
Abstract
Autophagy is a bulky catabolic process that responds to nutrient homeostasis and extracellular stress signals and is a conserved mechanism in all eukaryotes. When autophagy is induced, cellular components are sequestered within an autophagosome and finally degraded by subsequent fusion with a lysosome. During this process, the ATG12–ATG5 conjugate requires 2 different binding partners, ATG16L1 for autophagosome elongation and TECPR1 for lysosomal fusion. In our current study, we describe the crystal structures of human ATG5 in complex with an N-terminal domain of ATG16L1 as well as an internal AIR domain of TECPR1. Both binding partners exhibit a similar α-helical structure containing a conserved binding motif termed AFIM. Furthermore, we characterize the critical role of the C-terminal unstructured region of the AIR domain of TECPR1. These findings are further confirmed by biochemical and cell biological analyses. These results provide new insights into the molecular details of the autophagosome maturation process, from its elongation to its fusion with a lysosome.
Abbreviations:
- AFIM, ATG5 (5)-interacting motif
- AIR, ATG12–ATG5-interacting region
- ATG, autophagy-related
- ATG16N69, ATG16L1 N-terminal 69 residues
- buffer A, buffer containing 50 mM Tris-HCl, pH 8.0, 300 mM NaCl, and 1 mM TCEP
- DAPI, 4’, 6-diamidino-2-phenylindole
- FLuc, firefly luciferase
- FITC, fluorescein isothiocyanate
- FP, fluorescent polarization
- GAL4-BD, GAL4-DNA binding domain
- GFP, green fluorescent protein
- HR, helix rich
- ITC, isothermal titration calorimetry
- MR, molecular replacement
- PE, phosphatidylethanolamine
- PH, pleckstrin homology
- PtdIns3P, phosphatidylinositol 3-phosphate
- RLuc, Renilla luciferase
- r.m.s., root-mean-square
- SPR, surface plasmon resonance
- TECAIR, TECPR1 AIR
- TECPR1, tectonin β-propeller repeat containing 1
- Ubl, ubiquitin-like protein
- UFD, ubiquitin-fold domain
- VP16-AD, herpes simplex virus VP16 transcription activation domain
Introduction
Autophagy is a highly conserved lysosome (vacuole in yeasts)-dependent catabolic process in eukaryotes that is responsible for maintaining cellular homeostasis.Citation1,2 Nonselective autophagy has been studied for a long timeCitation3 and many types of selective autophagy involved in the clearance of aggregated proteins, damaged, or superfluous organelles, and invading pathogens have now been reported.Citation4-9 When macroautophagy (hereafter referred to as autophagy) is invoked, cellular cargoes are enveloped within a de novo generated double-membrane structure known as an autophagosome, and this event is orchestrated by many core machinery autophagy-related (Atg) proteins.Citation10 Two ubiquitin-like proteins (Ubls), Atg12 and Atg8 (LC3 in mammals), are involved in the early stage of autophagosome biogenesis and this process resembles the typical enzymatic cascade of the ubiquitination system.Citation11,12 After the C-terminal glycine residue of Atg12 is conjugated to a lysine residue of its substrate Atg5 through sequential reactions of the autophagic E1-enzyme Atg7 and E2-enzyme Atg10, the resulting Atg12–Atg5 (‘–’ depicts a covalent bond) conjugate induces the rearrangement of the catalytic cysteine residue of another E2-enzyme, Atg3, toward a threonine residue corresponding to the conserved asparagine residue in canonical E2-enzymes.Citation13 The conjugase activity of Atg12–Atg5 as an E3-enzyme for the lipid phosphatidylethanolamine (PE) modification of Atg8 is thereby enhanced.Citation13 In a separate process, to be activated by Atg7, the C-terminal residue of Atg8 is cleaved by the cysteine protease Atg4 to expose a glycine residue.Citation10 Through sequential delivery of the processed Atg8 via Atg7 and Atg3 forming thioester bonds,Citation14 the Atg8 finally forms a covalent bond to PE and the Atg8–PE conjugate then localizes to the phagophore (the precursor to the autophagosome) membrane.Citation15 The Atg12–Atg5 conjugate additionally binds to Atg16 (ATG16L1 in mammals) to form an oligomeric complex of approximately 350 kDa that promotes the elongation of the phagophore via a largely unknown mechanism.Citation16,17 In comparison to yeast Atg16 (), mammalian ATG16L1 has an additional 7 repeat WD40 domains, which facilitate the conjugation of LC3s to PE by recruiting other autophagic components onto the phagophore membrane and forming an approximately 800-kDa oligomeric complex.Citation18-20
Figure 1. Domain architecture of ATG5-interacting proteins. (Top) Atg16 from Saccharomyces cerevisiae consists of only 2 domains, Atg16N (yellow) and a coiled-coil (CC) region. (Middle) human ATG16L1 is a much larger protein consisting of ATG16N69 (purple), CC, and WD40 repeats. (Bottom) TECPR1 is a very large protein showing a complex domain composition including 2 dysferlin domains (DysF), several tectonin β-propeller repeats (TECPR), a pleckstrin homology (PH) domain, and TECAIR (green).
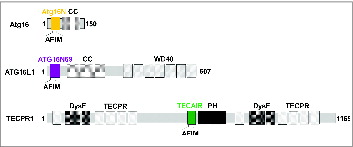
TECPR1 (tectonin β-propeller repeat containing 1) was recently characterized as a protein that functions in the autophagosome maturation process by promoting the fusion of autophagosomes with lysosomesCitation21,22 and that also plays a role in selective autophagy in the innate immune response in Shigella-infected cells.Citation23,24 TECPR1 consists of several repeating tectonin β-propeller repeats, 2 dysferlin domains, and an internal AIR (ATG12–ATG5-Interacting Region) domain followed by a pleckstrin homology (PH) domain (). In normal conditions, the AIR binds to the PH domain and induces autoinhibition to block the interaction between the PH domain and phosphatidylinositol 3-phosphate (PtdIns3P) molecules of the autophagosomal membrane and thereby regulate the autophagic function of TECPR1.Citation21,22 However, during autophagosome maturation the ATG12–ATG5 conjugate binds to AIR and the PH domain is subsequently liberated to attach to a PtdIns3P and thus tether the autophagosome to a lysosome. These 2 vesicles will then undergo SNARE-mediated fusion to form an autolysosome.Citation25 When the characteristics of TECPR1 were identified, 2 opposing findings about the colocalization of TECPR1 and ATG16L1 were reported.Citation21,23 TECPR1 forms a complex with the ATG12–ATG5 conjugate and exclusively with ATG16L1 in mammalian autophagosome maturation whereas it colocalizes with the ATG12–ATG5-ATG16L1 complex (“-” depicts a noncovalent interaction) to targeting bacterial pathogens in selective autophagy. Furthermore, there have been discrepancies between the results of previous TECPR1-deletion experiments; a normal autophagic flux was reported by Ogawa et al.Citation23 but autophagosome accumulation was described by Zhong and colleagues.Citation21
In our current study, we obtained clues regarding the switching process from phagophore elongation to autophagosome maturation based on the crystal structures of the ATG5-ATG16L1 N-terminal 69 residues (ATG16N69) and ATG5-TECPR1 AIR (TECAIR) complex. Our findings rule out the ternary complex formation of ATG12–ATG5-(ATG16L1-TECPR1), which is found in bacterial clearance.Citation24 Both ATG5 interacting partners have an α-helical structure and compete for the binding site of ATG5 via a conserved sequence W-x-x-x-I-x-x-x-L-x-x-R-x-x-x-Q/E, which we termed AFIM (ATG5 (Five)-Interacting Motif). Furthermore, we characterized the critical role of the C-terminal unstructured region of TECAIR. The significance of the AFIM sequence revealed by both crystal structures and the role of C-terminal unstructured region of TECAIR were evaluated by in vitro mutant analysis and in vivo autophagy flux measurements. Our current results provide new insights into the molecular mechanisms underlying autophagosome maturation, from its elongation to its fusion with a lysosome.
