
Abstract
P2RX7 is an ATP-gated ion channel, which can also exhibit an open state with a considerably wider permeation. However, the functional significance of the movement of molecules through the large pore (LP) and the intracellular signaling events involved are not known. Here, analyzing the consequences of P2RX7 activation in primary myoblasts and myotubes from the Dmdmdx mouse model of Duchenne muscular dystrophy, we found ATP-induced P2RX7-dependent autophagic flux, leading to CASP3-CASP7-independent cell death. P2RX7-evoked autophagy was triggered by LP formation but not Ca2+ influx or MAPK1-MAPK3 phosphorylation, 2 canonical P2RX7-evoked signals. Phosphoproteomics, protein expression inference and signaling pathway prediction analysis of P2RX7 signaling mediators pointed to HSPA2 and HSP90 proteins. Indeed, specific HSP90 inhibitors prevented LP formation, LC3-II accumulation, and cell death in myoblasts and myotubes but not in macrophages. Pharmacological blockade or genetic ablation of p2rx7 also proved protective against ATP-induced death of muscle cells, as did inhibition of autophagy with 3-MA. The functional significance of the P2RX7 LP is one of the great unknowns of purinergic signaling. Our data demonstrate a novel outcome—autophagy—and show that molecules entering through the LP can be targeted to phagophores. Moreover, we show that in muscles but not in macrophages, autophagy is needed for the formation of this LP. Given that P2RX7-dependent LP and HSP90 are critically interacting in the ATP-evoked autophagic death of dystrophic muscles, treatments targeting this axis could be of therapeutic benefit in this debilitating and incurable form of muscular dystrophy.
Abbreviations:
- 3-MA, 3-methyladenine
- ACTB, actin, β
- BECN1, Beclin 1, autophagy-related
- BzATP, 2′(3′)-O-(4-benzoylbenzoyl)adenosine 5′-triphosphate
- CASP, caspase
- DAPC, dystrophin associated protein complex
- DMD, Duchenne muscular dystrophy
- Dmdmdx, C57BL/10ScSn-Dmdmdx/J mouse model of DMD
- Dmdmdx p2rx7−/− double-mutant mouse model
- eATP, extracellular ATP
- EtBr, ethidium bromide
- GA, geldanamycin
- HSP90, heat shock protein 90
- HSPA2/HSP70, heat shock protein 2
- LDH, lactate dehydrogenase
- LP, large pore, P2RX7-dependent
- LY, Lucifer Yellow
- MAP1LC3B/LC3, microtubule-associated protein 1 light chain 3 β
- MAPK, mitogen-activated protein kinase
- P2RX7, purinergic receptor P2X, ligand-gated ion channel, 7
- PtdIns3K, phosphatidylinositol 3-kinase, class III
- Wt, C57BL/10ScSn wild-type mouse
Introduction
Duchenne muscular dystrophy is a severe and incurable inherited muscle disorder. It is characterized by a debilitating muscle wasting caused by altered mechanical stability of muscle cell membranes and altered signaling combined with inflammatory infiltrations (For a review see ref.Citation1). The membrane instability and signaling abnormalities are caused by the absence of DMD/dystrophin, the protein product of the mutant DMD gene. DMD orchestrates formation and function of the DMD-associated protein complex (DAPC), which links the cytoskeleton with the extracellular matrix and also anchors various signaling proteins. DAPC is lost from the dystrophic sarcolemma. Inflammatory cell infiltrations in Duchenne muscular dystrophy muscles are triggered by the danger-associated molecular patterns released as a result of sarcolemmal damage. Extracellular ATP (eATP) functions as one such endogenous danger signal operating through purinergic P2 receptors.Citation2 Cytoplasmic ATP levels in skeletal muscles exist at particularly high concentrations (5 to 10 mM).Citation3 When released in the dystrophic tissue, eATP is less efficiently eliminated because one of the lost DAPC members, SGCA (sarcoglycan, α [dystrophin-associated glycoprotein]), is an ATP-hydrolase.Citation4 Clearly, the environment of dystrophic muscles favors overactivation of P2 purinoceptors and this can be amplified by upregulated expression and function of P2RX7 (purinergic receptor P2X, ligand-gated ion channel, 7) directly in dystrophic Dmdmdx mouse myoblasts and myofibers.Citation5
P2RX7 is the predominant purinoceptor involved in eATP danger signaling: It is fully activated by significantly higher eATP concentrations than any other P2X receptor, at levels which normally exist in damaged tissue only. P2RX7 is an ATP-gated ion channel, activation of which triggers Ca2+ influx and MAPK1-MAPK3 (mitogen-activated protein kinase 1/3) phosphorylation. Additionally, in response to prolonged, high eATP stimulation, P2RX7 can exhibit a further open state with a considerably wider permeation to molecules of up to 900 Da that may be associated with cell death by apoptosis or necrosis.Citation6,7 P2RX7 activation has recently been shown to induce autophagy in various cell types.Citation8-11 Moreover, the latest studies have revealed that whereas chronic, high-level activation is cytotoxic to cells,Citation12 the low-level P2RX7 stimulation can provide metabolic advantages.Citation13 Despite their obvious functional implications, the exact permeation pathways through LPs, the physiological significance of the movement of large molecules across the membranes as well as the intracellular signaling cascades involved are not fully known and may differ in various cell types.
While studied extensively in immune cells, the significance of P2RX7 activation in skeletal muscles is largely unknown. At low eATP concentrations P2RX7 activation appears to affect proliferation and differentiation of myoblastsCitation14,15 while high eATP levels have been shown to be toxic to these cells.Citation14 Therefore, abnormalities in P2RX7 purinergic signaling found in dystrophic myoblasts and myotubes may have significant functional consequences. Indeed, in the mouse model for deficiency of DYSF/LGMD2B (dysferlin), the increased P2RX7 expression has been linked to the NLRP3 (NLR family, pyrin domain containing 3) inflammasome upregulation typical for the inflammatory response.Citation16 We have therefore set out to analyze the mechanism and effects of activation of P2RX7 in dystrophin-deficient myoblasts and myofibers. We show here that activation of P2RX7 on dystrophic myoblasts and myotubes resulted in the formation of LPs in cell membranes, autophagic flux, and cell death but not in apoptosis.
Macroautophagy (referred to as autophagy) is a highly conserved mechanism by which long-lived cellular constituents, organelles, and debris are sequestered within autophagosomes and targeted for lysosomal hydrolysis and subsequent reuse. This may occur in adaptation to stress stimuli such as nutrient deprivationCitation17 or as a housekeeping method of maintaining cellular homeostasis.Citation18 Autophagy plays an essential role in normal muscle function, controlling muscle mass,Citation19 adaptation to exercise,Citation20 and regulation of glucose metabolism.Citation21 Malfunctioning skeletal muscle autophagy therefore has severe consequences and has been implicated in various muscle diseases, including DMD (For a review see refs.Citation22,23).
The emerging roles for both autophagy and P2RX7 in muscle lead us to examine the involvement of P2RX7 in the autophagic pathway in diseased muscle. Here we demonstrate that, in muscles, HSP90 (heat shock protein 90) and HSPA2/HSP70 (heat shock protein 2) link P2RX7 LP formation to autophagy and that the canonical mechanisms of P2RX7 activation such as Ca2+ influx and MAPK1-MAPK3 phosphorylation are not essential for these effects. This work represents a novel mechanism of coordination of muscle cell homeostasis by extracellular nucleotides and is also the first demonstration that the mechanism of P2RX7-induced autophagy operates via heat shock proteins. This previously unrecognized pathway, in which autophagy stimulation through P2RX7 LP formation contributes to muscle disease, integrates previous separate observations that P2RX7Citation8 and HSP90Citation24 mediate autophagy induction and that manipulations of P2RX7,Citation5 heat shock proteins,Citation25 and autophagyCitation26 in dystrophic muscles can modulate the severity of this debilitating and lethal disease.
