
Abstract
ATM activation following DNA damage is a critical event which is required for efficient DNA repair and cell survival, yet signaling mechanisms controlling its activation are incompletely understood. The RhoGEF Net1 has previously been reported to control Rho GTPase activation and downstream cell survival outcomes following double strand DNA damage. However the role of Net1 isoforms in controlling ATM-dependent cell signaling has not been assessed. In the present work we show that expression of the Net1A isoform is specifically required for efficient activation of ATM but not the related kinase DNA-PK after ionizing radiation. Surprisingly Net1A overexpression also potently suppresses ATM activation and phosphorylation of its substrate H2AX. This effect does not require catalytic activity towards RhoA or RhoB, and neither Rho GTPase affects ATM activation, on its own. Consistent with a role in controlling ATM activation, Net1A knockdown also impairs DNA repair and cell survival. Taken together these data indicate that Net1A plays a plays a previously unrecognized, Rho GTPase-independent role in controlling ATM activity and downstream signaling after DNA damage to impact cell survival.
Abbreviations
| Net1 | = | neuroepithelial transforming gene 1 |
| RhoGEF | = | Rho guanine nucleotide exchange factor |
| ATM | = | ataxia-telangiectasia mutated |
| DNA-PK | = | DNA dependent protein kinase |
| DSB | = | double strand break |
| IR | = | ionizing radiation |
Introduction
Maintenance of genomic integrity is fundamental to cell survival. Thus organisms have evolved many mechanisms to recognize and repair damaged DNA. A particularly deleterious form of DNA damage are double strand breaks (DSBs), which trigger signaling cascades that establish cell cycle arrest and allow time for DNA repair.Citation1-3 Ataxia-telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK) are related kinases that phosphorylate distinct but overlapping sets of substrates and whose activation is essential for cells to respond to DNA DSBs.Citation4 Although they display significant homology in their kinase domains, ATM and DNA-PK are structurally unique and have distinct activation mechanisms. In the absence of DNA DSBs, ATM exists as an inactive dimer. However, following DNA damage, ATM is recruited to DNA breaks by the MRN complex, where it undergoes autophosphorylation at S1981 to stimulate its kinase activity.Citation5,6 DNA-PK is recruited to DNA DSBs by binding to its accessory proteins Ku70 and Ku80, which leads to its activation by autophosphorylation on S2056 and the T2609 cluster.Citation7-9 Although these mechanisms constitute the essential basis for activation of ATM and DNA-PK, it is clear that additional mechanisms modulate the activation state and downstream signaling capabilities of both kinases.Citation3,4,10
The neuroepithelial transforming gene 1 (Net1) is a Rho guanine nucleotide exchange factor that was originally identified as a transforming gene in NIH3T3 cells.Citation11 Two isoforms of Net1 exist in most cells, Net1 and Net1A, which differ in their N-terminal regulatory domains. These different regulatory domains confer distinct functions on Net1 proteins. For example, the longer Net1 isoform uniquely regulates mitotic progression while the shorter Net1A isoform contributes to cell adhesion, motility and extracellular matrix invasion.Citation12-15 Growing evidence indicates that Net1 proteins also control cell survival following the induction of DNA DSBs.Citation16-18 This function of Net1 isoforms is not well defined, as Net1 has been shown to control both cell survival and pro-apoptotic signaling pathways. Moreover, the signaling mechanisms by which Net1 proteins impact DNA damage responses are not well understood.
To better understand the role of Net1 in DNA damage responses, we investigated whether Net1 proteins control signaling pathways commonly activated by DNA DSBs. We observed that Net1 isoforms regulate the efficiency of ATM activation following ionizing radiation (IR)-induced DNA DSBs. We found that ATM activation was dependent on the Net1A isoform, and that ectopic expression of Net1A potently suppressed ATM activation and H2AX phosphorylation. The catalytic activity of Net1A towards its substrates RhoA and RhoB was not necessary for repression of H2AX phosphorylation by overexpressed Net1A. Importantly, Net1A expression was required for efficient DNA repair and cell survival. These results indicate that Net1A plays a Rho GTPase-independent role in modulating ATM-dependent signaling to control cell survival following DNA damage.