Results
Structure determination of the ATG5-ATG16N69 and ATG5-TECAIR complexes
To determine the crystal structures of the ATG5-ATG16L1 and ATG5-TECPR1 complexes, we initially tried to express full-length ATG5 alone and its binding partners ATG16L1 and TECPR1 separately. Although the ATG5 was expressed as a soluble protein, we faced an aggregation problem at the later stages of purification. The expression of full-length ATG16L1 and TECPR1 was also not straightforward, and the well-characterized ATG5-binding regions of ATG16L1 and TECPR1 did not behave well in solution when expressed alone. We thus co-expressed ATG16N69 and TECAIR with full-length ATG5 and were able to copurify these products successfully (details provided in Materials and Methods). The tight complex structures of the ATG5-ATG16N69 and ATG5-TECAIR are a likely explanation for their comigration at different chromatographic steps ( and Fig. S1).
Figure 2. Structure of the ATG5-ATG16N69 and ATG5-TECAIR complexes. (A) Ribbon diagram of the structure of ATG5-ATG16N69. UFD-1, UFD-2, and the HR domain in ATG5 are highlighted in yellow, pale cyan, and pale green, respectively. The N-terminal helix is colored orange and invisible residues (226–231) are denoted by cyan dots. The secondary structural elements are sequentially labeled. The N- and C-termini are also labeled. ATG16N69 is highlighted in purple, and its N- and C-terminal residues are indicated. (B) Molecular surface diagram showing the electrostatic potential of ATG5. Negatively and positively charged surfaces are colored red and blue, respectively. The invisible region is indicated by a transparent gray oval. The ATG16N69 protein highlighted in purple is shown as a ribbon diagram with a stick model of interacting residues with ATG5. For clarity, the residues are labeled in purple and the ‘a’ is appended to the residue number of ATG16N69. (C) Ribbon diagram of the ATG5-TECAIR structure. The color scheme for ATG5 is the same as in panel (A). The invisible residues (26–31 and 60–65) of ATG5 are shown as yellow dots. TECAIR is colored green and its N- and C-terminal residues are indicated. (D) Molecular surface diagram following the arrangements described in panel (B) with TECAIR colored green and shown as a ribbon diagram. The residues interacting with ATG5 are labeled in green and the ‘t’ is appended to the residue number of the TECAIR protein.
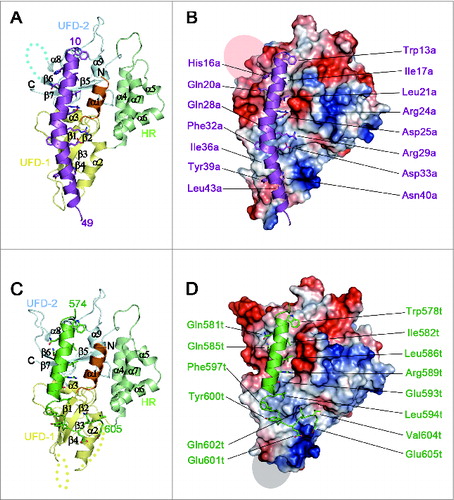
Human ATG5 consists of 2 ubiquitin-fold domains 1 and 2 (UFD-1 and UFD-2, respectively),Citation26 which are the equivalent of the ubiquitin-like domains A and B (UblA and UblB, respectively) of yeast Atg5,Citation27 and a helix rich (HR) domain exists between them (). The UFD contains 3 or 4 β-strands flanked by 2 α-helices in a similar arrangement to typical Ubl structures,Citation26-29 and the HR domain contains 4 α-helices (). Consequently, a total of 9 α-helices including the first helix α1 and 7 β-strands comprise the ATG5 structure with an approximate dimension of 65 × 45 × 45 Å. Forty out of 69 amino acid residues in the ATG16N69 fragment were present in the electron-density map and the rest were missing (Fig. S2A). The AIR domain of human TECPR1 has been reported as residues 566 to 610 of the protein.Citation21 For crystallization, however, the construct was manipulated to shorten the AIR domain to contain residues 573–610 because the front region was predicted to be disordered and generate twinned crystals. Although the new construct also produced twinned crystals, they were less severe. The crystal quality was dramatically improved by using a microseeding technique at 4°C (see Materials and Methods). Similar to ATG16N69, only the 32 residues of TECAIR directly interacting with ATG5 were seen on an electron-density map (Fig. S2B).
Overall structure of the ATG5-ATG16N69 and ATG5-TECAIR complexes
UFDs of human ATG5 show structural similarity with ubiquitin and Ubls (Fig. S3), such as ATG12, with Z scores of 8.5 (PDB code 3EHV)Citation30 and 8.7 (PDB code 4GDK).Citation26 By analysis using the DALI server,Citation31 the root-mean-square (r.m.s.) deviations between these UFDs are less than 2.2 Å. The inserted HR domain shows a relatively low structural similarity with other domains. The highest Z-score was found to be 3.7 against the Zn-sensing transcriptional regulator, ZntR in E. coli (PDB code 1Q08).Citation32
ATG16N69 and TECAIR bind to ATG5 by interacting noncovalently with UFD-1, UFD-2, and N-terminal helix α1 (). In the recently determined ATG12–ATG5-ATG16N triple complex structure,Citation26 it was also shown that 33 residues (from Gln11 to Ser43) of ATG16N are sufficient to bind to ATG5, and this is similar to our current structural finding of the absence of ATG12 conjugation. Additionally, ATG16N69 forms a crystallographic homodimer via its hydrophobic residues (Fig. S4A) and, intriguingly, the ATG12–ATG5-ATG16N complex also shows a crystallographic dimer in the exactly same manner (Fig. S4B) although it contains different protein components and has different crystalline packing.Citation26 The results of size exclusion chromatography plus multi-angle light scattering (SEC-MALS) analysis of our ATG5-ATG16N69 complex also revealed a clear dimer in solution (Fig. S1). This contrasts with the yeast Atg5-Atg16 complex structure,Citation27 which shows a relatively unstructured C-terminal helix in ATG16L1 providing an additional binding force for its association with ATG5 rather than self-dimerization (Fig. S5). We conclude from these findings that the N-terminal helix of human ATG16L1 is not only sufficient to interact with ATG5, but also drives self-dimerization even without the middle coiled-coil region, which is mandatory for dimerization of the Atg16/ATG16L1 protein from yeast and human.Citation33,34 In the case of TECAIR, however, the binding helix is more similar to that of yeast Atg16 (Fig. S5). The solution behavior of the ATG5-TECAIR complex is similar to that of yeast Atg5-Atg16N, which is a monomer (Fig. S1). Hence, it is not involved in self-dimerization but in the interaction with ATG5, as described below.