Results
P2RX7 activation triggers lytic pore formation and cell death in dystrophic myoblasts
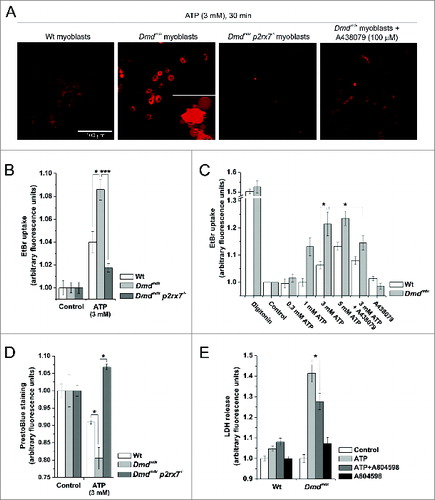
In cells expressing P2RX7 such as macrophages and dendritic cells, formation of LPs permeable to dyes such as ethidium bromide (EtBr) and cell death via apoptosis or necrosis are well-documented responses to prolonged stimulation with high concentrations of eATP.Citation27 When cultured as primary cells without serum and assayed under conditions favoring LP formation in macrophages,Citation28 myoblasts displayed dye uptake, membrane blebbing, cell shrinkage, and death consistent with the classical P2RX7 response usually found in macrophages ( and Video S1). At 3 mM ATP, EtBr uptake over a 30-min time course was significantly higher in dystrophic than wild-type (Wt) myoblasts, with no significant increase in fluorescence observed in Dmdmdx p2rx7−/− double-mutant myoblasts, demonstrating that this effect was P2RX7-dependent (). Pretreatment with 100 nM A438079 (specific P2RX7 antagonist) significantly reduced EtBr uptake in dystrophic and Wt myoblasts (). Subsequently, eATP-evoked cell death was evaluated using PrestoBlue cell viability assay and LDH release membrane integrity assay: eATP-mediated cell death was significantly enhanced in dystrophic vs Wt myoblasts and these effects were attenuated through P2rx7 ablation in Dmdmdx p2rx7−/− myoblasts or receptor antagonism using A804598 (). Interestingly, the mechanism of LP formation does differ between myoblasts and macrophages, which may reflect differences in P2RX7 expression levels between these cell types (see below), also explaining our previous inability to demonstrate P2RX7-dependent LP formation in myoblasts.Citation5
Figure 1. P2RX7 activation induces LP formation and cell death in dystrophic myoblasts. (A) Examples of EtBr fluorescence in wild-type (Wt) and dystrophic Dmdmdx myoblasts following 30 min exposure to 3 mM eATP in LP buffer (left panels). Right panels show EtBr uptake following the same treatment in myoblasts isolated from Dmdmdx p2rx7−/− double-mutant mice and Dmdmdx myoblasts after inhibition of P2RX7 by 30 min preincubation with 100 μM A438079. Inset in Dmdmdx panel shows classic ‘macrophage-like’ membrane blebbing following exposure of Dmdmdx myoblasts to eATP. (B) Summary data showing EtBr uptake dependence on P2RX7 expression in Wt, Dmdmdx and Dmdmdx p2rx7−/− myoblasts exposed to 3 mM eATP for 30 min (C). eATP dose response of EtBr uptake in Wt and Dmdmdx myoblasts exposed to indicated concentrations of ATP for 30 min, with or without preincubation with 100 μM A438079. Digitonin represents permeabilized positive control. (D) PrestoBlue fluorescence (cell viability) in Wt, Dmdmdx, and Dmdmdx p2rx7−/− myoblasts following 30 min exposure to 3 mM eATP. (E) LDH release from Wt, Dmdmdx, and Dmdmdx p2rx7−/− myoblasts following 30 min exposure to 3 mM eATP with or without the P2RX7-specific inhibitor, A804598 (100 nM). Mean +/− SE, n = 5, P < 0 .05* and 0.0001***.
P2RX7-dependent LP formation and autophagy induction are linked in dystrophic myoblasts
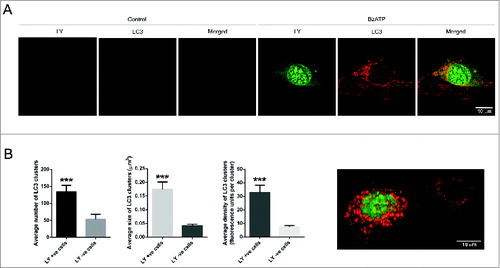
Phosphoproteomic analyses in dystrophic myoblasts indicated autophagy as a potential functional consequence of P2RX7 stimulation. To investigate whether the observed LP formation and cell death following eATP application were associated with autophagy, primary Dmdmdx myoblasts were exposed to 1 mM BzATP (a more selective P2RX7 agonist) for 25 min in the presence of 3 mM Lucifer Yellow (LY), an anionic marker of P2RX7-dependent LP formation. Significant increases in LY uptake observed in BzATP-treated dystrophic myoblasts indicated that both anionic and cationic pore pathwaysCitation29 are active in dystrophic myoblasts (). Immunolocalization of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), which is recruited to the membrane of phagophores (reviewed in ref.Citation30), revealed that cells displaying LP permeability had significantly increased number, size, and density of clusters containing both LY and LC3 (), indicating a previously unidentified link between LP formation and autophagy induction during the early stages of P2RX7-induced autophagy, prior to the initiation of autophagic cell death.
Figure 2. P2RX7-dependent LP formation is linked to autophagy induction in Dmdmdx myoblasts. (A) Representative images showing P2RX7-dependent increases in Lucifer Yellow uptake (LY, green) and autophagy induction (LC3 aggregation, red) in Dmdmdx myoblasts exposed to 1 mM BzATP. LY uptake was observed in cells displaying high levels of autophagy, where dye colocalized with LC3 immunofluorescence in distinct clusters. (B) ImageJ quantification of LC3 clusters number, size, and density in LY-positive and LY-negative cells using 3D compilations of z-stacked confocal images, with an example image shown. Mean +/- SE, n = 10, P = 0.0001***.
P2RX7 activation induces LC3-II formation independently of MAPK1-MAPK3 pathway activation in dystrophic myoblasts
To further investigate this autophagy response, primary myoblasts from either wild-type or Dmdmdx mouse muscle were exposed to 3 mM ATP for up to 60 min in physiological extracellular solution (i.e., normal sodium, calcium, and magnesium) and cell lysates from different time points were analyzed for LC3-II, a sensitive marker of autophagy induction.Citation31,32 The time-course experiments showed that dystrophic myoblasts displayed significantly heightened sensitivity to eATP-evoked LC3-II production compared to wild-type cells, with induction occurring 10 to 30 min following stimulation (). The use of Dmdmdx p2rx7−/− double-mutant myoblasts confirmed the LC3-II response to be P2RX7-dependent with no LC3-II being triggered in these cells following exposure to P2RX7 agonist BzATP (1 mM) (). Moreover, preincubation of dystrophic myoblasts with P2RX7 antagonist Brilliant Blue G (BBG, 10 μM) prior to stimulation with BzATP (1 mM) resulted in a significant reduction in LC3-II generation (, upper panels and ). LC3-II formation coincided with the latter half of the biphasic phospho-MAPK1-MAPK3 response we previously described in Dmdmdx myoblasts following P2RX7 activation.Citation5 The MAPK1-MAPK3 activation pathway is a positive regulator of autophagy induction.Citation33 To investigate whether the P2RX7-dependent LC3-II response occurred via this signaling mechanism, cells were preincubated with 100 μM of U0126 (MAP2K1/2 inhibitor), prior to agonist application. The observed extent of reduction in LC3-II levels indicated that MAPK1-MAPK3 signaling could not be identified as the main pathway of autophagy activation in Dmdmdx muscle cells ( lower panels and ).
Figure 3. eATP induces P2RX7-dependent autophagy in myoblasts. (A) Representative western blots showing increased P2RX7 expression levels in dystrophic (Dmdmdx) vs. wild-type (Wt) myoblasts, with a time-course of MAPK1-MAPK3 phosphorylation and autophagy induction (LC3-I to LC3-II shift) following exposure to 3 mM eATP. No detectable changes in mitophagy were observed as shown by COX4 expression levels. ACTB represents protein loading control. (B) Western blots of triplicate samples showing the dependency of autophagy induction on P2RX7 expression in myoblasts; note the lack of 17 to 14 kDa LC3 shift in Dmdmdx p2rx7−/- double-mutant myoblasts following 30 min exposure to 1 mM BzATP. (C) Western blots of triplicate samples showing different effects of inhibiting P2RX7 activation and MAPK1-MAPK3 phosphorylation on autophagy induction in Wt (left panel) and Dmdmdx (right panel) myoblasts. Cells were preincubated for 30 min with the P2RX7 antagonist Coomassie brilliant blue-G (BBG) or U0126 (MEK inhibitor) prior to 30 min 1 mM BzATP treatment. The classical P2RX7-MAPK1-MAPK3 activation cascade is involved in, but does not appear to be the sole pathway of autophagy induction in dystrophic myoblasts. Note: The phospho-MAPK1-MAPK3 is a multichannel (green/red) fluorescent blot, where yellow signal denotes increased MAPK1-MAPK3 phosphorylation. The left-hand lane in the Wt blot is a positive control. (D) Fold change in LC3-II levels in response to treatments shown in (C). Mean +/- SE, n =3, P < 0 .05* and 0.0001***.
