation.
Figure 3. Nuclear translocation is required for Cyclin B1 degradation. (A) Representative images of RPE Cyclin B1-eYFP and U2OS Cyclin B1-eYFP cells during ongoing Etoposide treatment. Scale bar: 20 μm. (B) RPE Cyclin B1-eYFP cells were treated with 1 μM Etoposide and the time between Cyclin B1-eYFP nuclear translocation and onset of degradation was determined for 67 individual cells. The average time ± standard deviation is indicated. (C) RPE Cyclin B1-eYFP cells were treated with 1 μM Etoposide, and Cyclin B1-eYFP nuclear translocation and degradation was assessed for 85 individual cells. (D) RPE Cyclin B1-eYFP cells were transfected with siRNA 48 h and 24 h prior to Etoposide addition, or treated with MG-132 or proTAME as indicated. Intracellular localization of Cyclin B1-eYFP was assessed in single cells at different time points after treatment with 1 μM Etoposide (‘−1 h’ time point). Inhibitors were added at the ‘0 h’ time point. (E) Representative images of U2OS Cyclin B1-eYFP cells treated as indicated. Scale bar: 20 μm. (F) Mitotic entry was assessed for Cyclin B1-eYFP-positive cells treated as in (E). Data are mean and standard deviation of 3 independent experiments. (G) Time-lapse quantification of cells treated as in (E). The error bars represent standard error of the mean signal of at least 8 positions. (H) The time point of nuclear translocation and the onset of degradation of Cyclin B1-eYFP were determined in single U2OS Cyclin B1-eYFP cells. Each line represents a single cell. Inhibitors were added 1 h after Etoposide treatment. (I) Representative images of RPE Cyclin B1-eYFP cells treated with 1 μM Etoposide from time point ‘−1 h’. After 1 h cells were treated as indicated. Scale bar: 20 μm. (J) The time point of nuclear translocation and the onset of degradation of Cyclin B1-eYFP were determined in single RPE Cyclin B1-eYFP cells. Each line represents a single cell. Inhibitors were added 1 h after Etoposide treatment.
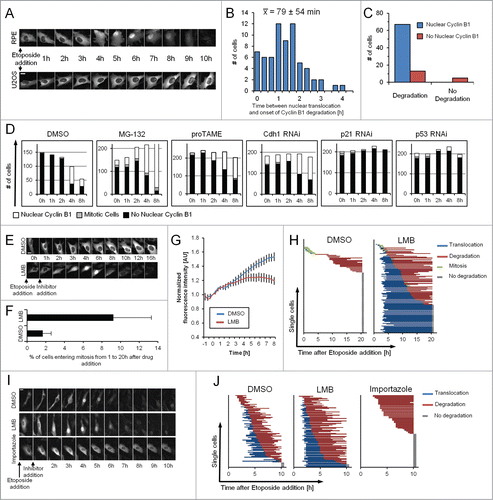
As APC/CCdh1 is predominantly active in the nucleus in G1 phase in an unperturbed cell cycle,Citation22 we reasoned that the observed nuclear translocation of Cyclin B1 in RPE cells during a DDR might be required for APC/CCdh1-mediated ubiquitination. Indeed, we find that addition of the Exportin 1 inhibitor LMB to Etoposide-treated U2OS cells leads to nuclear accumulation of Cyclin B1-eYFP and subsequent degradation in more than half of the cells (, and ). Interestingly, addition of LMB also induced mitotic entry in a subset of cells ( and ). However, in a small fraction of RPE cells Cyclin B1-eYFP degradation occurred without an apparent nuclear translocation (), indicating either that ubiquitination and degradation can also occur in the cytoplasm, or that nuclear translocation is rate-limiting in some cells. We therefore added Importazole, an inhibitor of Importin β to Etoposide-treated cells.Citation23 Addition of Importazole effectively blocked nuclear translocation, indicating that Cyclin B1 translocation during a DDR depends on active transport by Importin β (). Strikingly, when tracking single cells most cells did not show signs of Cyclin B1-eYFP degradation and only one cell lost Cyclin B1-eYFP during the time-course of the experiment ( and ). This indicates that nuclear import is crucial for effective Cyclin B1 degradation after DNA-damage checkpoint activation.