In the ATG5-ATG16N69 complex structure, 35 residues of ATG5 and 22 residues of ATG16N69 interact with each other with an interface area of 1082.6 Å2. Through complexation, the ΔiG value and ΔiG P-value are changed by -10.0 kcal mol−1 and 0.114, respectively, and the complexation significance score (CSS) is 1.0. Similarly, in the ATG5-TECAIR complex structure, 41 residues of ATG5 interact with 21 residues of AIR and form a 1213.9 Å2 interface area. The ΔiG and ΔiG P-value in this case are changed by -16.9 kcal mol−1 and 0.159, respectively, and the CSS is 1.0. These properties were evaluated using PDBePISA.Citation35 Interestingly, the same hydrophobic region of ATG5 is occupied by ATG16N69 or TECAIR through their helical segment () and the larger binding surface between ATG5 and TECAIR comes from the unstructured C-terminal portion of TECAIR, which is similar to that of yeast Atg5 ( and S5). These results indicate that ATG16L1 or TECPR1 form a complex with ATG5 using the same binding site and are therefore mutually exclusive.
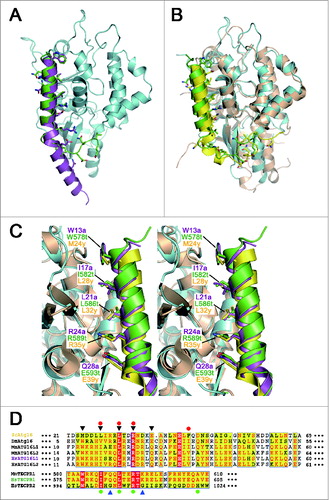
Figure 3. Comparisons among the ATG16 and TECPR family proteins. (A) Ribbon diagram showing the superposition of the ATG5-ATG16N69 and ATG5-TECAIR complexes. Human ATG5 is colored in cyan and ATG16L1 and TECPR1 are colored as in . (B) Ribbon diagram showing the superposition of yeast Atg5-Atg16 and ATG5-TECAIR complexes. Yeast Atg5 and Atg16 are colored gold and yellow, respectively. Coloring of the ATG5-TECAIR complex is as described panel (A). (C) Close-up stereo diagram of superposed AFIMs from yeast Atg16, human ATG16L1, and TECPR1. The key residues are shown using a stick model. (D) Sequence alignment of the ATG5-binding regions of ATG16s and TECPRs from different organisms (Sc, Saccharomyces cerevisiae; Dm, Drosophila melanogaster; Mm, Mus musculus; Hs, Homo sapiens). Shading indicates residues that are identical (red) or highly conserved (yellow) between species. The AFIM residues are denoted using black triangles (top), and 2 glycine residues in TECPR2 by blue triangles (bottom). The residues substituted in the mutation experiments are marked using a red-filled circle at the top for ATG16L1 and green-filled circle at the bottom for TECPR1. The sequence numbers for the aligned residues are also provided.
ATG16L1 and TECPR1 bind to ATG5 via a conserved motif within an α-helix
Although ATG16N69 and TECAIR exhibit structural differences in their overall binding to ATG5, they adopt a similar α-helical structure, which was originally described in the Saccharomyces cerevisiae Atg5-Atg16 (1–57) structure (). In this helix, 3 non-polar residues are oriented toward the hydrophobic cleft of ATG5 and charged residues make additional salt bridges near this cleft. The major interacting residues are Trp13a, Ile17a, Leu21a, Arg24a, and Gln28a of ATG16N69 and Trp578t, Ile582t, Leu586t, Arg589t, and Glu583t of TECAIR. In our current report, ‘a’ and ‘t’ are appended to the residue number of ATG16L1 and TECPR1 for clarity. From amino acid sequence alignment analysis (), we identified a shared region with properties of a motif for ATG5 binding by comparison with other species. We denoted this W-x3-I-x3-L-x2-R-x3-Q/E motif as AFIM, which can accommodate several residue perturbations but which has similar side chain sizes and chemical properties.
In the AFIM, tryptophan residues (Trp13a and Trp578t) make a hydrophobic cluster with Thr249, Pro250, and Trp253 of ATG5, and Ile (Ile17a and Ile582t) and Leu (Leu21a and Leu586t) residues are also adjusted to the hydrophobic biding pocket established by Val7, Ile243, Pro245, Thr249, Pro250, Trp253, and Leu258 of ATG5 (). In addition to the hydrophobic interactions, Arg24a and Arg589t form bipartite ionic interactions with the 2 main chain carbonyl oxygen atoms of ATG5: His241 from UFD-2 and Asp10 from N-terminal helix (). The only difference in the AFIM between ATG16N69 and TECAIR is in the last residues. Gln28a of ATG16N69 uses a side chain oxygen atom to make a polar interaction with a nitrogen atom of Arg41 of ATG5 (). The Glu593t of TECAIR uses both oxygen atoms to make an ionic interaction with the guanidium group of Arg41 (). To further dissect these interactions, we tried to generate mutants of ATG16N69 or TECAIR fused with a GST or MBP tag to perform in vitro affinity isolation assays. However, the fusion proteins were degraded probably due to their intrinsic instability and in the case of TECAIR this was much more severe. The importance of the key determinants was instead elucidated in a mammalian 2-hybrid assay (). Ile17a, Leu21a, and Arg24a of ATG16N69 and Ile582t, Leu586t, and Arg589t of TECAIR were tested in the first instance because they are highly engaged in the binding to ATG5 as part of the AFIM. When each of the hydrophobic Ile and Leu residues was mutated to the bulkier Trp residue, the binding between ATG5 and ATG16N69 or TECAIR was almost undetectable (). Also, when we mutated the positively charged Arg to Asp, the ionic interaction must have been disrupted and indeed no luciferase activity was detected (). These results revealed that the bulky mutation of hydrophobic residues and charge reversal of the Arg residue in ATG16L1 or TECPR1 prevents the interaction with ATG5 via steric hindrance and charge-charge repulsion. Hence, the appropriate size and charge of the side chain atoms of AFIM are requisite for the binding of ATG5.
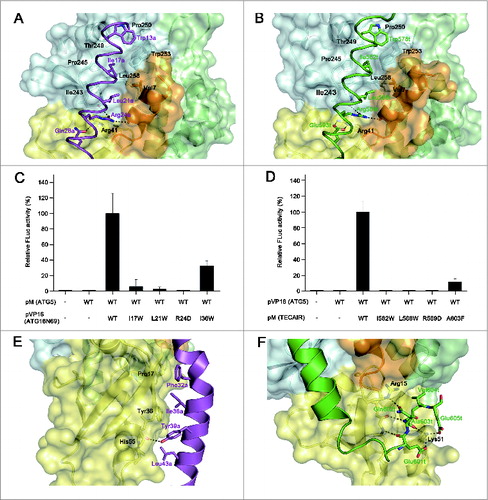
Figure 4. Details of the binding between ATG5 and its interaction partners, and mutagenesis mapping of the critical residues for these associations. (A) Magnified view showing the details of the interactions by the AFIM of ATG16L1. (B) Magnified view showing the details of the interaction by the AFIM of TECPR1. ATG5 is shown as a ribbon with a transparent molecular surface. The color scheme is the same as . Key residues for the interaction between ATG5 and its binding partners are shown using a labeled stick model. Black dashed lines designate hydrogen bonds. (C) Mammalian 2-hybrid analysis of the ATG5-ATG16N69 interaction. (D) Mammalian 2-hybrid analysis of the ATG5-TECAIR interaction. FLuc activities were normalized to RLuc activities and the normalized FLuc activity of the cells transfected with pM and pVP16 was arbitrarily set at 100%. Each bar and line represents the mean and standard deviation values of 3 independently performed transfections. (E) Magnified view showing the additional interaction of the C-terminal extended helical region of ATG16N69. (F) Magnified view showing the additional interaction of the C-terminal unstructured region of TECAIR.