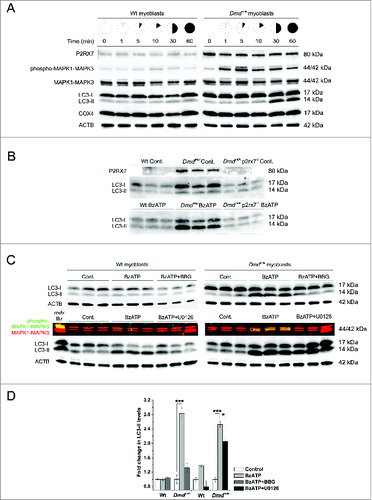
No noticeable mitophagy was observed under these conditions, as assessed using the COX4I1 (cytochrome c oxidase subunit IV isoform 1) markerCitation34 (). However, mitochondria shrank and retracted to a perinuclear localization following 30 min application of eATP (Fig. S1C), which coincided with an increase in reactive oxygen species production (Figs. S1A and B) and loss of mitochondrial membrane potential (see below). This was in agreement with our previous observations that DMD gene mutations affect energy metabolism in myoblasts.Citation35
P2RX7-mediated LC3-II induction occurs via increased autophagic flux in dystrophic myoblasts
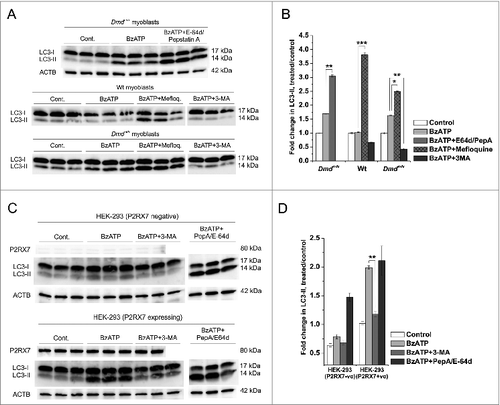
The induction of autophagy is a self-regulating process seeking equilibrium. Levels of LC3-II and therefore autophagosomes may decrease through lysosomal fusion at the same time as they are increased through stimulus, hence the turnover rather than simply the endogenous level of LC3-II is seen as a marker for autophagy induction.Citation36 The term autophagic flux is used to define the criteria for which a true increase in autophagy may be separated from a cessation of lysosomal activity or autophagosome-lysosome fusion.Citation37 Flux may be distinguished from accumulation using autophagy inhibitors and lysosomal protease inhibitors, where an increase in autophagic flux should be accompanied by an additional increase in LC3-II accumulation in the presence of lysosomal protease inhibitors.Citation32,38 In Dmdmdx myoblasts, the combination of lysosomal protease inhibitors, pepstatin A (PepA) and E-64d (both at 10 μg/ml) in conjunction with 1 mM BzATP stimulation produced significant increases in LC3-II accumulation when compared to 1 mM BzATP alone (, upper panel and ). Substituting PepA/E-64d with 1 μM mefloquine hydrochloride (Mefloq., another agent that blocks lysosomal proteases) also resulted in an increase in LC3-II accumulation in myoblasts (, lower panels and ). Preincubation with 5 mM 3-MA (autophagy inhibitor) abolished BzATP-evoked LC3-II production. As 3-MA is a class III phosphatidylinositol 3-kinase (PtdIns3K) inhibitor, this result indicates that P2RX7-mediated increases in autophagic flux in dystrophic myoblasts may occur via PtdIns3K-mediated pathways.
Figure 4. P2RX7 activation induces autophagic flux in dystrophic myoblasts. (A) Representative protein gel blots of triplicate samples showing LC3-II responses in dystrophic (Dmdmdx) and wild-type (Wt) myoblasts. LC3-II formation invoked following 30 min treatment with 1 mM BzATP was blocked by the autophagy inhibitor 3-methyladenine (3-MA), while lysosomal protease inhibitors E-64d/pepstatin A and mefloquine (Mefloq.), which prevent the degradation of autophagosomes, further enhanced LC3-II accumulation following BzATP treatment. (B) Fold changes of LC3-II conversion as a marker of autophagosome formation. A significant compound increase in LC3-II was observed in cells treated with 1 mM BzATP + E-64d/pepstatin A or mefloquine indicating that specific P2RX7 activation induces the formation of autophagosomes rather than blocks their degradation. Significant decreases in LC3-II observed in cells pretreated with 3-MA may indicate that P2RX7-dependent autophagosome formation occurs via a PtdIns3K-dependent mechanism. (C) P2RX7 expression confers eATP-dependent autophagic flux response to HEK-293 cells. Note no LC3-II response in untransfected (HEK-293-P2RX7 negative, upper panel) and very significant activation in P2RX7-transfected cells (HEK-293-P2RX7 expressing, lower panel) following 30 min treatment with 1 mM BzATP. This effect was blocked by autophagy inhibitor 3-MA (5 mM) and increased by lysosomal protease inhibitors PepA and E-64d (both at 10 μg/ml). (D) Fold changes of LC3-II responses observed in BzATP-treated HEK cells from (C). Mean +/- SE, n = 3, P < 0 .05*, 0.001** and 0.0001***.
To investigate the potential involvement of the recently described autophagic cell death pathway called autosis,Citation39 digoxin and digitoxigenin (both 10 μM) were used. These compounds, at the concentration that specifically inhibits autosis, had no effect on eATP evoked autophagic flux, LP formation, or cell death (Figs. S2A and B). This lack of response to inhibitors corresponded with our other data showing features opposite of what was found in autosis. Specifically, P2RX7 activation caused autophagic flux and cell detachment observed within minutes following stimulus, while in autosis, cells showed no changes until several hours poststimulation at which stage they died displaying increased substrate adherence.Citation39
We further investigated the ability of P2RX7 to invoke eATP-induced autophagic flux by comparing LC3-II flux in HEK-293 with that of HEK-293 cells stably transfected with mouse P2RX7 cDNA. Here also, application of 1 mM BzATP resulted in significant increases in LC3-II in transfected compared with control HEK-293 cells and this effect was again blocked and augmented by the autophagy inhibitor 3-MA and protease inhibitors PepA and E-64d, respectively (). To our knowledge, these represent the first demonstrations that the P2RX7 can act as a positive regulator of autophagic flux.
Phosphoproteomic analysis of the early purinoceptor signaling response in dystrophic myoblasts
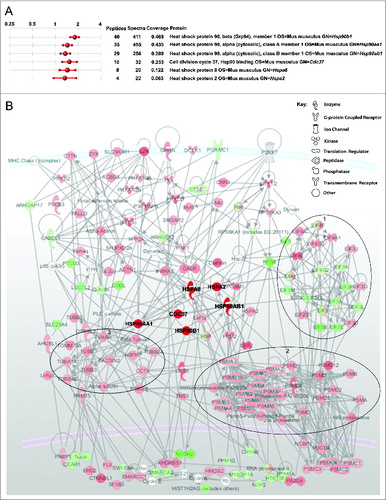
Putative P2RX7-evoked signaling cascades in dystrophic muscle cells were investigated using an iTraq-based mass spectrometry approach. Phosphoproteins extracted from wild-type and dystrophic myoblasts treated with 3 mM ATP for 10 min were analyzed using Spectrum Mill (Agilent) and inference values determined via the iQuantitator algorithm.Citation40 One thousand two hundred and seventy two unique phosphoproteins were identified, of which 263 showed significant differences in their phosphorylation status evoked by eATP. Pathway analysis was performed using MetaCore and IPA softwares. This approach identified significantly higher phosphorylation status in HSPA2, HSPA8 (heat shock protein 8), 3 HSP90 isoforms and in CDC37 (cell division cycle 37) members of the family of chaperone heat shock proteins in dystrophic myoblasts. HSPA2 and HSP90 involvement in autophagy has also been documentedCitation24,41 and, moreover, P2RX7 functionally interacts with these heat shock proteins as part of the P2RX7 signaling complex.Citation42 Therefore, having found heat shock proteins as potential downstream orchestrators of P2RX7 signaling in dystrophic vs wild-type myoblasts exposed to eATP () we investigated whether heat shock proteins mediate the observed P2RX7-dependent LC3-II response seen in dystrophic myoblasts.
Figure 5 (See previous page). Phosphoproteomic analysis of ATP treated dystrophic vs. normal myoblasts. (A) Sample of analysis report generated using iQuantitatorCitation40 showing fold increases in phosphoproteins detected in dystrophic vs normal myoblasts stimulated with 3 mM ATP for 10 min. Numbers of peptides and sequences detected are shown with percentage sequence coverage per protein. (B) IPA Path Designer software (Ingenuity) analysis of signaling cascade activation following 10 min 3 mM ATP treatment of dystrophic myoblasts. Molecules are represented as nodes relating to known functional class of molecule (see key); relationships between molecules (supported by at least one literature citation) are depicted as lines. The intensity of the molecular color denotes degree of up- (red) or downregulation (green). Heat shock proteins have been artificially highlighted here to aid interpretation of the illustration. Large oval outlines denote general clustered changes in phosphorylation status of related molecules: 1 = down regulation of translation, 2 = upregulation of proteasome, 3 = upregulation of tubulin rearrangements.
P2RX7-dependent LC3-II flux in dystrophic myoblasts requires large-pore formation and heat shock proteins but not calcium channel activity
Dystrophic muscle cells cultured in the presence of HSP90 inhibitors (75 nM geldanamycin [GA] or 75 mM 17-DMAG) or HSPA2 inhibitors (100 μM KNK437 or 100 nM VER155008) showed no LC3-II formation following P2RX7 stimulation with 1 mM BzATP (). This response was independent of calcium influx as it was not blocked by 10 μM BAPTA-AM (). In contrast, A438079 (antagonist of the P2RX7 LP formation)Citation43 completely inhibited BzATP-induced LC3-II flux ().