Results
Depletion of Net1 proteins attenuates ATM-dependent damage signaling after IR
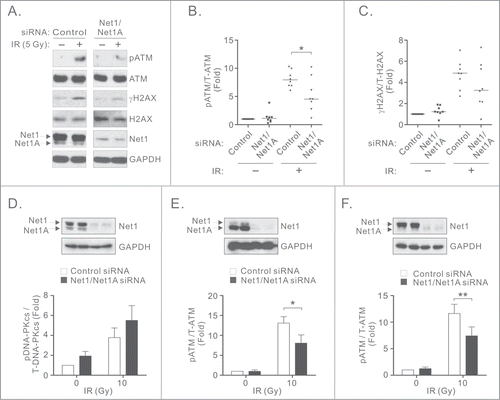
To determine whether Net1 controls signaling pathways activated immediately after induction of DNA DSBs, we tested whether knockdown of both Net1 isoforms in breast cancer cells affected the activation of ATM or DNA-PK. MCF7 cells were transfected with control siRNA or an siRNA targeting both Net1 isoforms, treated with IR, and tested for phosphorylation of ATM and its substrate H2AX (γH2AX). In control siRNA transfected cells we observed robust ATM phosphorylation on its activating site S1981, as well as phosphorylation of its downstream target H2AX 30 minutes after IR. However, knockdown of both Net1 isoforms significantly attenuated ATM phosphorylation after IR (). We also observed a trend towards reduced phosphorylation of H2AX in these cells (). Because DNA-PK can also phosphorylate H2AX in response to IR, we tested the effect of Net1 knockdown on its activation. Importantly, we observed that DNA-PK phosphorylation on its activating site S2056 was not significantly affected by Net1 knockdown, prior to and following IR exposure (). Thus, DNA-PK activation is not compromised by loss of Net1 expression and may compensate for the loss of ATM activity in Net1 knockdown cells. To determine whether other cancer cells require Net1 for efficient ATM activation, U2OS and BT474 cells were transfected with control or Net1 siRNAs and tested for IR-induced ATM activation. Similar to MCF7 cells, loss of Net1 expression compromised ATM activation in both of these cell lines (). These results indicate that Net1 isoforms contribute to IR-induced ATM activation in multiple cancer cell types.
Figure 1. Net1 proteins regulate ATM-mediated DNA damage signaling. (A) Net1 isoform expression is required for ATM activation. MCF7 cells were transfected with the indicated siRNAs and treated with 5 Gy IR. Thirty minutes after recovery the cells were lysed and tested by Western blotting using indicated antibodies. (B, C) Quantification of ATM (B) and H2AX (C) phosphorylation. Data shown are from 8 independent experiments. Bars are median values. Significance was determined by Student's T-test; * = P < 0.05. (D) MCF7 cells were transfected with control or Net1 targeting siRNAs. After 72 h the cells were irradiated and tested by Western blotting for phosphorylated and total DNA-PKcs. Data shown are the average of 3 independent experiments. Errors indicate standard error of the mean (SEM). (E) BT474 cells transfected with control or Net1 targeting siRNAs were treated with IR and then tested for ATM phosphorylation by Western blotting. Data shown are the average of 3 independent experiments. Errors are SEM; * = P < 0.05. (F) U2OS cells were transfected with control or Net1 targeting siRNAs and tested for phosphorylation of ATM after IR treatment. Data shown are the average of 3 independent experiments. Errors are SEM; ** = P < 0.005.