Contribution of additional interactions outside the AFIM
In addition to the AFIM, the remaining part of the helix also contributes to the interactions of ATG16L1 or TECPR1 with ATG5. Within the ATG5-ATG16N69 structure, the C-terminal portion of ATG16N69 contains several residues situated toward the interface, such as Phe32a, Ile36a, Tyr39a, and Leu43a (). The Pro17, Tyr36, and His55 residues of ATG5 contained in the β1 and β2, strands and α2 helix, respectively, interact with the above residues hydrophobically and only the hydroxyl group of Tyr39a in ATG16N69 forms a H-bond with the main chain on the β2 strand of ATG5. The residues that form a different side of the ATG16N69 region facing ATG5 are also hydrophobic (Ala31a′, Phe32a, Ile35a, Ty39a, Leu42a′ and Leu43a (the prime symbol [′] denotes the second subunit), but participate in dimerization (Fig. S4). By contrast, within the ATG5-TECAIR structure, the C-terminal portion of TECAIR winds up ATG5 via its unstructured tail and the interactions are composed of multiple H-bonds and salt bridges (). The main chain atoms of Glu601t and Ala603t of TECAIR form H-bonds with those of Leu38 on the β2 strand of ATG5, and the main chain oxygen atom of Gln602t interacts with the epsilon amino group of Lys51 on α2 helix. The Lys51 residue of ATG5 additionally interacts with Glu601t and Glu605t, and so highly participates in TECAIR binding. The side chain oxygen atom of Gln602t forms a H-bond with the guanidium group of Arg15 on the β1 strand of ATG5.
We further investigated these interactions using a mammalian 2-hybrid assay (). Ile36a of ATG16N69 and Ala603t of TECAIR were mutated to disrupt the interaction with ATG5. Indeed, mutations of Ile36a to the bulkier Trp residue and Ala603t to a bulkier Phe significantly reduced the binding efficiency through steric hindrance. However, this effect was not as strong as that caused by mutation of the AFIM. These results clearly show that the primary binding of ATG16L1 or TECPR1 to ATG5 is governed by the AFIM and that the outside C-terminal helical portion of ATG16N69 additionally contributes to its binding affinity for ATG5. Intriguingly, the C-terminal unstructured region of TECAIR interacts much more extensively with ATG5 () and its overall structural features are somewhat similar to yeast Atg16, but the details such as interacting residues and chain winding are different (). The length of the helical region in yeast Atg16 falls between that of ATG16L1 and TECPR1 (Fig. S5). Yeast Atg16 also shows a helical kink at nearly the same position as ATG16N69, and the Phe46 residue within the helical region of yeast Atg16 plays a critical role in the binding of yeast Atg5. In addition, 2 charged residues, Asp48 and Asn49, within the unstructured C-terminal region of Atg16 are involved in the Atg5 interaction, but are not as critical.Citation27 Indeed, the most structurally divergent region between human and yeast Atg5 is the α2 helix () which interacts extensively with the C-terminal unstructured region of human TECPR1, suggesting that the ATG5-TECPR1 interaction is much stronger than the ATG5-ATG16L1 interaction.
The pH dependency of binding between ATG5 and its interacting partners
To measure the binding constant between ATG5 and either ATG16L1 or TECPR1, we attempted different methods including isothermal titration calorimetry (ITC), fluorescent polarization (FP), and surface plasmon resonance (SPR). As noted above, the fusion-tagged ATG16N69 and TECAIR products are not stable in solution and we therefore designed and synthesized minimal binding peptides of both (ATG16N69 [A16: 36 residues from Phe10 to Lys45]; TECAIR [TEC: 36 residues from Thr575 to Val610]). Using these and fluorescein isothiocyanate (FITC)-labeled peptides, we attempted ITC and FP experiments. These were unsuccessful, however, as free ATG5 is also unstable in solution at high concentrations. Hence, the SPR immobilization technique was employed for biochemical characterization. Unexpectedly, the binding constant (KD, equilibrium dissociation constant) between ATG5 and A16 was found to be 50-fold higher than that between ATG5 and TEC at pH 7.4 (Fig. S6). In contrast, TECPR1 showed a higher affinity for ATG5 via its C-terminal unstructured region (). Intriguingly, the binding constants between ATG5 and its binding partners were comparable when we performed the same experiment at the slightly more acidic pH of 6.0 (Fig. S6). In particular, the dissociation rate constant (kd) between ATG5 and TEC peptide was observed to be the lowest among all of our current experimental data (Fig. S6). We are unsure of the exact pH outside the lysosomal membrane but there is a possibility that the pH in the membrane vicinity is lower than that of the cytosolic space. The binding constant calculated using shorter peptide analytes may not be exactly representative of the binding properties of the ATG5 interacting molecules, but the dramatic enhancement of binding between ATG5 and TECPR1 at a low pH suggests that during autophagosome maturation, ATG5 swaps its interacting partner from ATG16L1 to TECPR1. Due to the molecular interactions involved, this must be a unidirectional process.
Mutation of TECAIR causes an accumulation of autophagosomes
To investigate the effects of TECAIR mutations on autophagy flux, green fluorescent protein (GFP)-tagged LC3, a well-known autophagy marker, was expressed in cells. Autophagy was induced by the addition of rapamycin and punctate GFP-LC3 fluorescent signals indicating recruitment of LC3 protein to phagophores and/or its presence on autophagosomal membranes were detected in the cytosol (). In TECPR1 WT transfected COS-7 cells, rapamycin treatment induced autophagosome formation followed by immediate disappearance of the GFP-LC3 signal via lysosome fusion (). However, in TECPR1 mutant (I582W, L586W, R589D, and A603F) transfected cells, ATG12–ATG5 conjugate interactions with TECPR1 mutants seemed to be perturbed, which might be due to a significant reduction in the ability of these mutants to bind ATG5 (). Hence, lysosomal fusion with autophagosomes would be disrupted, and indeed autophagosomes were found to accumulate in the cytosol when autophagy was induced by the addition of rapamycin (). Consistent with these binding data, mutations in the AFIM produced a more severe accumulation of autophagosomes than the A603F mutation (). Electron microscopy further revealed that the number of autophagosomes was increased in cells transfected with mutant TECPR1 genes ( and Fig. S7). These data are very consistent with the results of the luciferase assay and structural analysis, and lend further support to our working model ().
Figure 5. Accumulation of autophagosomes in the TECPR1 mutant cells. (A) COS-7 cells were transfected with the indicated plasmids encoding Flag-tagged TECPR1 wild-type (WT), I582W, L586W, R589D, and A603F mutant. After 48 h of transfection, the cells were transduced for 24 h with GFP-LC3 and then incubated for a further 2 h in the absence or presence of 500 nM rapamycin (-Rapa or +Rapa). Cells were then fixed, permeabilized, and stained with 4’,6-diamidino-2-phenylindole (DAPI). Representative images of GFP-LC3 (green) and DAPI (blue) fluorescence are shown. Scale bar = 10 μm. (B) The fluorescent intensity of GFP-LC3 in the cytoplasm of >50 cells for each experimental group was quantified and expressed as a percentage of the value for the control group. Data are the means ± SEM from 3 independent experiments.