Figure 6. Heat shock proteins and LP formation mediate P2RX7-dependent LC3-II shift independently of calcium influx in dystrophic myoblasts. (A and B) Representative immunoblots of LC3-II and BECN1 levels in dystrophic myoblasts stimulated with 1 mM BzATP following preincubation with 2 sets of HSP90 and HSPA2 inhibitors, KNK437 and geldanamycin (A) and 17-DMAG and VER155008 (B). Graphs in (A) and (B) denote fold changes in LC3-II and BECN1 expression shown in adjacent blots (T/C = Treated/Control). P2RX7 channel activity and LP formation effects were assessed using BAPTA-AM (A) and A438079 (P2RX7 pore-specific antagonist, B), respectively. (C) Inhibition of HSP90 or HSP 70 caused a dose-dependent increase in BECN1 expression in dystrophic myoblasts. The Dmdmdx myoblasts were treated with indicated concentrations of GA or KNK437 for 16 h (left panel), or with 75 nM GA for indicated time periods (right panel) and then lysed. Nonlinear curve fitting function of GRAPHPAD Prism V6.01 was used to determine EC50 of 12.72 nM for GA. (D) Inhibition of HSP90 results in targeting of BECN1 to aggresomes in dystrophic myoblasts. The Dmdmdx myoblasts were stimulated with 1 mM BzATP with or without pretreatment with 75 nM GA, as indicated. Control cells showed puncta of BECN1 staining (green signal) separate from the Proteostat aggresome marker (red) while in cells pretreated with GA there was colocalization of BECN1 with aggresomes (yellow signal). Mean +/− SE, n = 3, P < 0 .05* and 0.001**.
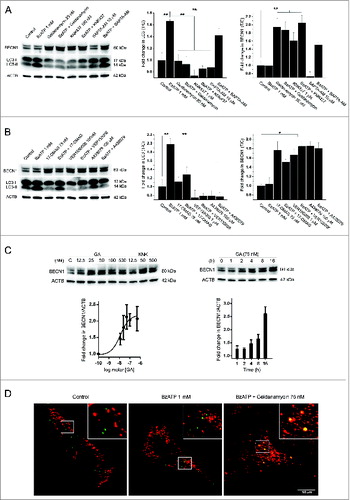
HSP90 has previously been shown to functionally interact with BECN1/Beclin 1 in macrophages, where GA treatment inhibits LC3-II responses through degradation of BECN1.Citation24 Therefore, we investigated the effect of the HSP inhibitors GA and KNK437 on BECN1 expression in dystrophic myoblasts. Contrary to findings in macrophages, where inhibition of HSP90 but not HSPA2 reduced BECN1 expression,Citation24 dystrophic myoblasts showed dose-dependent increases in BECN1 levels following HSP90 or HSPA2 inhibition, with EC50 12.72 nM for GA ().
This was unexpected as increases in BECN1 are normally associated with increased autophagy. Therefore, we performed an aggresome assay in cells treated with BzATP and GA to test whether, in the absence of the chaperone activity of HSP90, BECN1 might form aggregates. Indeed, clear colocalization of BECN1 with aggresomes was observed in cells pretreated with GA (), indicating that HSP90 inhibition in dystrophic myoblasts causes targeting to and accumulation of nonfunctional BECN1 in aggresomes.
P2RX7 LP formation in myoblasts is autophagy-dependent, HSP90-dependent, HSPA2-independent and a trigger for autophagy in myoblasts but apoptosis in macrophages
Preincubation with 5 mM 3-MA for 1 h prior to 30 min 3 mM ATP application blocked all EtBr uptake in myoblasts () and also significantly reduced LDH release (not shown). The HSP90 inhibitor GA, but not the HSPA2 inhibitor KNK437, prevented P2RX7 LP formation in myoblasts. In contrast, these effects were not observed in macrophages, indicating cell-specific responses ().
Figure 7. Mechanisms of eATP-induced LP formation and cell death differ between myoblasts and macrophages (MØs). (A) Images of EtBr fluorescence in Wt and Dmdmdx myoblasts and Wt MØs following 30 min exposure to 3 mM eATP with or without preincubation with the autophagy inhibitor 3-methyladenine (3-MA, 5 mM), the HSP90 inhibitor GA (75 nM) or the HSPA2 inhibitor KNK437 (100 nM). (B) EtBr uptake in Wt and Dmdmdx myoblasts following 30 min exposure to 3 mM eATP with and without 30 min preincubation with autophagy inhibitors 5 mM 3-MA, or 0.2 μM wortmannin (left panel), or following preincubation with HSPA2 inhibitors, KNK437 and VER155008, HSP90 inhibitors, geldanamycin (GA) and 17-DMAG, or the P2RX7 LP-specific antagonist A438079. Mean +/- SE, n = 3, P < 0 .05* and 0.001**. (C) Time-lapse live cell confocal microscopy-based visualization of mitochondrial membrane potential (red) and CASP3-CASP7 activity (green) in wild-type (Wt) and dystrophic (Dmdmdx) myoblasts and wild-type macrophages (MØs) exposed to 3 mM ATP for the indicated time. One μM Staurosporine treatment represents positive control for apoptosis. eATP induced cumulative increases in CASP3-CASP7 activity in MØs but not in myoblasts.
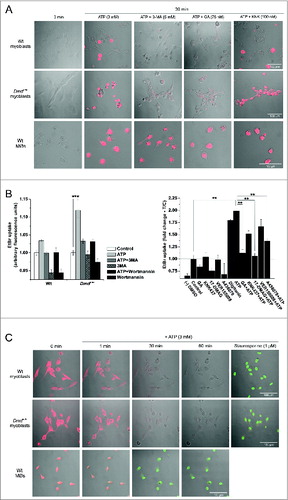
P2RX7 stimulation in macrophages induces loss of mitochondrial membrane potential and CASP3-CASP7 (caspase 3-caspase 7)-dependent cell death.Citation44 Therefore, we have examined whether, in myoblasts, the same process can coexist with autophagy. Time-lapse live cell images were collected during continuous incubations in tetramethylrhodamine ethyl ester perchlorate (TMRE, 200 nM) to monitor mitochondrial membrane potential and DEVD-NucView488 (5 μM) to monitor CASP3-CASP7 activity. Exposure to 3 mM eATP caused loss of mitochondrial membrane potential noticeable within 1 or 2 min in both macrophages and myoblasts. Thereafter, during the period of 10 to 30 min >80 % of macrophages displayed progressively increasing CASP3-CASP7 activity (), exactly as previously described.Citation44 In contrast, this CASP3-CASP7 response was not observed in either wild-type or dystrophic myoblasts (). No signal was observed in myoblasts even after 2 h incubation with eATP (data not shown). One μM staurosporine was sufficient to induce CASP3-CASP7 activation in >90 % of wild-type and dystrophic myoblasts within 2 h of addition (), confirming these cells capacity to undergo canonical apoptosis and indicating differences in P2RX7-induced cell death cascades between myoblasts and macrophages.
Dystrophic myotubes also respond to eATP with P2RX7-dependent LP formation, LC3-II induction, and cell death
Myoblasts, as the proliferating precursors of muscle cells, differ significantly in their biology and pathology from differentiated, multinucleated, contractile muscle-forming myotubes. Therefore, we sought to establish whether the effects of P2RX7 activation found in dystrophic myoblasts, are indeed retained by myotubes. Exposure to 3 mM eATP for 30 min induced significant EtBr fluorescence in wild-type and dystrophic myotubes, with dystrophic cells displaying increased responses (). The Dmdmdx p2rx7−/− myotubes did not show LP formation following eATP stimulation (, lower panels and , left panel), confirming eATP responses as P2RX7-specific. Significant reductions in EtBr fluorescence were observed when dystrophic myotube cultures were preincubated with 5 mM 3-MA (autophagy inhibitor), 75 nM GA (HSP90 inhibitor) () or 100 μM A438079 (). P2rx7 ablation also significantly reduced LDH release from myotubes following eATP treatment (, left panel), as did preincubation with 3-MA and A804598 (P2RX7 antagonist) (, right panel). Moreover, dystrophic myotubes displayed significantly increased BzATP-evoked LC3-II generation compared to wild-type cells, a response not seen in Dmdmdx p2rx7−/− myotubes ().