Cyclin B1 nuclear translocation marks the restriction point for terminal cell cycle exit
DNA damage-dependent activation of APC/CCdh1 was shown to promote permanent cell cycle exit in untransformed cells.Citation6-9 Similarly, Kikuchi and colleagues found that the loss of Cyclin B1 after DNA damage is associated with a concurrent increase in cellular senescence.Citation19 In line with these findings, we never observed a single cell that regained expression of Cyclin B1-eYFP once having degraded it. Furthermore, we never detected recurrence of Cyclin B1-eYFP expression or mitosis when following cells for up to 5 d after damage (). Moreover, whereas the G2 population of U2OS cells retains both Cyclin B1-eYFP and checkpoint recovery competence, RPE cells lose Cyclin B1-eYFP over time and do not enter mitosis after combined treatment with the checkpoint kinase inhibitors KU60019 and UCN-01 once Cyclin B1-eYFP is degraded (). Taken together, this shows that RPE cells have lost the competence to resume cell division after APC/CCdh1 activation.
Figure 4. Single RPE cells are not able to recover after Cyclin B1 nuclear translocation. (A) Representative montage of an RPE Cyclin B1-eYFP cell growing on a fibronectin-coated micropattern treated with 1 μM Etoposide for 48 h and followed for an additional 114 h after Etoposide was washed out. Scale bar: 50 μm. (B) RPE Cyclin B1-eYFP cells and U2OS Cyclin B1-eYFP cells were treated for 5, 10 or, 15 h with 1 μM Etoposide or mock treated. Subsequently, damaged cells were forced into mitosis using KU60019 and UCN-01. Nocodazole was added to block cells in mitosis. The percentage of Cyclin B1-eYFP positive cells and the percentage of mitotic cells were determined by flow cytometry. The graphs denote averages of 3 independent experiments. Error bars represent standard errors. (C) Time-lapse microscopy quantifications of RPE Cyclin B1-eYFP cells treated with 1 μM Etoposide at time point 0 and caffeine addition at the indicated time points. The error bars represent standard error of the mean signal of at least 8 positions. (D) The percentage of Cyclin B1-eYFP-positive cells passing through mitosis was assessed from (C). (E) The time point of nuclear translocation and the onset of degradation of Cyclin B1-eYFP were determined in single cells treated as in (C). Each line represents a single cell. The dotted lines indicate presence of caffeine. (F) Representative images of cells in (C) treated with Caffeine. Mitotic cell is marked by asterisk. (G) RPE Cyclin B1-eYFP cells were treated with 1 μM Etoposide and caffeine was added after 4 h. Cyclin B1-eYFP intracellular localization and cell fate was assessed for 96 individual cells.
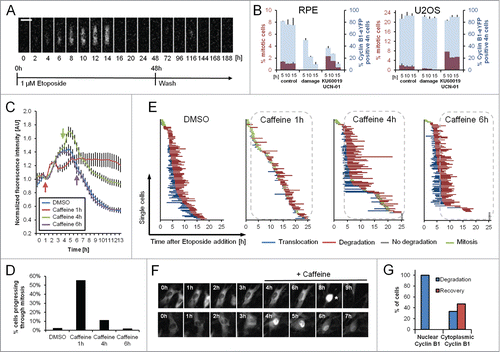
To assess when single cells become determined to permanently exit the cell cycle we monitored live cells and blocked ATM and ATR kinase activity by addition of caffeine at different time points after Etoposide addition. Addition of caffeine shortly after damage stimulated mitotic entry and sustained Cyclin B1-eYFP expression over time. In contrast, caffeine had limited effect on both Cyclin B1-eYFP levels and forced mitotic entry when added 6 hours after Etoposide ( and ). When analyzing single cells we found that addition of caffeine 1 hour after Etoposide had a dramatic effect on both mitotic entry and timing of Cyclin B1-eYFP degradation, showing that ATM/ATR activity initiates these events (). However, when added 4 or 6 hours after Etoposide, at a time when nuclear translocation of Cyclin B1-eYFP had occurred in most cells, forced mitotic entry was only apparent in cells where no signs of nuclear translocation or degradation of Cyclin B1-eYFP were present ( and ). Moreover, we never observed mitotic entry for cells that had already translocated Cyclin B1-eYFP to the nucleus at the time of caffeine addition ().