The Net1A isoform is specifically required for ATM activation
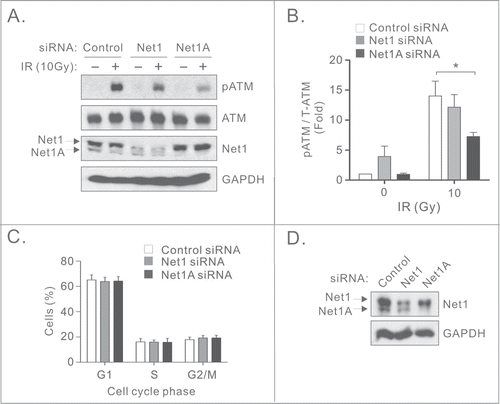
Two isoforms of Net1 exist in most cells, Net1 and Net1A, which are predominantly localized in the nucleus. The Net1 isoform has been shown to control mitotic progression while Net1A mainly regulates RhoA-dependent cell motility.Citation12-14 To test whether specific isoforms of Net1 contributed to ATM activation, MCF7 cells were transfected with control or Net1 isoform specific siRNAs,Citation13 exposed to IR and tested for ATM activation. We observed that knockdown of Net1A, but not Net1, significantly reduced the activation of ATM activation following IR (). Interestingly, knockdown of the Net1 isoform resulted in a slight increase in ATM activation in non-irradiated cells (), which is consistent with DNA damage occurring due to disruption of mitotic progression.Citation14 Because ATM activation is attenuated during mitosis,Citation19,20 we assessed whether knockdown of Net1 isoforms inhibited ATM activation secondary to effects on the cell cycle. However, we observed that loss of expression of individual Net1 isoforms did not affect cell cycle distribution, indicating that the requirement for Net1A in ATM activation is not a secondary consequence of cells accumulating in G2/M ().
Figure 2. The Net1A isoform is specifically required for ATM activation. (A) Knockdown of Net1A, but not Net1, compromises ATM activation. MCF7 cells were transfected with a control siRNA, or siRNAs specific for individual Net1 isoforms. Three days later the cells were treated with IR. After 30 min the cells were harvested for Western blotting using the indicated antibodies. (B) Quantification of ATM activation. Data shown are the average of 3 independent experiments. Errors are SEM. * = P < 0.05. (C) Net1 isoform knockdown does not affect cell cycle distribution. MCF7 cells were transfected with indicated siRNAs. After 3 days the cells were collected, fixed, and analyzed for cell cycle distribution by flow cytometry. Shown is the average of 3 independent experiments. Errors are SEM. (D) Representative Western blot of siRNA transfected cells used for flow cytometry.
Net1A overexpression suppresses ATM and γH2AX foci formation independent of Rho GTPase activation
Since Net1A knockdown blocked efficient ATM activation in MCF7 cells, we investigated whether Net1A overexpression also affected ATM signaling. For these experiments we measured the localization of phosphorylated ATM (pATM) or H2AX (γH2AX) to IR induced nuclear foci (IRIF). Surprisingly, we observed that overexpression of Net1A, but not the control protein β-galactosidase containing a nuclear targeting signal (NLS-β-Gal), dramatically inhibited the formation of pATM IRIF (). This was unlikely to be due to direct interaction of Net1A with ATM, as ATM did not co-immunoprecipitate with transfected Net1A ().
Figure 3. Net1A overexpression suppresses H2AX-IRIF formation independent of Rho GTPase activation. (A) Net1A overexpression suppresses pATM accumulation in irradiation induced nuclear foci (IRIF). MCF7 cells were transfected with Myc-epitope tagged NLS-β-Gal or HA epitope-tagged wild-type Net1A. After 48 h, the cells were treated 10 Gy IR, fixed and stained for pATM. Shown are representative cells from 3 independent experiments. (B) Quantification of pATM in transfected cells. pATM was measured for at least 100 cells/experiment. Data shown are the average of 3 independent experiments. Errors are SEM. *** = P < 0.001. (C) ATM does not interact with HA-Net1A. MCF7 cells were transfected with HA-Net1A or HA-Net1ΔN, treated or not with IR, and tested for interaction with endogenous ATM by co-immunoprecipitation. Shown is a representative experiment from 3 independent experiments. (D) Catalytic activity is not required for γH2AX suppression by overexpressed Net1A. MCF7 cells were transfected with NLS-β-Gal, wild-type Net1A, catalytically-inactive Net1A L267E, or constitutively active Net1A lacking the N-terminal domain (Net1ΔN). After 48 h, the cells were treated 10 Gy IR, fixed and stained for γH2AX. Shown are representative cells from 3 independent experiments. (E) Quantification of suppression of γH2AX IRIF by Net1A. γH2AX was measured for at least 100 cells per experiment. Data shown are the average of 3 independent experiments. Errors are SEM. *** = P < 0.001. (F) MCF7 cells were transfected with NLS-β-Gal, wild type Net1A, constitutively active RhoA (L63RhoA), or constitutively active RhoB (V14RhoB). After 48h the cells were treated 10 Gy IR, fixed and stained for γH2AX. Shown are representative cells from 3 independent experiments. (G) Quantification of γH2AX in Rho GTPase transfected cells. γH2AX was measured for at least 100 cells per experiment. Data shown are the average of 3 independent experiments. Errors indicate SEM. *** = P < 0.001.