Figure 8. eATP induces P2RX7-mediated LP formation and autophagy in dystrophic myotubes. (A) EtBr fluorescence in Wt, Dmdmdx, and Dmdmdx p2rx7−/− myotubes following 30 min exposure to 3 mM eATP with and without preincubation with the autophagy inhibitor 3-MA (5 mM) or with the HSP90 inhibitor geldanamycin (GA, 75 nM). Note the absence of response in Dmdmdx p2rx7−/− myotubes lacking P2RX7. Digitonin (50 μg/ml) represents permeabilized positive controls. (B) Summary data representing EtBr uptake in Wt and Dmdmdx myotubes shown in (A). eATP-dependent increases in EtBr fluorescence were observed in both Wt and Dmdmdx myotubes with Dmdmdx displaying significantly higher uptake relative to Wt cells. In both Wt and Dmdmdx myotubes, preincubation with P2RX7 antagonist A438079 (100 μM), 3-MA (5 mM) or GA (75 nM) produced significant reductions in eATP-induced EtBr fluorescence. Three-MA alone blocked EtBr background signal in unstimulated cells. (C) Summary data of differences in LDH release from Wt, Dmdmdx and Dmdmdx p2rx7−/− myoblasts following 30 min exposure to 3 mM eATP, and Wt and Dmdmdx in the absence or presence of autophagy and P2RX7 inhibitors (3-MA [5 mM] and A804598 [100 nM], respectively). (D) Representative western blots showing P2RX7 and LC3-II levels in Wt, Dmdmdx and Dmdmdx p2rx7−/− double-mutant myotubes incubated for 30 min with 1 mM BzATP. Graph depicts fold change in LC3-II levels in Dmdmdx and Dmdmdx p2rx7−/− myotubes compared to Wt myotubes following 30 min exposure to 1 mM BzATP. Means +/− SE, n = 5, P < 0 .05*, 0.001** and 0.0001***.
![Figure 8. eATP induces P2RX7-mediated LP formation and autophagy in dystrophic myotubes. (A) EtBr fluorescence in Wt, Dmdmdx, and Dmdmdx p2rx7−/− myotubes following 30 min exposure to 3 mM eATP with and without preincubation with the autophagy inhibitor 3-MA (5 mM) or with the HSP90 inhibitor geldanamycin (GA, 75 nM). Note the absence of response in Dmdmdx p2rx7−/− myotubes lacking P2RX7. Digitonin (50 μg/ml) represents permeabilized positive controls. (B) Summary data representing EtBr uptake in Wt and Dmdmdx myotubes shown in (A). eATP-dependent increases in EtBr fluorescence were observed in both Wt and Dmdmdx myotubes with Dmdmdx displaying significantly higher uptake relative to Wt cells. In both Wt and Dmdmdx myotubes, preincubation with P2RX7 antagonist A438079 (100 μM), 3-MA (5 mM) or GA (75 nM) produced significant reductions in eATP-induced EtBr fluorescence. Three-MA alone blocked EtBr background signal in unstimulated cells. (C) Summary data of differences in LDH release from Wt, Dmdmdx and Dmdmdx p2rx7−/− myoblasts following 30 min exposure to 3 mM eATP, and Wt and Dmdmdx in the absence or presence of autophagy and P2RX7 inhibitors (3-MA [5 mM] and A804598 [100 nM], respectively). (D) Representative western blots showing P2RX7 and LC3-II levels in Wt, Dmdmdx and Dmdmdx p2rx7−/− double-mutant myotubes incubated for 30 min with 1 mM BzATP. Graph depicts fold change in LC3-II levels in Dmdmdx and Dmdmdx p2rx7−/− myotubes compared to Wt myotubes following 30 min exposure to 1 mM BzATP. Means +/− SE, n = 5, P < 0 .05*, 0.001** and 0.0001***.](/cms/asset/cffd9511-08b5-4143-8f54-ea072b7ecdce/kaup_a_994402_f0008_oc.jpg)
Discussion
Several studies, including our own, have previously shown increased P2RX7 expression and activity in dystrophic muscles of the Dmdmdx mouse model of DMD.Citation5,15,45 Here we document the functional significance of P2RX7 activation in muscle cells, which while having some resemblance to the canonical macrophage response, showed a very significant difference in cell death pathways. In contrast to macrophages, eATP triggered oxidative stress but no mitophagy nor apoptosis in myoblasts. Specifically, the absence of eATP-induced CASP3-CASP7 activationCitation44 in myoblasts and the lack of protection from eATP by autophagy inhibitors in macrophages, are the most striking. In mouse microgliaCitation8 and in human retinal pigmented epithelial cells,Citation46 P2RX7 signaling has been reported to negatively regulate autophagic flux through the impairment of lysosomal functions. Yet increases in flux have been reported in monocytes and their derived macrophages following P2RX7 activation during mycobacterial infection,Citation9 thus supporting our data and emphasizing cell-specific and functional differences in the way in which purinergic receptors can affect autophagy.
P2RX7 is an ATP-gated ion channel, activation of which triggers Ca2+ influx and MAPK1-MAPK3 phosphorylation but this receptor can exhibit a further open state with a considerably wider permeation,Citation7 allowing uptake of dyes such as EtBr and LY used in this study. Our data agrees with previous observations that LP formation can vary in different cells and we also confirmed that assay conditions play an important role.Citation47 Crucially, using specific antagonists we have shown that it is the LP formation that triggers autophagy in muscle cells. Moreover, LY entering through the LP was targeted to LC3-positive organelles, presumably autophagosomes. It is tempting to speculate that the same may apply to specific biomolecules. However, no physiologically-relevant compounds, which enter cells via the LP, have been identified so far. In addition, there was a reciprocal effect where LP formation in myoblasts but not macrophages was blocked by the autophagy inhibitor 3-MA. These differences may be related to the significantly lower levels of receptor expression in dystrophic myoblasts, which are <25 % of that found in the macrophage cell line J774 or the stably transfected HEK cells (Figs. S3A and B). However, a muscle-specific signaling mechanism should also be considered.
We investigated the mechanism involved in this previously unrecognized link between P2RX7 pore formation and autophagy using combinations of specific agonists and antagonists. First, we found that the autophagic response in dystrophic myoblasts does not require the extracellular Ca2+ influx associated with the ion channel function of the P2RX7. Furthermore, while the MAPK1-MAPK3 activation pathway plays some role, as shown by the partial inhibition of autophagic flux with U0126, this was not the most significant signaling pathway in autophagy triggered by P2RX7 stimulation. A recent study in tumor cells has identified a novel signaling axis following activation of P2RX7 that involved PRKA/Protein kinase, AMP-activated, AKT1S1/AKT1 substrate 1, and MTOR/mechanistic target of rapamycin.Citation11 Given the data showing autophagy stimulation via AMPK agonist and MTOR blocker administration to ameliorate Dmdmdx pathology,Citation48 the existence of such a pathway in muscles should be investigated.
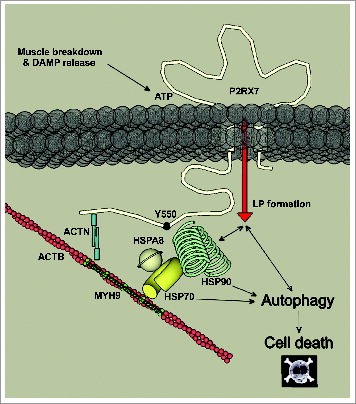
However, we focused our attention on a novel pathway discovered in proteomic analyses used to identify downstream orchestrators of P2RX7 signaling in dystrophic myoblasts. Proteomics pointed toward chaperone heat shock proteins and indeed, pharmacological inhibitors of HSPA2 and HSP90 blocked LC3-II formation in myoblasts exposed to eATP. When heterologously expressed in HEK cells, P2RX7 has been shown to physically interact with a large complex of proteins, including HSPA2 and HSP90Citation42 and these proteins are known to associate with each other via a protein called STIP1.Citation49 HSP90 associated with the P2RX7 complex is tyrosine-phosphorylated and decreasing tyrosine phosphorylation using geldanamycin produced increased sensitivity of P2RX7 to agonist and membrane blebbing, thus suggesting a nonchaperone function of HSP90 as a negative regulator of P2RX7.Citation50 Here we confirmed that this is a physiological interaction that also occurs in primary myoblasts and myotubes. Moreover, in muscle cells this interaction is associated with a novel downstream event in the plethora of pathways triggered by this receptor, namely signaling between the P2RX7 LP and autophagy (). P2RX7 has a long C terminus, distinguishing it from other receptors of the P2X family and which is essential in pore formation.Citation51 It would be interesting to establish whether this P2RX7-HSP90 is a direct interaction, in which case it is worthwhile identifying the specific P2RX7 domain(s) involved. Alterations in this binding region(s) may be responsible for differences in P2RX7 function in specific cells and may also have a role in pathologies. Previous studies show that P2RX7Y550F substitution is associated with an increase in the level of tyrosine phosphorylation of HSP90 but without affecting the amount of HSP90 protein interacting with the receptor.Citation50 This might indicate that P2RX7 interaction with heat shock proteins is indirect and it has been suggested that it may involve ACTB/Actin, β, which is present in the P2RX7 signaling complex.Citation42 However, ACTB disruption by cytochalasin D or latrunculin A does not alter LP functions.Citation52 Moreover, our finding that only HSP90 but not HSPA2 antagonist blocked P2RX7 pore formation, while both blocked autophagic flux triggered by P2RX7 activation points to functional specialization of these two HSP proteins in the P2RX7 signaling complex. Clearly, further studies should identify the exact interactions between proteins of this complex. These are likely to vary in different cells and this has important implications for understanding the function of P2RX7 and also for designing targeted pharmacological treatments.