Thus, although DNA damage-dependent cell cycle exit requires APC/CCdh1 activation, it is only ATM/ATR-dependent until nuclear translocation of Cyclin B1. Therefore, Cyclin B1 nuclear translocation marks the restriction point for losing checkpoint recovery competence and for subsequent APC/CCdh1-dependent permanent cell cycle exit in G2 phase.
APC/CCdh1 activation is temporally decoupled from checkpoint activation
Although the DDR is activated within minutes in all cells, we observed large differences between the times of onset of Cyclin B1-eYFP nuclear translocation and degradation in individual cells, suggesting that Cyclin B1 degradation may depend on the cell cycle phase (). As Cyclin B1-eYFP levels are a direct readout of cell cycle progression, we grew cells on fibronectin-coated micropatterns, which enabled us to accurately quantify fluorescence in single U2OS or RPE cells over long periods of time.Citation18
Figure 5. The time point of Cyclin B1 degradation is decoupled from DDR activation. (A) RPE Cyclin B1-eYFP cells were treated with 1 μM Etoposide and the time point of onset of Cyclin B1-eYFP degradation was assessed. The red bar indicates cells that did not initiate degradation during the time of the experiment. (B) Representative montages of single U2OS Cyclin B1-eYFP and RPE Cyclin B1-eYFP cells on fibronectin-coated micropatterns during an unperturbed cell cycle (control) and during treatment with 1 μM Etoposide (damage). Time between images is 1 h. Scale bar: 50 μm. (C) Single-cell quantifications of time-lapse images as in (B) for U2OS Cyclin B1-eYFP cells and RPE Cyclin B1-eYFP cells under treatment with 1 μM Etoposide from time point ‘0 h’. The green track indicates a cell undergoing mitosis. The dashed orange lines denote the average of the displayed cells. (D) RPE Cyclin B1-eYFP cells were grown on fibronectin-coated micropatterns and treated with 1 μM Etoposide. The Cyclin B1-eYFP levels at the time of Etoposide addition are plotted versus the time point of maximum Cyclin B1-eYFP levels (‘time point of Cyclin B1-eYFP degradation’). (E and H) In silico synchronizations of single-cell quantifications as in (C) in unperturbed conditions (E) or under treatment with 1 μM Etoposide (H). Data are from a single experiment in parallel wells, allowing for direct comparison of fluorescence levels. Cells in (E) are synchronized in silico to mitosis. Graphs (H) show tracks of cells with near-background levels at the time of Etoposide addition. RPE Cyclin B1-eYFP cells were synchronized in silico for to the maximum level of Cyclin B1-eYFP reached. (F) The time point of apparent nuclear translocation of Cyclin B1-eYFP before mitosis was assessed in RPE Cyclin B1-eYFP and U2OS Cyclin B1-eYFP cells. (G) Time-lapse microscopy quantifications of the nuclear to cytoplasm ratio of single RPE Cyclin B1-eYFP and U2OS Cyclin B1-eYFP cells. Cells were treated with 1 μM Etoposide or mock treated with DMSO at time point ‘−1 h’. LMB was added at time point ‘0 h’. Mean and standard error of at least 15 cells per condition are depicted. (I) Quantification of DNA content (DAPI) vs. estimated time. Cells were sorted for DAPI levels and the predominant localization of Cyclin B1 was assessed for each cell.