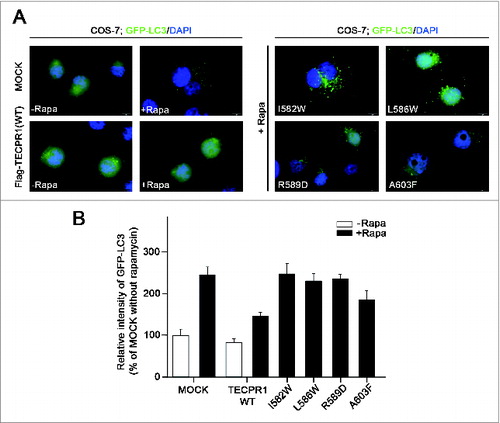
Figure 6. Electron microscopy image of TECPR1 mutant cells. (A) TEM images of control and wild-type cells in the absence or presence of 500 nM rapamycin (-Rapa or +Rapa). (B) Cells expressing I582W, L586W, R589D, and A603F mutant TECPR1 protein in presence of 500 nM rapamycin. Several autophagosomes appeared in the cytosol. Scale bars = 1 μm. Autophagosomes and autolysosomes are indicated by white and black arrows, respectively. See Figure S7 for the quantification of TEM images.
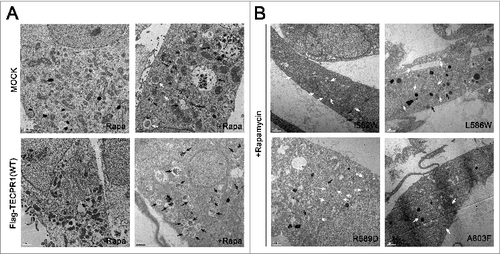
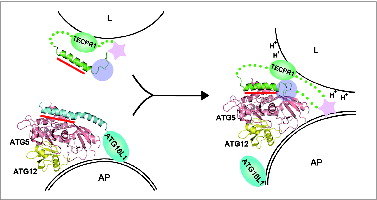
Figure 7. A schematic model for autophagosome maturation via the interaction between ATG5 and TECPR1. The autophagosomal membrane binding mode of the ATG12–ATG5-ATG16 complex is still unclear.Citation49,58,59 The primary ATG5-binding region is the AFIM within the α-helix in both ATG16L1 and TECPR1 and is indicated by the red bar. Additional interactions involving the C-terminal unstructured loop of TECPR1 denoted by the transparent purple circle may play a critical role in the interaction swap with ATG16L1. Furthermore, the lower pH near the lysosomal membrane may trigger tighter binding between ATG5 and TECPR1. The pleckstrin homology domain indicated by the pink star has been proposed to be embedded within TECPR1 before fusion and then exposed upon ATG5-TECPR1 complex formation for an additional interaction with PtdIns3P to enable membrane fusion.Citation21 AP, autophagosome; L, lysosome.
Discussion
Details of the molecular functions of much of the ATG core machinery participating in autophagosome elongation processes have been revealed by structural studies, biochemical analyses, and in vivo experiments from yeast to human.Citation13,26,36-44 Despite this, however, autophagosome maturation is still poorly understood and the recently discovered TECPR1 protein is one of the few factors identified to date that mediates the fusion of autophagosomes and lysosomes.Citation21,22 TECPR1 was initially characterized in association with TRAPP vesicle-tethering, the CCT chaperonin, and the ATG12–ATG5-ATG16L1 complex by extensive proteomic analysis, suggesting its involvement in phagophore elongation and autophagosome assembly.Citation45 Two subsequent independent studies reported critical roles of TECPR1 in the efficient autophagic targeting of bacterial pathogens and in the fusion between autophagosomes and lysosomes.Citation21-24 However, some of the observations in those 2 studies conflicted, most notably the colocalization of TECPR1 with ATG16L1 and accumulation of autophagosomes under TECPR1-deficient condition.Citation21,23 Our elucidated crystal structure clearly reveals that ATG5 cannot accommodate both ATG16L1 and TECPR1 simultaneously. However, according to Ogawa et al., TECPR1 interacts with WIPI2, a yeast Atg18 homolog and PtdIns3P-interacting protein, suggesting that it might be possible that ATG5, ATG16L1, and TECPR1 can all colocalize in the phagophore, but not via direct interactions. Our cell biology experiments herein employed TECPR1 mutants, which are defective in ATG5 binding (), and we found good agreement between our results and those reported previously using TECPR1-depletion.Citation21 Mutations in the AFIM of TECPR1 increase the number of autophagosomes when autophagy is induced by rapamycin (). Hence, our structural data and findings from our mutant studies support the experimental results of Zhong and colleagues.Citation21
ATG16L1 and TECPR1 interact with ATG5 primarily through an α helix containing the W-x3-I-x3-L-x2-R-x3-Q/E motif, or AFIM, which is conserved among different species (). However, the primary sequence is not sufficient to enable this as the helical structure is necessary for binding to the ATG5 cleft. For example, TECPR2 has a similar motif, W-x3-I-x3-L-x2-R-x3-I as the AFIM (), but was reported not to interact with the ATG12–ATG5 conjugateCitation21 and in our hands showed very weak binding affinity (data not shown). There are 2 helix-breaking glycine residues (Gly1003 and Gly1010) within the TECPR2 sequence, and the Q/E in the AFIM that enables H-bonding with ATG5 in TECPR1 is replaced with a hydrophobic isoleucine residue (Ile1012) in TECPR2 (). Permutated peptides comprising the sequence ‘I-x3-L-x2-R-x3-Q’ were previously synthesized and assayed for their binding affinity to ATG5, which revealed the critical role for Ile, Leu, and Arg of ATG16L1.Citation46 The binding affinity is in the micromolar range, but there must be tighter binding if the first tryptophan residue in AFIM is included since it is one of the prominent interacting residues with ATG5 (). Our binding constant analysis produced quite complicated results, but a pH dependency between ATG5 and its binding partners was clearly evident (Fig. S6). It is well-known that influenza hemagglutinin undergoes a pH-induced conformational change when it approaches the endosomal membrane.Citation47,48 Although we could not pursue the conformational change of ATG5-interacting partners in our current experiments, the tighter binding we observed between ATG5 and TEC peptide at low pH suggests a possible mechanism for regulating the fusion between autophagosomes and lysosomes.