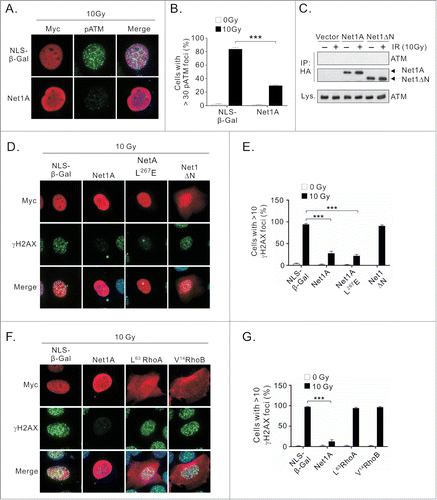
The cellular activity of Net1A has hitherto been shown to be dependent on its ability to stimulate the activity of RhoA or RhoB. However, because both knockdown and overexpression of Net1A suppressed ATM activation, we hypothesized that Net1A may play a scaffolding role to control ATM activation. To test this, MCF7 cells were transfected with NLS-β-Gal, wild type Net1A, catalytically-inactive Net1A (L267E), or an N-terminally truncated form of Net1A that constitutively activates RhoA (Net1ΔN).Citation21 ATM signaling after IR exposure was then assessed by staining cells for phosphorylated H2AX (γH2AX). In these assays we observed that expression of both wild type and catalytically-inactive Net1A (Net1A L267E) efficiently repressed γH2AX IRIF formation, while expression of constitutively active Net1ΔN was without effect (). These experiments indicate that the ability of Net1A to suppress DNA damage signaling was not dependent on its ability to activate RhoA or RhoB, which are the only 2 GTPase substrates of Net1A.Citation18,21 To confirm that Rho protein signaling did not affect ATM activation, we tested whether expression of constitutively active RhoA (L63RhoA) or RhoB (V14RhoB) suppressed γH2AX IRIF formation. We observed that γH2AX IRIF formation was not suppressed by expression of active forms of either Rho GTPase, indicating that RhoA and RhoB do not control H2AX phosphorylation (). Taken together, these data indicate that overexpressed Net1A suppresses ATM activation and phosphorylation of its downstream substrate H2AX independent of its ability to activate RhoA or RhoB.
Net1A is required for DNA double strand break repair and cell survival
As ATM activation is required for efficient DNA repair, we measured the extent of DNA damage occurring in Net1A depleted cells by neutral comet assay, before and after IR. We observed that loss of Net1A expression resulted in an increase in the amount of damaged DNA in non-irradiated cells, as well as in cells four hours after irradiation (). These data suggest that Net1A expression is required for efficient DSB repair.