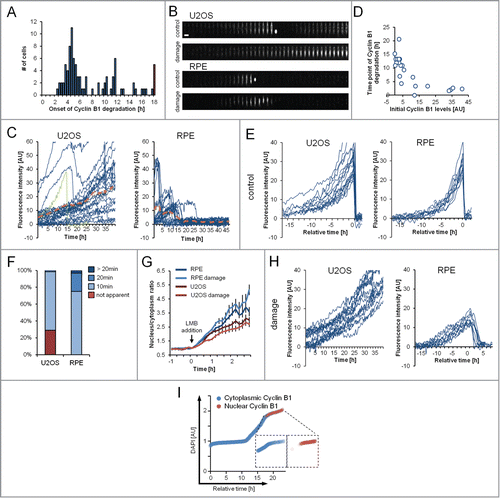
Whereas on Western blot, U2OS cells reach a steady state of Cyclin B1 after DNA damage, Cyclin B1-eYFP levels vary dramatically over time in individual cells ( and ). In fact, a majority of U2OS cells continuously increase Cyclin B1-eYFP to protein levels that are higher compared to what is observed during unperturbed growth. Similar to U2OS cells, no RPE cell sustains a steady-state of Cyclin B1-eYFP over time (). After an initial increase, RPE cells degrade Cyclin B1-eYFP over a 15-hour time-period. Interestingly, the time point of degradation correlates with the Cyclin B1-eYFP level at the time point of the DNA damage insult, that is cells with high Cyclin B1-eYFP level initiate degradation earlier than cells with low Cyclin B1-eYFP level (). As a consequence, Cyclin B1-eYFP levels are prevented from exceeding the levels observed during mitotic entry. Thus, Cyclin B1-eYFP degradation is temporally decoupled from initiation of the DDR in a manner that correlates with the initial levels of Cyclin B1-eYFP, indicating a cell cycle-dependent control of Cyclin B1 degradation.
To allow better comparison between individual cells, we next synchronized cells from a single experiment containing both unperturbed and Etoposide-treated RPE and U2OS cells in silico. When synchronizing unperturbed cells in mitosis, both RPE and U2OS cells showed similar dynamics and similar levels of Cyclin B1-eYFP (). However, in U2OS cells Cyclin B1-eYFP accumulation starts earlier and with larger variability between individual cells (), and we note that nuclear translocation of Cyclin B1-eYFP is less apparent compared to RPE cells both in unperturbed and DNA damage conditions ( and ). We next focused on cells with a low level of Cyclin B1-eYFP at the time of Etoposide addition. U2OS cells required approximately 30 hours before reaching similar Cyclin B1-eYFP level as during unperturbed mitotic entry, suggesting that Cyclin B1 accumulation is slower after initiation of a DDR (). When synchronizing RPE cells with initially low levels of Cyclin B1-eYFP at their maximal level of fluorescence, we note that degradation is initiated at similar Cyclin B1-eYFP levels in all cells (). Thus, indicating that in RPE cells Cyclin B1 is allowed to accumulate up to a distinct threshold before degradation is initiated. As this threshold corresponds to Cyclin B1 levels at the S/G2 transition in unperturbed growth ( and ) we reasoned that Cyclin B1 degradation is initiated when cells exit S phase. To test this hypothesis, we assessed the cell cycle timing of Cyclin B1 nuclear translocation during DNA damage by sorting single RPE cells according to their DNA content and determined Cyclin B1 localization by immunofluorescence. Strikingly, we find that Cyclin B1 nuclear localization occurs only in cells in G2 phase (). Thus, Cyclin B1 accumulation continues after DNA damage and Cyclin B1 nuclear translocation and subsequent degradation is only initiated when Cyclin B1 has reached a distinct threshold level in G2 phase.
Discussion
Terminal cell cycle exit is decoupled from DDR activation
In principle, DNA repair should be initiated as soon as a lesion is recognized, whereas a cell cycle block only needs to be activated late in interphase to prevent mitotic entry. By examining Cyclin B1 dynamics in single RPE cells we show that APC/CCdh1 activation is temporally decoupled from the DNA damage insult. While at a population level Cyclin B1 expression remains stable during the first hours after DNA damage, we find that no single cell maintains steady Cyclin B1 levels over time. Rather Cyclin B1 levels continue to rise and degradation is only initiated once a certain threshold is reached. Cells containing very low levels of Cyclin B1-eYFP initially accumulate it to levels indicative of entering G2 phase. At the same time, only cells with 4n DNA content initiate Cyclin B1 degradation, showing that terminal cell cycle exit occurs in G2 phase, but not in S phase.
Similarly, the higher the Cyclin B1-eYFP content the more rapidly degradation is initiated. Thus, cells late in G2 phase exit the cell cycle faster than cells in early G2 phase, suggesting that G2 activities directly feed into the decision for cell cycle exit. As terminal cell cycle exit can be seen as a last resort of the DDR to avoid mitotic entry and thereby propagation of mutations, incorporation of G2 activities in the decision process suggests an elegant mechanism for timely cell cycle exit only when necessary.