Figure 9. Schematic depicting known interactions at the C terminus of P2RX7 involving ACTB, ACTN (actinin, α), MYH9 (myosin, heavy polypeptide 9, non-muscle), and heat shock proteins mediating autophagic cell death in dystrophic myoblasts following P2RX7 activation. HSPA2, HSPA8, and HSP90 have been shown to bind P2RX7 C-terminal tail, where Y550 residue plays an important role in mediating phosphorylation status of HSP90.Citation42,50 In myoblasts, P2RX7 activation following ATP released from damaged or degenerating muscles result in activation of HSP90-mediated LP formation and autophagic flux, culminating in CASP3-CASP7 independent cell death. Inhibition of HSP90 or of autophagy prevented LP formation in myoblasts, indicating involvement of HSP90-mediated autophagy in this process. Inhibition of HSPA2 resulted in inhibition of autophagy but not LP formation or cell death.
BECN1 is a component of multiple PtdIns3K complexes that are involved in autophagy and vacuolar protein sorting pathwaysCitation53 and interacts functionally with HSP90.Citation24 Therefore, we have investigated the effect of HSP inhibition on BECN1 expression. Contrary to findings in macrophages, where inhibition of HSP90 with GA reduced LC3-II responses through proteasomal degradation of BECN1,Citation24 there was a dose-dependent increase in BECN1 levels in response to HSP90 or HSPA2 antagonists in dystrophic myoblasts. The failure of chaperone and/or proteasome machineries can trigger aggresome formation.Citation54 Indeed, we found that the increase in BECN1 following HSP inhibition in dystrophic myoblasts was due to its accumulation in aggresomes. Skeletal muscles, being a postmitotic tissue, are vulnerable to misfolded and dysfunctional proteinsCitation55 and both human and mouse dystrophic muscles have been shown to increasingly accumulate proteins damaged through reactive oxygen species.Citation56 In dystrophic muscle cells, aggregation of BECN1 in the absence of chaperone proteins coincided with an increase in reactive oxygen species production (Fig. S1), resembling defective CFTR-driven aggresome sequestration of BECN1 in cystic fibrosis.Citation57
Autophagy is a highly controlled process: Regulatory coordination between heat shock proteins and autophagy has recently been uncovered.Citation58 Interestingly, the outcomes varied significantly depending on the mode of activation; increased HSPA2 levels prevented starvation-induced autophagy, while heat stress exposure-induced HSPA2 increases were associated with increases in autophagy.Citation58 It is intriguing to consider that this complex interaction between 2 systems of cellular homeostasis may also involve P2RX7 activation. Indeed, P2RX7 was recently found to be a key modulator of metabolic oxidative stress-mediated autophagy in experimental nonalcoholic steatohepatitis and this process also involved HSPA2.Citation59 Thus, a very similar mechanism is active in 2 different models of human diseases affecting liver and skeletal muscles.
There is also crosstalk between autophagy and apoptosis, which although not well understood, clearly facilitates a controlled stress response.Citation60 In many cases autophagy precedes apoptosis and several proteins (e.g. BECN1) play important roles in both pathways. Given that P2RX7 activation is known to trigger apoptosis in many cell types and apoptosis in myoblasts has been documented and reported to be a mediator of cell death in dystrophic muscles,Citation61 we have examined whether these processes can coexist in myoblasts. However, we found that although myoblasts possess the capacity for responding to apoptotic stimuli, eATP acting on P2RX7 receptors triggered CASP3-CASP7-independent cell death in myoblasts.
The results reported here indicate that P2RX7 LP-mediated autophagy may be the determining factor in the eATP-evoked cell death cascade in dystrophic myoblasts. Although specific parameters of autophagic cell death remain the topic of a debate,Citation62,63 our findings that (i) myoblast cell death occurs without CASP activation, (ii) induction of autophagic flux is observed, and (iii) cell death is suppressed using inhibitors of autophagy did fit fully with the autophagic cell death conditions stipulated by Shen et al.Citation64
Under physiological conditions, autophagy is responsible for recycling of cytoplasmic contents. Under stress, cells use autophagy as an element of homeostatic mechanisms maintaining sufficient macromolecular and energy provisions. Our understanding of the molecular mechanisms of the “basal autophagy” in skeletal muscles has only just started to develop,Citation22 but autophagy maintaining cell homeostasis appears integral to muscle physiological growth. Its activation could also be essential to the processes of muscle repair. Therefore, the following scenario could be envisaged: Activation of P2RX7 by brief exposure to ATP and/or by low-level danger signals (eATP) released in mild tissue damage would be fully modulated by ecto-ATPases and thus could result in a self-limiting stimulation of autophagy leading to recirculation of damaged organelles and would aid muscle growth and regeneration through beneficial, low level, tonic P2RX7 stimulation. In contrast, as a ‘danger receptor’ P2RX7 becomes upregulated only on muscle cells which are in danger, and in a chronic damage state (e.g., in dystrophinopathy and dysferlinopathy) large amounts of ATP and P2RX7 upregulation could lead to prolonged, chronic stimulation and overactivation of authophagic pathways, inducing autophagic muscle death. Furthermore, SGCA, a specialized ecto-ATPase activated by high eATP levels,Citation4 is lost from the sarcolemma of dystrophic muscles. This could contribute to the creation of a vicious circle of pathology through further overactivation of P2RX7 (for a review see ref.Citation65).
However, the involvement of autophagy in DMD pathology is unclear. Some studies indicate a positive role: De Palma et al. report significantly lower levels of LC3-II in total muscle protein extracts from both Dmdmdx leg and diaphragm muscles compared with Wt animals and beneficial effects from autophagy augmentation by limited food intake.Citation66 However, a more recent study reports that autophagy is impaired in Dmdmdx mouse leg but not in diaphragm muscles.Citation67 Pauly et al. report that autophagy stimulation via AMPK agonist administration ameliorates dystrophic features in Dmdmdx diaphragms.Citation48 Rapamycin (an autophagy activator via MTOR blockade) also significantly improves the dystrophic phenotype.Citation68 In contrast, in another study, phosphoinositide 3-kinase-AKT (thymoma viral proto-oncogene)-MTOR activation rather than blockade ameliorates pathology in Dmdmdx utrn−/− mouse.Citation69 We also have examined LC3-II in tibialis anterior and diaphragms of control and dystrophic Dmdmdx mice at the same age and with the antibody used by De Palma et al. There was a vast intragroup variability between muscle samples but we found no significant difference in LC3-II levels between normal and dystrophic muscles (Fig. S4). It is worth noting that simple comparison of total LC3-II levels can be inaccurate as a measure of autophagic status and measurements of autophagic flux would provide a more informative assessment.Citation31,32,37,38 The importance of elucidating the true nature of the autophagic status is highlighted by autophagic vacuolar myopathies, where levels of autophagic markers are constitutively high through lack of proper autophagosome and lysosome clearance and yet autophagy is deemed defective and its restoration brings therapeutic benefits.Citation70 Therefore, autophagy appears both down- and upregulated in different muscle disorders with its upregulation proving beneficial in mouse models of Collagen VICitation71 and DuchenneCitation66 muscular dystrophy while its inhibition gives positive outcomes in LAMA2/laminin α2-deficientCitation26,and DMD- and UTRN-deficient mouse muscle.Citation69 Separation of muscle from nonmuscle effects might explain some of the discrepancies. Indeed, all studies to date that looked at autophagy in isolated muscle cells have documented negative effects of increased autophagy levels (summarized in Fig. S5). This fits with our finding that overactivation of P2RX7 leading to increased autophagic flux resulted in cell death of both myoblasts and myotubes.
However, in macrophagesCitation72,73 and dendritic cellsCitation74 increased autophagy decreases inflammatory mediator release and increased inflammatory mediator releases coincide with reduced autophagy (Fig. S5).Citation75 Thus, autophagy may have contrasting functions in muscle and immune cells. It is important to consider this possibility as dystrophic muscles are a hub of cellular interactions, chiefly involving mature and proliferating muscle cells and infiltrating immune cells. Therefore, when manipulating autophagy levels any perceived benefit derived from a global inhibition of autophagy should take into account effects that may relate to a disease model, all cell types involved and in vitro or in vivo scenarios.
Tonic versus excitotoxic activation profiles may govern the roles of both autophagy and P2RX7 activation in muscle disease, where ‘a little’ is good and even essential but ‘too much’ is inevitably bad. Tonic stimulation of P2RX7 promotes myoblast proliferation and differentiation,Citation14,15 whereas high-level, stimulation promotes excitotoxic cell death.Citation5,14
It appears that high eATP acting through P2RX7 overexpressed on dystrophic muscle cells could activate a number of mechanisms, including LP opening and efflux of the intracellular content, signaling cascades, induction of cell death via autophagy, and inhibition of regeneration. Therefore, blockade of this receptor should have a more significant effect than treatments addressing each of these pathways separately. In addition, the antiinflammatory effects of blocking the P2RX7-NLRP3 inflammasome formation on infiltrating cells could ameliorate the disease symptoms further still.