Figure 4. Net1A is required for DNA repair and cell survival. (A) Net1A knockdown delays DNA repair after IR. MCF7 cells were transfected with control or Net1A siRNAs. Three days later the cells were treated with 10 Gy IR. Four h later the cells were tested for DNA damage by comet assay. Shown is a representative experiment from 3 independent experiments. (B) Quantification of comet assays. Results are the average of 3 independent experiments. At least 100 cells were analyzed per sample. Errors are SEM; * = P < 0.05, ** = P < 0.005. (C) Representative Western blot of siRNA-transfected cells used for comet assays. (D) Net1A knockdown reduces cell survival after IR. MCF7 cells were transfected with control or Net1A siRNAs. Three days later the cells were treated with indicated doses of IR, replated and allowed to grow for 2 weeks. Shown are representative plates from 3 independent experiments. (E) Quantification of survival fraction assays. Results are the average of 3 independent experiments. Errors are SEM; * = P < 0.05. (F) Representative Western blot of siRNA-transfected cells used for survival assays.
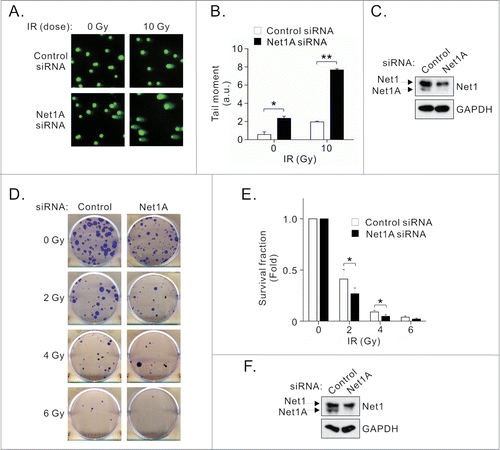
Failure to repair DNA DSBs results in reduced cell survival, and it is clear that defects in ATM signaling are associated with increased sensitivity to radiation induced cell death.Citation22-24 Thus, we tested the functional importance of Net1A in cell survival after exposure to IR by colony formation assay. We observed that knockdown of Net1A decreased cell survival in response to IR (). These data indicate that loss Net1A expression adversely impacts cell survival, most likely by reducing ATM activation and DNA repair.
Discussion
In the present work we have found that the Net1A isoform controls ATM activation, DNA repair and cell survival after gamma irradiation. Importantly, we observed that Net1A controls ATM signaling in a Rho GTPase-independent manner, as catalytically-inactive Net1A suppressed H2AX phosphorylation as efficiently as wild type Net1A, and expression of constitutively active Net1A or its substrates RhoA and RhoB did not suppress H2AX phosphorylation. These findings are reminiscent of the role of the longer Net1 isoform in mitotic control, as it also did not require Rho GTPase activation to promote mitotic progression.Citation14 This work also suggests that Rho GTPase-independent functions of Net1 isoforms may be more common than previously thought.
The ultimate role of Net1 proteins in DNA damage signaling has been somewhat unclear. Previously we found that simultaneous knockdown of both Net1 isoforms in HeLa cells caused increased cell death after DNA DSBs caused by IR or bacterial cytolethal distending toxin.Citation17 However, others have observed that Net1 controls RhoB activation after IR to promote cell death.Citation18 In the present work we have found that Net1 isoforms contribute to ATM activation in multiple cell types. We also observed that Net1A knockdown resulted in a reduced rate of DNA repair and impaired survival after gamma irradiation. Based on these data and previously published work, we suggest that Net1A controls both pro-survival (ATM and p38 MAPK/MAPKAP2) and apoptotic (RhoB) pathways in response to IR, and that the balance between these signaling pathways will ultimately determine the survival outcome for the cell. Further work will be required to assess whether this model is correct.
Treatment with IR is an important therapeutic approach in breast cancer, and resistance to IR induced cell death allows for cancer recurrence. Net1 isoforms have been reported to be overexpressed in many human cancers, including breast cancer.Citation15,Citation25-27 As Net1A controls ATM activation in the absence of activity towards Rho GTPases, these data suggest that relatively modest changes in the level of Net1A expression may have a profound effect on the response of cancer cells to DNA damage caused by IR or other cancer therapeutics. Interestingly, Rac1 has recently been shown to control DNA damage responses.Citation28 Since Rac1 also controls Net1A function, it may be that Net1A mediates Rac1 effects on DNA damage signaling. Delineation of the mechanism by which Net1A controls ATM activity may therefore provide novel avenues to sensitize cancer cells to DNA damaging agents such as IR.