Nuclear translocation of Cyclin B1 marks a restriction point for terminal cell cycle exit
Upon successful repair of damaged DNA, cells should be allowed to resume cell division. However, not all cells will reenter the cell cycle. Several studies show that this permanent cell cycle exit depends on APC/CCdh1 activation.Citation6-9 Indeed, chicken DT40 Cdh1−/− cells fail to maintain a DNA damage-induced G2 arrest.Citation24 However, we find that the actual point-of-no-return occurs already earlier. Once all damaged DNA is repaired, the upstream ATM and ATR kinases lose their activating stimuli, which will lead to their eventual inactivation. We show that simulating repair by blocking ATM/ATR activity can induce recovery only in cells that had not previously translocated Cyclin B1 to the cell nucleus. Thus, after Cyclin B1 nuclear translocation, cell cycle exit is no longer dependent on the presence of DNA damage signaling. As a consequence, depending on its initial cell cycle state, a cell has a set time to repair damaged DNA, before Cyclin B1 nuclear translocation in G2 phase marks the restriction point for terminal cell cycle exit.
Interestingly, both Cyclin B1 nuclear localization and APC/CCdh1 activation have to be tightly connected to prevent checkpoint override. APC/CCdh1 activation and Cyclin B1 degradation were reported to be dependent on p21.Citation6 In contrast, APC/CCdh1 activation appears to be a general response to DNA damage that also occurs in cells that do not terminally exit the cell cycle due to a defective p21 response.Citation9,15 Our data resolves this discrepancy as it suggests that a critical role of p21 is to sequester Cyclin B1 in the nucleus,Citation7 which then, in turn, is required for Cyclin B1 degradation. In fact, transformed cells exclude Cyclin B1 from the nucleusCitation26 and accumulate it to high levels during ongoing DNA damage.Citation27 Under these conditions forced nuclear translocation of Cyclin B1 actually induces checkpoint override or Cyclin B1 degradation.Citation8 Notably, this data highlights an intriguing analogy to the unperturbed cell cycle, where Cyclin B1 nuclear translocation leads to a restriction point for mitotic entry.Citation28-30 Thus, Cyclin B1 nuclear translocation marks a decision point – leading to mitosis or terminal cell cycle exit.
Conclusion
We suggest that cell cycle exit is decoupled from the initiation of the DDR in a manner that depends on a cell's initial cell cycle phase and, thus, correlates to its Cyclin B1 levels. Once a certain threshold level is reached in G2 phase Cyclin B1 nuclear translocation occurs and cell fate becomes determined toward permanent cell cycle exit.
Materials and methods
Cell culture
U2OS cell lines were cultured in Dulbecco's modified eagle medium (DMEM) + GlutaMAX (Invitrogen) supplemented with 6% heat-inactivated fetal bovine serum (FBS, HyClone) and 1% Penicillin/Streptomycin (HyClone). RPE cell lines were cultured in DMEM/Nutrient mixture F-12 (DMEM/F-12) + GlutaMAX (Invitrogen) supplemented with 10% FBS and 1% Penicillin/Streptomycin. HEK293T cells for viral particle production were cultured in DMEM + GlutaMAX (Invitrogen) supplemented with 10% FBS and 1% Penicillin/Streptomycin. All cell lines were cultured in an ambient-controlled incubator at 37°C with 5% CO2. For live-cell imaging experiments, cells were cultured in Leibowitz's L-15 medium (Invitrogen) supplemented with 10% FBS and 1% Penicillin/Streptomycin.
RNAi
For RNA interference experiments SMARTpool ON-TARGET plus siRNAs targeting CDKN1A (p21), TP53 (p53), FRZ1 (Cdh1), CDC20 (Cdc20), BTRC (β-TrCP), ZC3HC1 (NIPA), or CCNB1 (Cyclin B1) were purchased from Dharmacon. Cells were seeded at a density of 45000 cells/ml and transfected with siRNA using HiPerFect (Qiagen) and OptiMEM (Invitrogen) at 48 h and 24 h before analysis of phenotypes. siRNAs were employed at a final concentration of 20 nM.