As such, P2RX7 targeting may represent a more viable avenue for therapy. The outcome of some preclinical trials with broad P2 purinoceptor antagonistsCitation76 and our own studies with CBBCitation5 and in Dmdmdx p2rx7−/− double-mutant mice (manuscript submitted) may suggest that more specific, ‘drug-like’ P2RX7 antagonists may bring therapeutic gains in dystrophic muscles by reducing inflammation and increasing muscle cell survival even under conditions where eATP levels remain high. Moreover, a recent study has highlighted the potential therapeutic benefits of specifically inhibiting P2RX7 LP formation in the treatment of chronic pain.Citation77 Therapies combining selective anti-inflammatory drugsCitation78 with P2RX7 antagonists could result in improved efficacy and reduced side effects compared to current DMD management with glucocorticoids.
In summary, our results in dystrophic myoblasts and myotubes demonstrate a novel mechanism of autophagic cell death, which is triggered specifically by P2RX7 LP formation and requires interactions with heat shock proteins. Moreover, we reveal reciprocity in this mechanism in muscles but not in macrophages, whereby P2RX7 LP formation requires autophagy and is HSP90-dependent but HSPA2-independent. Finally, we show that molecules entering through the LP can be targeted to autophagosomes. Targeting this P2RX7-autophagy axis could be of therapeutic benefit in this debilitating and life-shortening form of muscular dystrophy.
Materials and Methods
Animals
C57BL10ScSn, Dmdmdx and Dmdmdx p2rx7−/− double-mutant 4-mo-old male mice were used in accordance with approvals granted by the institutional Ethical Review Board and the Home Office (UK). To generate Dmdmdx p2rx7−/− double-mutant animals (manuscript submitted) a female Dmdmdx mouse was crossed with a male p2rx7 knockout mouse (Jackson lab s/n: 005576).Citation79 Following selective breeding, animals were genotyped by PCR using specific primer sets to the Dmdmdx mutationCitation80 and the neomycin cassetteCitation79 and a colony of homozygous double-mutant Dmdmdx p2rx7−/− double-mutant animals was established. All mice were bred and maintained in a controlled environment (12 h light-dark cycle, 19–23°C ambient temperature, 45–65% humidity). They were fed a standard pellet diet (Economy Rodent Breeder Diet, Special Diet Services, Witham, UK) and allowed water ad libitum. Mice were killed by CO2 inhalation and organs dissected and flash frozen in liquid nitrogen prior to protein extraction.
Cell cultures
Wild-type and dystrophic myoblast cell lines derived from adult male “immorto” mice were as previously described.Citation5,81 Myoblasts and myotubes were cultured as primary cells without immortalization stimulus. Culture media consisted of KnockOut DMEM (Invitrogen, 10829–018) supplemented with 20% v/v KnockOut serum replacement (KSR; Invitrogen, 10828–028), 10% v/v donor horse serum (Sera Labs, S-222-FSI) and 2 mM L-glutamine (Sigma, G7513). Myoblast to myotube differentiation was induced by withdrawing KSR and reducing donor horse serum to 5% v/v. Peritoneal macrophages were isolated by peritoneal cavity lavage with 5ml ice cold Dulbecco's phosphate-buffered saline (DPBS; Sigma, D8537) containing 2% KSR and were maintained under the same conditions as myoblast cultures. J774 (J774A.1 cell line) mouse macrophages were purchased from American Type Culture Collection (TIB-67) and cultured as per myoblasts. Primary myoblasts were obtained from freshly dissected muscles from 4 month-old male mice as previously described.Citation5 HEK-293 cells stably transfected with mouse P2RX7 cDNA (courtesy of Dr. Friedrich Koch-Nolte, Hamburg) and HEK-293 control cells were cultured in media containing 10% v/v Fetal Bovine Serum (Sigma, F9665) and 2 mM L-glutamine (Sigma, G7513).
Antibodies and reagents
The following antibodies were used: anti-P2RX7 (Synaptic Systems, 177003) 1:1000, anti-MAPK1-MAPK3 (Cell Signaling Technology, 9102) 1:2000, anti-phospho-MAPK1-MAPK3 (Cell Signaling Technology, 9106) 1:1000, anti-ACTB (Sigma, A2066) 1:1000, anti-LC3B (Sigma, L7543) 1:1000, anti-GAPDH (Sigma, G9545) 1:1000, anti-COX4/COXIV (Abcam, ab14744) 1:000, anti-BECN1 (Santa Cruz Biotechnology, 48381) 1:200. Other chemicals used were as follows: P2RX7 antagonists A438079 and A804598 (Tocris Bioscience, 2972 and 4473, respectively) and Brilliant Blue G (BBG, Ascent Scientific, ASC-389), HSPA2 and HSP90 inhibitors, VER155008, geldanamycin and 17-DMAG (Tocris Biosciences, 3803, 1368 and 2610, respectively), Proteostat aggresome detection kit (Enzo Life Sciences, ENZ-51035-K100), HSPA2 inhibitor KNK437 (Merck Millipore, 373260), the cell permeable, fluorogenic CASP3-CASP7 substrate DEVD-Nucview488 (Cambridge Biosciences, BT10403) and the mitochondrial membrane potential-sensitive probe tetramethylrhodamine ethyl ester (TMRE; Sigma, 87917), digoxin and digitoxigenin (Sigma, D6770 and D9404, respectively).
Western blotting
Proteins were extracted from adherent cells by scraping in 100 μl ice-cold extraction buffer containing 1 x LysisM (Roche, 04719956001), 1x cOmplete ULTRA Mini EDTA-free protease inhibitor cocktail tablet (Roche, 05892791001), 1 x PhosSTOP phosphatase inhibitor cocktail tablet (Roche, 04906845001), 2 mM sodium orthovanadate (Sigma, S6508). Cells were disrupted by passing 10 times through a 25 g needle followed by centrifugation for 30 s at 800 g. Total proteins from frozen tissues were extracted in ice-cold extraction buffer (as above) using 50 passes between glass Teflon homogenizers (Fisher Scientific, 11592453). Samples were centrifuged (800 g for 3 min at 4°C) to remove debris and concentrations determined using the Bicinchoninic acid assay kit (Sigma, B9643). Twenty to 40 μg protein was mixed with Laemmli buffer at 1:1 v/v ratio with 2.5% v/v β-mercaptoethanol, heated for 5 min at 95°C, then chilled on ice prior to gel loading. Samples were separated on Any-KD Mini-protean TGX ready gels (Bio-Rad, 456–9034) and electroblotted onto Hybond-P membranes (GE Healthcare, RPN303F). Blots were blocked in 5% w/v nonfat milk powder in 1x Tris buffered saline (TBST; 50 Mm Tris, 150 mM NaCl, 0.01% v/v Tween-20, Sigma T1503, S7653 and P1379, respectively), for 1 h prior to probing with primary antibody diluted in the same blocking buffer (overnight at 4°C or 2 h at room temperature), then washed (3 times) with 1 x TBST for 10 min and incubated with the appropriate horseradish peroxidase-conjugated secondary antibody; anti-mouse 1:10:000 (Sigma, A4416), anti-rabbit 1:5000 (Sigma, A6154) overnight at 4°C or 1 h at room temperature. Specific protein bands were visualized using Luminata Classico or Forte chemiluminescent substrates (Merck Millipore, WBLUC0500 and WBLUF0500, respectively), images were obtained using a ChemiDoc MP system (Bio-Rad, Hertfordshire, UK). Alexa Fluor 488 anti-mouse and 555 anti-rabbit secondary antibodies (Life Technologies, A-11034 and A-21429, respectively) were used for fluorescence detection of MAPK1-MAPK3 activation and images obtained using a LI-COR Odyssey Fc imaging system (Cambridge, Cambridgeshire, UK). Densitometric analyses were performed using the integrated density measurement function of ImageJ software.Citation82 All experiments were repeated at least 3 times in triplicate with similar results obtained throughout.
LC3-II induction assay
Cells were seeded at numbers providing 70% confluency in 6-well plates (Nunc, 10119831) in 2 ml of medium. After 24 h, growth medium was replaced with one containing 10% V/V KSR. Cells were incubated for a further 16 h prior to application of agonists and antagonists. ATP (3 mM) and the semispecific P2RX7 agonist BzATP (1 mM; Sigma, B6396) were applied for 30 min, with or without 30 min preincubation with P2RX7 antagonists A804598 (100 nM; Tocris, 4473) or Brilliant Blue G (BBG, 10 μM; Ascent Scientific, ASC-389). Where indicated, cells were preincubated for 30 min with autophagy inhibitors: 3-methyladenine (3-MA, 5 mM; Sigma, M9281), mefloquine hydrochloride (Mefloq., 1 μM, Bioblocks, QUO24–1), the lysomal protease inhibitors: Pepstatin A and E-64d (both at 10 μg/ml; Sigma 77170 and E8640, respectively), or the cardiac glycoside autosis inhibitors digoxin and digitoxigenin (5 μM; Sigma, D6770 and D9404, respectively). Cells were washed in 1 x PBS, then scraped in 100 μl ice-cold lysis buffer (as above) and lysed by 10 passes through a 25 g needle. Samples were centrifuged at 800 g for 30 s to remove insoluble materials.