Based on our current structural data, in which the conjugation of ATG12 was omitted in both the ATG5-ATG16N69 and ATG5-TECAIR structures, we speculate that there is no role of ATG12 in the interaction between ATG5 and either ATG16L1 or TECPR1 because it is known to operate on the opposite site of the AFIM (Fig. S4B). Also, it has been reported that ATG12 conjugation does not induce a conformational change in ATG5, and that ATG12 functions as a binding module for ATG3.Citation49 However, it is very possible that TECPR1 binds to an ATG12 conjugated form of ATG5 because the ATG12–ATG5-ATG16 complex mediates ATG8–PE formation on the phagophore membrane. Interestingly, the structure of Atg16 from yeast falls between that of ATG16L1 and TECPR1 (Fig. S5). Indeed, amino acid sequence alignments of human ATG16L1 and yeast Atg16 show some homology in the AFIM, but nowhere else (). Yeast Atg16 possesses a long coiled-coil structure as a tetramerCitation50 whereas human ATG16L1 has an additional domain, a WD40 repeat 7-bladed β-propeller (), and its major oligomeric state is suggested to be an octamer.Citation18 The structurally determined N-terminal regions in yeast Atg16 and human ATG16L1 are a monomer and dimer, respectively, which may underlie the different oligomeric nature of the full-length proteins. Further structural studies on the full-length proteins will be needed to clarify the role of the N-terminal region in the correct formation of tetrameric yeast Atg16 and octameric human ATG16L1. TECPR1 also possesses tectonin β-propeller repeats () but their exact role is not yet clear. A previous report has shown that TECPR1 has binding affinity for PtdIns3P through its PH domain right after TECAIR () and interestingly, the lipid interaction of TECPR1 was found to be in concert with the ATG12–ATG5 association because TECPR1 alone does not interact with PtdIns3P.Citation21 Hence, it has been suggested that TECAIR regulates the lipid binding of TECPR1 ()Citation21,22 and the structural information on full-length and/or a defined domain of TECPR1 will be indispensable for further functional characterization. However, our current structure and cell biology data for TECAIR provide a good and detailed molecular dissection of the fusion event between autophagosomes and lysosomes, which will help to guide future investigations.
Materials and Methods
Plasmid construction
Full-length cDNA of human ATG5 was purchased from the Korea Human Gene Bank. The gene for the N-terminal domain of human ATG16L1 was obtained from a human cDNA library (TaKaRa). The gene for the AIR domain of TECPR1 (residue from 566 to 610) was chemically synthesized (Integrated DNA Technologies, Inc.). The ATG5 gene was amplified by PCR using oligonucleotides containing the restriction enzyme sites, BamHI and XhoI for forward and reverse primers, respectively. The amplified PCR products were digested by these restriction enzymes and ligated into the first multiple cloning site after the His6-tag of the pETDuet-1 vector (Novagen, 71146-3). This plasmid was then transformed into E. coli strain BL21(DE3). The ATG16N69 gene was amplified by PCR with primers harboring the restriction enzyme sites NdeI and KpnI. This product was cloned into the second multiple cloning site of the His6-ATG5 containing pETDuet-1 vector using these restriction enzyme sites, and also transformed into E. coli BL21(DE3). The TECAIR constructs were cloned in the same way as ATG16N69 but using NdeI and XhoI restriction sites. The QuikChange site-directed mutagenesis method (Stratagene) was used to prepare the constructs for expressing mutant ATG16N69 and TECAIR.
Protein expression and purification
The ATG5-ATG16N69 and ATG5-TECAIR complexes were expressed and purified using the procedure described below. For the co-expression of His6-ATG5 with ATG16N69 or the AIR domain, E. coli harboring the corresponding expression plasmids were cultured with 1 L of Luria-Broth (casein peptone, 10 g/L; yeast extract 5 g/L; sodium chloride, 5 g/L) at 37°C. When absorbance at 600 nm reached 0.7, 0.4 mM of IPTG (Calbiochem, 420322) was added and the cultures were maintained at 18°C for 24 h. Cell harvesting was performed by centrifugation at 6,300 g for 30 min, and resuspension with 10 ml of buffer A (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1 mM TCEP [Sigma-Aldrich, C4706]). The cells were then ruptured by sonication in the presence of 0.1 mM PMSF (USB, 20203) and protease inhibitor cocktail (Roche, 05892791001), and clarified by centrifugation for 1 h at 27,200 g. The filtered supernatant fraction was loaded onto an Ni-NTA column (GE Healthcare, 17-5248-02), washed with 20 volumes of buffer A, and eluted by gradually increasing the concentration of imidazole (USB, 17525) up to 500 mM. The sample was the applied to a HiTrap Q column (GE Healthcare, 17-5156-01) for anion exchange chromatography. The eluent was further purified by gel filtration chromatography using Superdex75 16/600 (GE Healthcare, 28-9893-33).
Crystallization
Both ATG5-ATG16N69 and ATG5-TECAIR complexes were concentrated to 9–13 mg/ml and crystallized at 20°C by hanging drop vapor diffusion.Citation51 The initial crystal of the ATG5-ATG16N69 complex was obtained within 1 or 2 d in 50 mM Tris-HCl, pH 8.5, 100 mM KCl, 10 mM MgCl2, 30% (v/v) PEG 400 (Hampton Research, HR2-603). Improved crystals were obtained in the range of pH 8.3-8.7 and 26–32% (v/v) PEG 400 within 2–4 d. The initial crystal of the ATG5-TECAIR complex was obtained using 100 mM MES-imidazole buffer (Molecular Dimensions, MD2-100-100; mixture of 55.5% MES and 44.5% imidazole), pH 6.5, 120 mM alcohols (Molecular Dimensions, MD2-100-73; mixture of 1,6-hexanediol, 1-butanol, 1,2-propanediol, 2-propanol, 1,4-butanediol, 1,3-propanediol), 12.5% (v/v) of MPD (2-methyl-2,4-pentanediol; Molecular Dimensions, MD2-100-24), 12.5% (w/v) PEG 1000 (Molecular Dimensions, MD2-100-5), and 12.5% (w/v) PEG 3350 (Molecular Dimensions, MD2-100-9). For better crystallization, microseeding was used. To make the seeding solution, drops containing microcrystals were mixed with the same solution as the mother liquor except that it had a 10% higher precipitant concentration. After vortexing, the seeding solution was serially diluted from 10−1 to 10−6 fold, and 1 μl aliquots were added to 5 μl of protein and 4 μl of the mother liquor mixture. Finally, the precipitant concentration of the mother liquor was 5∼10% lower than that initially. The crystals were grown in an appropriately diluted solution at 4°C within 2–4 d.
Data collection and structure determination
Crystals were flash-frozen in liquid nitrogen after sequential transfer in mother liquor containing 5–15% (w/v) glycerol (USB, 16374). Diffraction data were collected at the Photon Factory AR-NW12A beamline, KEK, Japan using an ADSC Quantum 210 detector at a 1.0000 Å wavelength. The collected data were processed using HKL2000 software.Citation52 The phases of the ATG5-ATG16N69 complex were obtained by molecular replacement (MR) with the partial structure of the 2 UFDs of Atg5 from Kluyveromyces marxianus (PDB code 3VQI)Citation53 using PHASER software,Citation54 and the phases of the ATG5-TECPR1 complex were obtained by MR with the refined structure of ATG5-ATG16N69. Automatic model buildings were performed using PHENIX,Citation55 and COOT was used for manual model rebuilding.Citation56 The structures were refined with PHENIX.REFINE.Citation55 Detailed statistics are presented in .
Table 1. Data collection and refinement statistics
Mammalian 2-hybrid analyses
The human ATG5 gene was cloned into the pM and pVP16 vectors (Clontech, 630305) using the BamHI and HindIII restriction sites. Wild-type and mutant ATG16N69 and TECAIR were also cloned into the pVP16 and pM vectors, respectively.Citation57 HeLa cells were cultured in Dulbecco's modified Eagle's medium (Hyclone) containing 10% fetal bovine serum (Hyclone, SH30919.03) and 1% penicillin/streptomycin (Hyclone, SV30010). HeLa cells were then transiently cotransfected using Lipofectamine 2000 (Invitrogen, 11668-019) with 4 plasmids: (1) the reporter plasmid pFR-Luc (Clontech, 211344), which encodes the firefly luciferase (FLuc) encoding cDNA downstream of a basic transcriptional promoter (TATATA) with 5 tandem repeats of the yeast Gal4 binding sites; (2) the reporter plasmid pRL-CMV (Promega, E2261), which encodes Renilla luciferase (RLuc) cDNA and serves to control for variations in the efficiencies of transfection and protein recovery; (3) an effector plasmid that expresses the GAL4-DNA binding domain (GAL4-BD; pM) or GAL4-BD-fused effector protein; (4) an effector plasmid that expresses the herpes simplex virus VP16 transcription activation domain only (VP16-AD; pVP16) or the VP16-AD-fused effector protein. The interaction between TP53 and T antigen was used as a positive control. Two days after transfection, cells were harvested and dual luciferase activities were measured using the Luciferase assay kit (Promega, E1960).