Inhibitors and plasmids
The inhibitors used in this study were employed at the following concentrations: MG-132 at 10 μM (Sigma Aldrich), proTame at 15 μM (R&D systems), Leptomycin B at 10 ng/ml (Sigma Aldrich), Importazole at 66 μM (Sigma Aldrich), UCN-01 at 300 nM (Sigma Aldrich), KU60019 at 10 μM (Tocris Bioscience), Nocodazole at 250 ng/ml (Sigma Aldrich), S-Trityl-L-cysteine (STLC) at 10 μM (Tocris Bioscience), caffeine at 5 mM (Sigma Aldrich), Etoposide at 0.125 to 8 μM (Sigma Aldrich), and Neocarzinostatin (NCS) at 0.125 to 8 nM (Sigma Aldrich). Based on our titrations, 1 μM Etoposide was sufficient to induce damage to cells at all cell cycle stages and it was the lowest concentration where both U2OS and RPE cells displayed a robust checkpoint arrest (; Fig. S1B). Therefore, we used 1 μM Etoposide in our experiments.
Live-cell microscopy and micropatterns
For live-cell imaging of cell populations, 18000 cells were seeded in 96-well imaging plates (BD Falcon) 16 h before imaging on either a DeltaVision Spectris imaging system (Applied Precision) using a 20×, NA 0.7 objective, or on a ImageXpress system (Molecular Devices) using a 20×, NA 0.45 objective.
For experiments involving micropatterns, cells were detached using PBS containing 0.2% EDTA and seeded on fibronectin-coated micropatterns (CYTOO) at a density of 60000 cells/ml 5 h before starting the experiment. After 30 min cover slips were washed with PBS to remove unattached cells.
Images were processed and analyzed using ImageJ. For time-lapse microscopy quantification of populations the background was subtracted for every image series using rolling ball background subtraction. Furthermore, the image series were manually screened and series with fluorescent aggregates were excluded from the analysis. Subsequently, integrated fluorescent intensities were calculated and values were normalized for each series on the time point before inhibitor addition. Mean values of at least 8 individual positions per condition were calculated.
Immunofluorescence
Cells were grown in 96-well imaging plates (BD Falcon), fixed using 3.7% formaldehyde (Sigma Aldrich) for 5 min at room temperature, permeabilized by ice-cold methanol for 2 min, blocked with 2% bovine serum albumin (Sigma Aldrich) in PBS supplemented with 0.1% Tween-20 (PBST) and incubated with primary antibodies for 2 h at room temperature. After washing with PBST, the cells were incubated with AlexaFluor-conjugated secondary antibodies for 1 h. DNA was stained with DAPI (0.5 μg/ml; Sigma Aldrich). The following antibodies were used: Cyclin B1 V152 (1:400; #4135 Cell Signaling), ɣH2AX (1:400; #9781 Cell Signaling). Images were acquired on an ImageXpress system (Molecular Devices) using a 40× NA 0.6 or a 60× NA 0.7 objective.
Immunoblotting
For Western blot analysis 1.5 × 106 U2OS or RPE cells were cultured in 10-cm dishes. DNA damage was induced using Etoposide (1 μM and 5 μM) and samples were collected 4 h, 8 h, 24 h and 48 h after damage. Cells were washed using PBS and lysed in 500 μl of sample buffer (BioRad). 15 μl of each sample were loaded, run on 4–15% polyacrylamide gels (BioRad) and transferred to a PVDF membrane (BioRad) for immunoblotting. The following antibodies were used: Cyclin B1 GNS-1 (1:500; sc-245 Santa Cruz), Cyclin B1 V152 (1:1000; #4135 Cell Signaling), Cyclin A2 H-432 (1:1000; sc-751 Santa Cruz), Plk1 F-8 (1:500; sc-17783 Santa Cruz), Cdk1 POH1 (1:1000; #9116 Cell Signaling), Cdk2 78B2 (1:1000; #2564 Cell Signaling), p21 12D1 (1:1000; #2947 Cell Signaling), p53 DO-1 (1:500; sc-126 Santa Cruz), β-tubulin 9F3 (1:1000; #2128S Cell Signaling), GAPDH (1:25000; G9545 Sigma Aldrich), GFP (1:1000; ab290 abcam). Secondary antibodies conjugated to horseradish peroxidase (GE Healthcare) were diluted at 1:5000.