Materials and Methods
Cell culture and transfection
MCF7 breast cancer cells and U2OS osteosarcoma cells were maintained in Dulbecco's modified Eagle medium (DMEM) (Sigma) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). BT474 cells were grown in Roswell Park Memorial Institute (RPMI) medium plus 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. All cells were grown in a humidified 10% CO2 incubator. Cells were irradiated with gamma rays from a cesium source (JL Shepherd and Associates), at the indicated doses. For plasmid transfections, cells were seeded in 6-cm dishes 1 day before transfection. Cells were transfected with DNA using deacetylated polyethylenimine (PEI) 2200 reagent (provided by Dr. Guangwei Du, UT-HSC). For siRNA transfections, cells were transfected with 20 nM of siRNA duplexes using INTERFERin (Polyplus) according to the manufacturer's instructions. The siRNA target sequences used were described previously.Citation13 Analyses were performed 24–48 h after transfection of plasmids and 72 h after siRNA transfection.
Plasmids and antibodies
Wild type Net1A, Net1A L267E and Net1ΔN were as described.Citation29 NLS-β-Galactosidase was provided by Dr. Rebecca Berdeaux (UT-HSC), and was subcloned into pCMV5M by PCR. Myc-L63RhoA was described previously.Citation30 V14RhoB cDNA was obtained from Guthrie cDNA resource center. The following primary antibodies were used: mouse anti-Net1 (sc-50392), mouse anti-GAPDH (sc-27724), rabbit anti-Myc (sc-789) Santa Cruz Biotechnologies; rabbit anti-HA (600-401-384), Rockland Immunochemicals; mouse anti-γH2AX (S139) (05-636), mouse anti-phospho-ATM (Ser1981) (05-740) Millipore; mouse anti-DNA-PKcs (MS423-P1) Neomarkers; rabbit anti-phospho-ATM (ab81292), phospho-DNA-PKcs (Ser2056) (18192) Abcam; and rabbit anti-H2AX (2595) (Cell Signaling Technologies).
Western blot analyses
Cells were washed with PBS and lysed in 2x Laemmi sample buffer or SDS lysis buffer (2.0% SDS, 20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 80 mM β-glycerophosphate, 1 mM Na3VO4, 20 mM NaF, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, 1 mM PMSF). Whole cell lysates were sonicated and boiled in Laemmi sample buffer for 5 min. Equal amounts of protein were resolved by SDS-PAGE, transferred to PVDF membrane (GE Healthcare), and incubated with the appropriate primary antibodies. Band intensities were quantified using ImageJ software (NIH).
Immunofluorescence microscopy
MCF7 cells were grown glass coverslips. Cells were fixed with 4% paraformaldehyde (Thermo Fisher Scientific) in PBS for 20 min at room temperature. Cells were then permeabilized in 0.5% Triton X-100 for 10 min, blocked with 5% bovine serum albumin for 30 min, and incubated with primary antibody for 1h at 37°C. Cells were washed and incubated with Alexa-Fluor 488 anti-mouse or Alexafluor 594 anti-rabbit antibody (Molecular probes) plus DAPI (1 μg/ml) for 1h at room temperature, and mounted onto glass slides with FluorSave reagent (EMD Millipore). Cells were visualized using a Zeiss Axiophot epifluorescence microscope and images were captured using Axiovision software (Carl Zeiss). Mean fluorescence image intensities were quantified using Image J software.