Fluorescence analysis of autophagy
The full-length human TECPR1 gene containing a C-terminal 3×Flag tag was cloned into the pcDNA6.1 vector using the HindIII and XhoI restriction sites. The TECPR1 mutants, I582W, L586W, R589D, and A603F, were generated by PCR mediated site-directed mutagenesis. COS-7 cells were cultured under a humidified atmosphere of 5% (v/v) CO2 at 37°C in Dulbecco's modified Eagle's medium (Gibco, 12100-046) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, 12483-020). For DNA transfection, the cells were transfected for 2 d with appropriate vectors using polyethyleneimine (Sigma-Aldrich, 764582). For fluorescence analyses, COS-7 cells were transduced for 24 h with GFP-LC3 according to the manufacturer's protocol (Premo Autophagy Sensor [LC3B-FP] system [Invitrogen, P36235]). For autophagy induction, 500 nM rapamycin (Sigma-Aldrich, R0395) was added in the cells. The cells were fixed, permeabilized, stained with DAPI (5 μg/ml), and then examined by fluorescence microscopy. Fluorescence images were acquired with an Olympus BX53 fluorescence microscope equipped with an Olympus DP70 digital camera. The fluorescent intensity of GFP-LC3 in the perinuclear region of >50 cells in each group was quantified using Image Gauge V4.0 software.
Electron microscopy
After DNA transfection, the cells were fixed with 2.5% (v/v) glutaraldehyde (Ted Pella, 18426) dissolved in 150 mM cacodylate buffer (Ted Pella, 18851) and then exposed for 1 h at 4°C to 2% (w/v) osmium tetroxide (Ted Pella, 18459) in the same buffer. En bloc staining was then performed with 2% (w/v) uranyl acetate (Ted Pella, 19481) aqueous solution. The cells were dehydrated in an ethanol series, infiltrated with Spurr's resin (PELCO, 18300-4221) series, and polymerized at 60°C for 8 h. The embedded cells were cut with a diamond knife on an ultramicrotome (MTX-L, RMC). The sections were mounted directly on 150 mesh copper grids (Gilder, G200) and stained with 2% (w/v) uranyl acetate in 50% (v/v) methanol for 20 min and Reynold's lead citrate for 10 min. The grids were examined with a Tecnai F20 (FEI, Netherlands) at 200 kV.
SPR analysis
Molecular interactions between ATG5 and peptides derived from ATG16L1 and TECPR1 were evaluated using SPR spectrometry (SR7500DC system, Reichert Analytical Instrument). Purified ATG5 was immobilized on a carboxymethyl dextran sensor chip as a ligand. A 0.2 mg/ml concentration of ATG5 was applied over the biochip for 3 min (30 μl/min) to enable binding. Subsequently, 2 different washing solutions (10 mM PBS, pH 7.4[10 mM phosphate buffer, pH 6.0, 150 mM NaCl] and 0.5 mM TCEP) were applied to the biochip for an additional 3 min (30 μl/min) for molecular dissociation analysis. The association and dissociation rate constants were determined in 2 different pH buffers by fitting the binding profiles with the Scrubber 2.0 software provided by the manufacturer.
Accession codes
Atomic coordinates and structure factor files have been deposited in the Protein Data Bank under the accession codes 4TQ0 for ATG5-ATG16N69 and 4TQ1 for ATG5-TECAIR complex, respectively.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplemental data for this article can be accessed on the publisher's website.
Supplemental_Files.pdf
Download PDF (1.4 MB)Acknowledgments
We thank the staff at NW12 beamline, Photon Factory, Japan and at 5C beamline, Pohang Accelerator Laboratory, Korea and Qing Zhong (UC Berkeley) for providing the full-length TECPR1 gene.
Funding
This work was supported by National Research Foundation of Korea (NRF) grants from the Korean government (MEST) (NRF-2011-0028168 to H.K.S.; NRF-2012-Global Ph.D. Fellowship to J.H.K.).
References
- Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci 2005; 118:7-18; PMID:15615779; http://dx.doi.org/10.1242/jcs.01620
- Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr 2007; 27:19-40; PMID:17311494; http://dx.doi.org/10.1146/annurev.nutr.27.061406.093749
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Yorimitsu T, Klionsky DJ. Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell Biol 2007; 17:279-85; PMID:17481899; http://dx.doi.org/10.1016/j.tcb.2007.04.005
- Massey AC, Zhang C, Cuervo AM. Chaperone-mediated autophagy in aging and disease. Curr Top Dev Biol 2006; 73:205-35; PMID:16782460; http://dx.doi.org/10.1016/S0070-2153(05)73007-6
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14; PMID:21179058; http://dx.doi.org/10.1038/nrm3028
- Kim BW, Hong SB, Kim JH, Kwon do H, Song HK. Structural basis for recognition of autophagic receptor NDP52 by the sugar receptor galectin-8. Nat Comms 2013; 4:1613; PMID:23511477; http://dx.doi.org/10.1038/ncomms2606
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012; 482:414-8; PMID:22246324; http://dx.doi.org/10.1038/nature10744
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458-67; PMID:19491929; http://dx.doi.org/10.1038/nrm2708
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792-802; PMID:20418806; http://dx.doi.org/10.1038/emboj.2010.74
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859-64; PMID:18704115; http://dx.doi.org/10.1038/embor.2008.163
- Sakoh-Nakatogawa M, Matoba K, Asai E, Kirisako H, Ishii J, Noda NN, Inagaki F, Nakatogawa H, Ohsumi Y. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol 2013; 20:433-9; PMID:23503366; http://dx.doi.org/10.1038/nsmb.2527
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; http://dx.doi.org/10.1242/jcs.01131
- Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165-78; PMID:17632063; http://dx.doi.org/10.1016/j.cell.2007.05.021
- Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 1999; 18:3888-96; PMID:10406794; http://dx.doi.org/10.1093/emboj/18.14.3888
- Mizushima N, Yoshimori T, Ohsumi Y. Role of the Apg12 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2003; 35:553-61; PMID:12672448; http://dx.doi.org/10.1016/S1357-2725(02)00343-6
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679-88; PMID:12665549; http://dx.doi.org/10.1242/jcs.00381
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092-100; PMID:18321988; http://dx.doi.org/10.1091/mbc.E07-12-1257
- Fujita N, Saitoh T, Kageyama S, Akira S, Noda T, Yoshimori T. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J Biol Chem 2009; 284:32602-9; PMID:19783656; http://dx.doi.org/10.1074/jbc.M109.037671
- Chen D, Fan W, Lu Y, Ding X, Chen S, Zhong Q. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol Cell 2012; 45:629-41; PMID:22342342; http://dx.doi.org/10.1016/j.molcel.2011.12.036
- Chen D, Zhong Q. A tethering coherent protein in autophagosome maturation. Autophagy 2012; 8:985-6; PMID:22617511; http://dx.doi.org/10.4161/auto.20255