Generation of cell lines
The U2OS Cyclin B1-eYFP and RPE Cyclin B1-eYFP cells were generated as previously described.Citation18,31 Briefly, the open reading frame of eYFP lacking the start codon was inserted into the CCNB1 locus by adeno-associated virus-mediated homologous recombination. The targeting construct was designed to contain 959 bp of homology with the CCNB1 locus in the 5′ of the STOP codon and 1253 bp 3′ of the STOP codon. The construct was cloned into a rAAV backbone and the resulting clones were verified by test digest prior to transfection of HEK293T cells to produce viral particles. Transduced and control U2OS and RPE cells were harvested with trypsin/EDTA 96 h after transduction, washed, resuspended in PBS, and filtered through a cell strainer to avoid cell aggregates (BD Falcon). Positive clones were obtained by sorting cells in a FACSAria III (Becton Dickinson) sorter as described previously by Mata and colleagues.Citation32
Flow cytometry
Cells were harvested with trypsin/EDTA, resuspended and fixed with 70% ethanol. After fixation, cells were incubated with an antibody against Histone H3 pS10 (1:200; #3377 Cell Signaling) and a secondary antibody conjugated to AlexaFluor647 (Invitrogen). Finally, nucleic acids were stained using 40 μg/ml propidium iodide. Cells were analyzed on a FACSCanto II (Becton Dickinson).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Files
Download Zip (831.2 KB)Acknowledgments
We thank Camilla Sjögren and Nico Dantuma for critical comments on the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Additional information
Funding
References
- Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J. Rapid destruction of human Cdc25A in response to DNA damage. Science 2000; 288:14259; PMID: 10827953; http://dx.doi.org/10.1126/science.288.5470.1425
- Karlsson-Rosenthal C, Millar JB. Cdc25: mechanisms of checkpoint inhibition and recovery. Trend Cell Biol 2006; 16:285-92; PMID: 16682204; http://dx.doi.org/10.1016/j.tcb.2006.04.002
- Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 2009; 21:245-55; PMID: 19230643; http://dx.doi.org/10.1016/j.ceb.2009.01.018
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20:1803-15; PMID: 11313928; http://dx.doi.org/10.1038/sj.onc.1204252
- St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, Dastidar J, DaCosta A, Mattia M, Manfredi JJ. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Molecular Cell 2004; 16:725-36; PMID: 15574328; http://dx.doi.org/10.1016/j.molcel.2004.11.002
- Wiebusch L, Hagemeier C. p53- and p21-dependent premature APC/C-Cdh1 activation in G2 is part of the long-term response to genotoxic stress. Oncogene 2010; 29:3477-89; PMID: 20383190; http://dx.doi.org/10.1038/onc.2010.99
- Charrier-Savournin FB, Chateau MT, Gire V, Sedivy J, Piette J, Dulic V. p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Mol Biol Cell 2004; 15:3965-76; PMID: 15181148; http://dx.doi.org/10.1091/mbc.E03-12-0871
- Jin P, Hardy S, Morgan DO. Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J Cell Biol 1998; 141:875-85; PMID: 9585407; http://dx.doi.org/10.1083/jcb.141.4.875
- Lossaint G, Besnard E, Fisher D, Piette J, Dulic V. Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional. Oncogene 2011; 30:4261-74; PMID: 21532626; http://dx.doi.org/10.1038/onc.2011.135
- Baus F, Gire V, Fisher D, Piette J, Dulic V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J 2003; 22:3992-4002; PMID: 12881433; http://dx.doi.org/10.1093/emboj/cdg387
- Gillis LD, Leidal AM, Hill R, Lee PW. p21Cip1/WAF1 mediates cyclin B1 degradation in response to DNA damage. Cell Cycle 2009; 8:253-6; PMID: 19158493; http://dx.doi.org/10.4161/cc.8.2.7550
- Stern B, Nurse P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trend Genet: TIG 1996; 12:345-50; PMID: 8855663; http://dx.doi.org/10.1016/0168-9525(96)10036-6
- Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. JCell Biol 2009; 185:193-202; PMID: 19364923; http://dx.doi.org/10.1083/jcb.200812045
- Lindqvist A, de Bruijn M, Macurek L, Bras A, Mensinga A, Bruinsma W, Voets O, Kranenburg O, Medema RH. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J 2009; 28:3196-206; PMID: 19713933; http://dx.doi.org/10.1038/emboj.2009.246
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008; 134:256-67; PMID: 18662541; http://dx.doi.org/10.1016/j.cell.2008.05.043
- Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 2011; 30:1079-92; PMID: 21317870; http://dx.doi.org/10.1038/emboj.2011.27
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9:297-308; PMID: 18285803; http://dx.doi.org/10.1038/nrm2351
- Akopyan K, Silva Cascales H, Hukasova E, Saurin AT, Mullers E, Jaiswal H, Hollman DA, Kops GJ, Medema RH, Lindqvist A. Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol Cell 2014; PMID: 24582498; http://dx.doi.org/10.1016/j.molcel.2014.01.031
- Kikuchi I, Nakayama Y, Morinaga T, Fukumoto Y, Yamaguchi N. A decrease in cyclin B1 levels leads to polyploidization in DNA damage-induced senescence. Cell Biol Int 2010; 34:645-53; PMID: 20222868; http://dx.doi.org/10.1042/CBI20090398
- Nakayama Y, Yamaguchi N. Role of cyclin B1 levels in DNA damage and DNA damage-induced senescence. Int Rev Cell Mol Biol 2013; 305:303-37; PMID: 23890385; http://dx.doi.org/10.1016/B978-0-12-407695-2.00007-X
- Lee J, Kim JA, Barbier V, Fotedar A, Fotedar R. DNA damage triggers p21WAF1-dependent Emi1 down-regulation that maintains G2 arrest. Mol Biol Cell 2009; 20:1891-902; PMID: 19211842; http://dx.doi.org/10.1091/mbc.E08-08-0818
- Gutierrez GJ, Tsuji T, Chen M, Jiang W, Ronai ZA. Interplay between Cdh1 and JNK activity during the cell cycle. Nat Cell Biol 2010; 12:686-95; PMID: 20581839; http://dx.doi.org/10.1038/ncb2071
- Soderholm JF, Bird SL, Kalab P, Sampathkumar Y, Hasegawa K, Uehara-Bingen M, Weis K, Heald R. Importazole, a small molecule inhibitor of the transport receptor importin-beta. ACS Chem Biol 2011; 6:700-8; PMID: 21469738; http://dx.doi.org/10.1021/cb2000296
- Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, Saya H. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J 2001; 20:6499-508; PMID: 11707420; http://dx.doi.org/10.1093/emboj/20.22.6499
- Kleiblova P, Shaltiel IA, Benada J, Sevcik J, Pechackova S, Pohlreich P, Voest EE, Dundr P, Bartek J, Kleibl Z, et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J Cell Biol 2013; 201:511-21; PMID: 23649806; http://dx.doi.org/10.1083/jcb.201210031
- Smeets MF, Mooren EH, Begg AC. The effect of radiation on G2 blocks, cyclin B expression and cdc2 expression in human squamous carcinoma cell lines with different radiosensitivities. Radiother Oncol: J Eur Soc Ther Rad Oncol 1994; 33:217-27; PMID: 7716262; http://dx.doi.org/10.1016/0167-8140(94)90357-3
- Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. JCell Biol 1996; 134:963-70; PMID: 8769420; http://dx.doi.org/10.1083/jcb.134.4.963
- Gavet O, Pines J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol 2010; 189:247-59; PMID: 20404109; http://dx.doi.org/10.1083/jcb.200909144
- Lindqvist A. Cyclin B-Cdk1 activates its own pump to get into the nucleus. J Cell Biol 2010; 189:197-9; PMID: 20404105; http://dx.doi.org/10.1083/jcb.201003032
- Santos SD, Wollman R, Meyer T, Ferrell JE Jr. Spatial positive feedback at the onset of mitosis. Cell 2012; 149:1500-13; PMID: 22726437; http://dx.doi.org/10.1016/j.cell.2012.05.028
- Collin P, Nashchekina O, Walker R, Pines J. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol 2013; 15:1378-85; PMID: 24096242; http://dx.doi.org/10.1038/ncb2855
- Mata JF, Lopes T, Gardner R, Jansen LE. A rapid FACS-based strategy to isolate human gene knockin and knockout clones. PloS One 2012; 7:e32646; PMID: 22393430; http://dx.doi.org/10.1371/journal.pone.0032646