Neutral Comet assays
MCF7 cells were transfected with siRNAs and then untreated or treated with 10 Gy IR. Cells were harvested 4h after irradiation. Comet assays were carried out using a Comet Assay Kit (Trevigen) according to the manufacturer's instructions. Briefly, 1 × 105 cells were combined with LMAgarose at a ratio 1:10 (v/v). 50 μl of the mixture was pipetted onto a comet slide. The slides were placed at 4°C for 10 min and then immersed in lysis buffer at 4°C for 1h. Following electrophoresis, slides were washed with 70% ethanol and air dried for 30 min. Samples were then stained with SYBR green and images were captured using 20× magnification. Images were saved as bitmap files and quantified using TriTek CometScore™ Freeware v1.5.
Flow cytometry analysis
Cells were collected 72 h after siRNA transfection, washed with PBS and fixed with 70% ethanol overnight at −20°C. Cells were then washed 3 times with PBS and incubated with propidium iodide (50 μg/ml) and RNase A (1 μg/ml) for 30 min at room temperature. Cell cycle distributions were detected using an LSR Fortessa cell analyzer system (BD Biosciences) and analyzed with BD FACSDiva software.
Clonogenic survival assays
MCF7 cells were treated with indicated doses of IR, 72h after transfection of siRNAs. After 1 h recovery, the cells were replated in triplicate into 6 well dishes and incubated for 10–14 days to allow colony formation. Colonies were stained with 0.5% crystal violet in 20% ethanol and colonies containing at least 50 cells were counted.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank the Frost, Clark, Cunha, Dessauer and Denicourt lab meeting groups for critical analysis of this work.
Additional information
Funding
References
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol. Cell. 2007; 28:739-45; PMID:18082599; http://dx.doi.org/10.1016/j.molcel.2007.11.015
- Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 2009; 21:245-55; PMID:19230643; http://dx.doi.org/10.1016/j.ceb.2009.01.018
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/10.1038/nat-ure08467
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499-506; PMID:12556884; http://dx.doi.org/10.1038/nature01368
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003; 22:5612-21; PMID:14532133; http://dx.doi.org/10.1093/emboj/cdg541
- Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, Chen DJ. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 2002; 16:2333-8; PMID:12231622; http://dx.doi.org/10.1101/gad.1015202
- Douglas P, Sapkota GP, Morrice N, Yu Y, Goodarzi AA, Merkle D, Meek K, Alessi DR, Lees-Miller SP. Identification of in vitro and in vivo phosphorylation sites in the catalytic subunit of the DNA-dependent protein kinase. Biochem J 2002; 368:243-51; PMID:12186630; http://dx.doi.org/10.1042/BJ20020973
- Soubeyrand S, Pope L, Pakuts B, Hache RJ. Threonines 2638/2647 in DNA-PK are essential for cellular resistance to ionizing radiation. Cancer Res 2003; 63:1198-201; PMID:12649176
- Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle 2007. 6:931-42; PMID:17457059
- Chan AM, Takai S, Yamada K, Miki T. Isolation of a novel oncogene, NET1, from neuroepithelioma cells by expression cDNA cloning. Oncogene 1996; 12:1259-66; PMID:8649828
- Carr HS, Morris CA, Menon S, Song EH, Frost JA. Rac1 controls the subcellular localization of the Rho guanine nucleotide exchange factor Net1A to regulate focal adhesion formation and cell spreading. Mol Cell Biol 2013; 33:622-34; PMID:23184663; http://dx.doi.org/10.1128/MCB.00980-12
- Carr HS, Zuo Y, Oh W, Frost JA. Regulation of focal adhesion kinase activation, breast cancer cell motility, and amoeboid invasion by the RhoA guanine nucleotide exchange factor Net1. Mol Cell Biol 2013; 33:2773-86; PMID:23689132; http://dx.doi.org/10.1128/MCB.00175-13