- Ogawa M, Yoshikawa Y, Kobayashi T, Mimuro H, Fukumatsu M, Kiga K, Piao Z, Ashida H, Yoshida M, Kakuta S, et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe 2011; 9:376-89; PMID:21575909; http://dx.doi.org/10.1016/j.chom.2011.04.010
- Ogawa M, Sasakawa C. The role of Tecpr1 in selective autophagy as a cargo receptor. Autophagy 2011; 7:1389-91; PMID:21795850; http://dx.doi.org/10.4161/auto.7.11.17151
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomeslysosomes. Cell 2012; 151:1256-69; PMID:23217709; http://dx.doi.org/10.1016/j.cell.2012.11.001
- Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12∼ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 2013; 20:59-66; PMID:23202584; http://dx.doi.org/10.1038/nsmb.2431
- Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F. Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem 2007; 282:6763-72; PMID:17192262
- Jeong YJ, Jeong BC, Song HK. Crystal structure of ubiquitin-like small archaeal modifier protein 1 (SAMP1) from Haloferax volcanii. Biochem Biophys Res Commun 2011; 405:112-7; PMID:21216237; http://dx.doi.org/10.1016/j.bbrc.2011.01.004
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem 2012; 81:203-29; PMID:22524316; http://dx.doi.org/10.1146/annurev-biochem-060310-170328
- Falini G, Fermani S, Tosi G, Arnesano F, Natile G. Structural probing of Zn(II), Cd(II) and Hg(II) binding to human ubiquitin. Chem Commun 2008:5960-2; PMID:19030552; http://dx.doi.org/10.1039/b813463d
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res 2010; 38:W545-9; PMID:20457744; http://dx.doi.org/10.1093/nar/gkq366
- Changela A, Chen K, Xue Y, Holschen J, Outten CE, O’Halloran TV, Mondragon A. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science 2003; 301:1383-7; PMID:12958362; http://dx.doi.org/10.1126/science.1085950
- Fujioka Y, Noda NN, Nakatogawa H, Ohsumi Y, Inagaki F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem 2010; 285:1508-15; PMID:19889643; http://dx.doi.org/10.1074/jbc.M109.053520
- Parkhouse R, Ebong IO, Robinson CV, Monie TP. The N-terminal region of the human autophagy protein ATG16L1 contains a domain that folds into a helical structure consistent with formation of a coiled-coil. PLoS One 2013; 8:e76237; PMID:24086718; http://dx.doi.org/10.1371/journal.pone.0076237
- Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol 2007; 372:774-97; PMID:17681537; http://dx.doi.org/10.1016/j.jmb.2007.05.022
- Noda NN, Ohsumi Y, Inagaki F. ATG systems from the protein structural point of view. Chem Rev 2009; 109:1587-98; PMID:19236009; http://dx.doi.org/10.1021/cr800459r
- Metlagel Z, Otomo C, Takaesu G, Otomo T. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc Natl Acad Sci U S A 2013; 110:18844-9; PMID:24191030; http://dx.doi.org/10.1073/pnas.1314755110
- Hong SB, Kim BW, Lee KE, Kim SW, Jeon H, Kim J, Song HK. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol 2011; 18:1323-30; PMID:22056771; http://dx.doi.org/10.1038/nsmb.2165
- Noda NN, Satoo K, Fujioka Y, Kumeta H, Ogura K, Nakatogawa H, Ohsumi Y, Inagaki F. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol Cell 2011; 44:462-75; PMID:22055191; http://dx.doi.org/10.1016/j.molcel.2011.08.035
- Taherbhoy AM, Tait SW, Kaiser SE, Williams AH, Deng A, Nourse A, Hammel M, Kurinov I, Rock CO, Green DR, et al. Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol Cell 2011; 44:451-61; PMID:22055190; http://dx.doi.org/10.1016/j.molcel.2011.08.034
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298-302; PMID:17986448; http://dx.doi.org/10.1074/jbc.C700195200
- Hong SB, Kim BW, Kim JH, Song HK. Structure of the autophagic E2 enzyme Atg10. Acta Crystallogr Sect D Biol Crystallogr 2012; 68:1409-17; PMID:22993095; http://dx.doi.org/10.1107/S0907444912034166
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107-32; PMID:21801009; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005
- Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 2014; 20:460-73; PMID:23725295; http://dx.doi.org/10.1089/ars.2013.5371
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; http://dx.doi.org/10.1038/nature09204
- Zhao Z, Zhang Z, Li Y, Zhou M, Li X, Yu B, Wang R. Probing the key interactions between human Atg5 and Atg16 proteins: a prospective application of molecular modeling. ChemMedChem 2013; 8:1270-5; PMID:23804289; http://dx.doi.org/10.1002/cmdc.201300256
- Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994; 371:37-43; PMID:8072525; http://dx.doi.org/10.1038/371037a0
- Zhou Y, Wu C, Zhao L, Huang N. Exploring the early stages of the pH-induced conformational change of influenza hemagglutinin. Proteins 2014; 82:2412-28; PMID:24854389; http://dx.doi.org/10.1002/prot.24606
- Noda NN, Fujioka Y, Hanada T, Ohsumi Y, Inagaki F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep 2013; 14:206-11; PMID:23238393; http://dx.doi.org/10.1038/embor.2012.208
- Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 2002; 277:18619-25; PMID:11897782; http://dx.doi.org/10.1074/jbc.M111889200
- Bolanos-Garcia VM, Chayen NE. New directions in conventional methods of protein crystallization. Prog Biophys Mol Biol 2009; 101:3-12; PMID:20018205; http://dx.doi.org/10.1016/j.pbiomolbio.2009.12.006
- Minor W, Tomchick D, Otwinowski Z. Strategies for macromolecular synchrotron crystallography. Structure 2000; 8:R105-10; PMID:10801499; http://dx.doi.org/10.1016/S0969-2126(00)00139-8
- Yamaguchi M, Noda NN, Yamamoto H, Shima T, Kumeta H, Kobashigawa Y, Akada R, Ohsumi Y, Inagaki F. Structural insights into Atg10-mediated formation of the autophagy-essential Atg12-Atg5 conjugate. Structure 2012; 20:1244-54; PMID:22682742; http://dx.doi.org/10.1016/j.str.2012.04.018
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr 2007; 40:658-74; PMID:19461840; http://dx.doi.org/10.1107/S0021889807021206
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr Sect D Biol Crystallogr 2012; 68:352-67; PMID:22505256; http://dx.doi.org/10.1107/S0907444912001308
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr 2004; 60:2126-32; PMID:15572765; http://dx.doi.org/10.1107/S0907444904019158
- Luo Y, Batalao A, Zhou H, Zhu L. Mammalian two-hybrid system: a complementary approach to the yeast two-hybrid system. BioTechniques 1997; 22:350-2; PMID:9043710
- Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, Martens S. Mechanism and functions of membrane binding by the Atg5-Atg12Atg16 complex during autophagosome formation. EMBO J 2012; 31:4304-17; PMID:23064152; http://dx.doi.org/10.1038/emboj.2012.278
- Kaufmann A, Beier V, Franquelim HG, Wollert T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell 2014; 156:469-81; PMID:24485455; http://dx.doi.org/10.1016/j.cell.2013.12.022