- Menon S, Oh W, Carr HS, Frost JA. Rho GTPase-independent regulation of mitotic progression by the RhoGEF Net1. Mol Biol Cell 2013; 24:2655-67; PMID:23864709; http://dx.doi.org/10.1091/mbc.E13-01-0061
- Dutertre M, Gratadou L, Dardenne E, Germann S, Samann S, Lidereau R, Driouch K, de la Grange P, Auboeuf D. 2010. Estrogen regulation and physiopathologic significance of alternative promoters in breast cancer. Cancer Res 70:3760-70; PMID:20406972
- Dubash AD, Dubash AD, Guilluy C, Srougi MC, Boulter E, Burridge K, Garcia-Meta R. The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS One 2011; 6:e17380; PMID:21390328
- Guerra L, Carr HS, Richter-Dahlfors A, Masucci MG, Thelestam M, Frost JA, Frisan T. A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS One 2008; 3:e2254; PMID:18509476; http://dx.doi.org/10.1371/journal.pone.0002254
- Srougi MC, Burridge K. The nuclear guanine nucleotide exchange factors Ect2 and Net1 regulate RhoB-mediated cell death after DNA damage. PLos One 2011; 6:e17108; PMID:21373644
- Zhang W, Peng G, Lin SY, Zhang P. DNA damage response is suppressed by the high cyclin-dependent kinase 1 activity in mitotic mammalian cells. J Biol Chem 2011; 286:35899-905; PMID:21878640; http://dx.doi.org/10.1074/jbc.M111.267690
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014; 344:189-93; PMID:24652939; http://dx.doi.org/10.1126/science.1248024
- Alberts AS and Treisman R. Activation of RhoA and SAPK/JNK signalling pathways by the RhoA-specific exchange factor mNET1. EMBO J 1998; 17:4075-85; PMID:9670022; http://dx.doi.org/10.1093/emboj/17.14.4075
- Fang Z, Fang Z, Kozlov S, Mckay MJ, Woods R, Birrell G, Sprung CN, Murrell DF, Wangoo K, Teng L, et al. Low levels of ATM in breast cancer patients with clinical radiosensitivity. Genome Integr 2010; 1:9; PMID:20678261; http://dx.doi.org/10.1186/2041-9414-1-9
- Renwick A, Thompson D, Seal S, Kelly P, Chaqtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006; 38:873-5; PMID:16832357; http://dx.doi.org/10.1038/ng1837
- Tommiska J, Bartkova J, Heinonen M, Hautala L, Kilpivaara O, Eerola H, Aittomaki K, Hofstetter B, Lukas J, von Smitten K, et al. The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancer. Oncogene 2008;27:2501-6; PMID:17982490; http://dx.doi.org/10.1038/sj.onc.1210885
- Lahiff C, Schilling C, Cathcart MC, Mulligan N, Doran P, Muldoon C, Murray D, Pidgeon GP, Reynolds JV, Macmathuna P, et al. Prognostic significance of neuroepithelial transforming gene 1 in adenocarcinoma of the oesophagogastric junction. Br J Surg 2014; 101:55-62; PMID:24375299; http://dx.doi.org/10.1002/bjs.9373
- Shen SQ, Li K, Zhu N, Nakao A. Expression and clinical significance of NET-1 and PCNA in hepatocellular carcinoma. Med Oncol 2008; 25:341-5; PMID:18214716; http://dx.doi.org/10.1007/s12032-008-9042-6
- Tu Y, Lu J, Fu J, Cao Y, Fu G, Kang R, Tian X, Wang B. Over-expression of neuroepithelial-transforming protein 1 confers poor prognosis of patients with gliomas. Jpn J Clin Oncol 2010; 40:388-94; PMID:20304779; http://dx.doi.org/10.1093/jjco/hyp186
- Fritz G, Kaina B. Rac1 GTPase, a multifunctional player in the regulation of genotoxic stress response. Cell Cycle 2013; 12:2521-22; PMID:23907156; http://dx.doi.org/10.4161/cc.25807
- Qin H, Carr HS, Wu X, Muallem D, Tran NH, Frost JA. Characterization of the biochemical and transforming properties of the neuroepithelial transforming protein 1. J Biol Chem 2005; 280:7603-13; PMID:15611121; http://dx.doi.org/10.1074/jbc.M412141200
- Frost JA, Xu S, Hutchison MR, Marcus S, Cobb MH. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol Cell Biol 1996; 16:3707-13; PMID:8668187