Phosphoprotein mass spectrometry analysis
Cells were seeded at 70% confluency in 600-cmCitation2 plates (Nunc, 166508) in 135 ml of medium for 24 h prior to the addition of 3 mM ATP for 10 min. Cells were washed in 1 x DPBS (Sigma, D8537), then scraped into 3 ml ice cold lysis buffer (as above) and homogenized by 10 passes through a 25 g needle. Samples were centrifuged at 800 g for 30 s to remove insoluble materials, prior to phosphoprotein purification per the manufacturer's instructions (Phosphoprotein Purification Kit, Pierce, 88512). Samples were then desalted using maxi dialysis tubes (3 ml, 3.5 kDa; Generon, DO35) floating in cold, double-distilled H2O for 2 h, then for a further 16 h in fresh dialysis solution. Sample pH was adjusted to pH 6.0 using triethylammonium (Sigma, 90357), then samples were denatured in 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP; Sigma C4706) in triethylammonium (Sigma, 90357) at 60°C for 1 h. Cysteine residues were blocked in 55 mM iodoacetamide Sigma, I1149) at RT for 30 min. Samples were trypsin digested in 1 μg trypsin (Promega, V5111) per 1 μg protein at 30°C for 15 min. Samples were then iTraq labeled from 114–117 (Sciex, 4374321) dissolved in EtOH for 1 h at RT, then subjected to isoelectric focusing in 24 well, 12.5% w/v polyacrylamide gel strips (Agilent Technologies, 5188–6424) using an OFFGEL fractionating system (Agilent 3100 Fractionator, Agilent Technologies, Massy, Paris, France) for 48 h. Separated samples were analyzed by ESI-LC-MS (Agilent 6200 TOF, Agilent Technologies, Massy, Paris, France) and phosphopeptide fragments identified by interrogating the Mascot database (Matrix Science) using Spectrum Mill software (Agilent Technologies). Fold changes and significance levels were determined using the iQuantitatorCitation40 algorithm and pathway analysis carried using pathway designer functions of Metacore (Thomson Reuters) and IPA (Ingenuity, Qiagen) software.
Large pore, CASP3-CASP7, and cell viability assays
Myoblasts and peritoneal macrophages were plated onto 3 cm dishes or 96-well plates (Nunc, 11810765 and 10212811, respectively) and cultured for 24 h in normal growth medium, followed by 16 h in low-serum medium (10% v/v KSR). For dye uptake assays, cells were incubated in the Large Pore (LP) buffer (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 10 mM Na-HEPES) containing 5 μM ethidium bromide (EtBr; Sigma E1510) or 3 mM Lucifer Yellow (LY; Sigma, L0259) dyes for LP assays or 5 μM Nucview488 (Cambridge Biosciences, BT10403) and 200 nM TMRE (Sigma, 87917) for CASP3-CASP7 assays. Following addition of agonists, dye uptake was analyzed using LSM 510 Meta microscope (Zeiss, Cambridge, Cambridgeshire, UK) with heated stage and 40 x immersion lens and using a POLARstar Optima plate reader (BMG Labtech, Aylesbury, Buckinghamshire, UK) for 96-well plates. Time-lapse images and fluorescence measurements were taken at 15 min intervals from 0 to 60 min. At the end of each time course, 50 μg/ml digitonin (Sigma, D141) was added to nonselectively permeabilize all cells. Cell viability was assessed using LDH release (Sigma, TOX7) and PrestoBlue (Life Technologies, A-13261) assay kits as per the manufacturers’ instructions.
For immunolocalization of LC3 with dyes in LP assays, dystrophic myoblasts were grown on coverslips, incubated in the LP buffer containing 3 mM LY and treated with 1 mM BzATP for 25 min, after which cells were washed 3 times in 1 x PBS, then fixed in ice-cold 4% paraformaldehyde for 30 min. N.B. LY can be covalently linked to surrounding biomolecules by aldehyde, allowing dye localization in cellular compartments of fixed cells. Following 3 further washes in PBS, cells were permeabilized in 0.1% Triton X-100 in PBS, blocked with serum specific to the host of the secondary antibody origin (10%; Sera Labs, S-707-HIS) for 30 min at RT, then incubated with anti-LC3 antibody (Sigma, L7543) overnight at 4°C. Cells were then washed 3 times in 1 x PBS prior to secondary antibody (Alexa Fluor) application for 1 h at room temperature. Coverslips were then washed and mounted on slides using mounting medium (Ibidi, 5000), and images acquired using a Zeiss LSM 5.10 confocal microscope (Zeiss, Cambridge, Cambridgeshire, UK). Images were analyzed using ImageJ software's particle analysis plugin.
Aggresome assays
Myoblasts were cultured on coverslips as per the LP assay. Cells were pretreated with 75 nM GA prior to 1 mM BzATP treatment for 25 min, washed 3 times in 1 x PBS and fixed in ice-cold 4% paraformaldehyde for 30 min. Following 3 further washes in PBS, cells were permeabilized in 0.1% Triton X-100 in PBS, blocked with goat serum (10%; Sera Labs, S-707-HIS) in PBS for 30 min at room temperature, then incubated with primary BECN1 antibody overnight at 4°C. Cells were then washed 3 times in 1 x PBS prior to secondary antibody (Alexa Fluor 488 anti-mouse; Life Technologies, A-11029) application for 1 h at room temperature. Aggresomes were detected using the Proteostat aggresome detection kit (Enzo Life Sciences, ENZ-51035-K100) according to the manufacturer's instructions. Coverslips were mounted on slides using FluorPreserve mounting media and images acquired using a Zeiss LSM 5.10 confocal microscope. Images were analyzed using ImageJ software's particle analysis and colocalization plugins.
Statistical analysis
Results are reported as means +/− SEM where n refers to number of independent experiments (3 to 5). For statistical analysis, a one-way analysis of variance (ANOVA) was performed using the post-hoc Tukey test (Microcal Origin 7.0). Differences were considered statistically significant at P <0 .05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
KAUP_A_994402_supplementary.zip
Download Zip (20.4 MB)Acknowledgments
We thank Drs F Koch-Nolte (University Medical Center, Hamburg) for providing HEK(P2RX7) cells, A Young (CRUK, Cambridge) for sharing autophagy expertise, D Coletti (Paris 6) for permission to use his skull cell graphic and Y Geng, Y Choi and W Michowski (Dana Farber, Boston) for critical reading of the manuscript.
Funding
This work was supported by the Interreg IV (TC2N) grant to DV and DCG, Duchenne Parents Project (NL) to DCG and Muscular Dystrophy Association USA to CY and DCG.
References
- Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol 2013; 201:499-510; PMID:23671309; http://dx.doi.org/10.1083/jcb.201212142
- Hou W, Zhang Q, Yan Z, Chen R, Zeh Iii HJ, Kang R, Lotze MT, Tang D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis 2013; 4:e966; PMID:24336086; http://dx.doi.org/10.1038/cddis.2013.493
- Hetherington HP, Spencer DD, Vaughan JT, Pan JW. Quantitative (31)P spectroscopic imaging of human brain at 4 Tesla: assessment of gray and white matter differences of phosphocreatine and ATP. Magn Reson Med 2001; 45:46-52; PMID:11146485; http://dx.doi.org/10.1002/1522-2594(200101)45:1%3c46::AID-MRM1008%3e3.0.CO;2-N
- Sandona D, Gastaldello S, Martinello T, Betto R. Characterization of the ATP-hydrolysing activity of alpha-sarcoglycan. Biochem J 2004; 381:105-12; PMID:15032752; http://dx.doi.org/10.1042/BJ20031644
- Young CN, Brutkowski W, Lien CF, Arkle S, Lochmuller H, Zablocki K, Górecki DC. P2´7 purinoceptor alterations in dystrophic mdx mouse muscles: relationship to pathology and potential target for treatment. J Cell Mol Med 2012; 16:1026-37; PMID:21794079; http://dx.doi.org/10.1111/j.1582-4934.2011.01397.x
- Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2´7). Science 1996; 272:735-8; PMID:8614837; http://dx.doi.org/10.1126/science.272.5262.735
- Browne LE, Compan V, Bragg L, North RA. P2´7 receptor channels allow direct permeation of nanometer-sized dyes. J Neurosci 2013; 33:3557-66; PMID:23426683; http://dx.doi.org/10.1523/JNEUROSCI.2235-12.2013
- Takenouchi T, Nakai M, Iwamaru Y, Sugama S, Tsukimoto M, Fujita M, Wei J, Sekigawa A, Sato M, Kojima S, Kitani H, et al. The activation of P2´7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J Immunol 2009; 182:2051-62; PMID:19201858; http://dx.doi.org/10.4049/jimmunol.0802577
- Biswas D, Qureshi OS, Lee WY, Croudace JE, Mura M, Lammas DA. ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol 2008; 9:35; PMID:18627610; http://dx.doi.org/10.1186/1471-2172-9-35
- Kawano A, Tsukimoto M, Mori D, Noguchi T, Harada H, Takenouchi T, Kitani H, Kojima S. Regulation of P2´7-dependent inflammatory functions by P2´4 receptor in mouse macrophages. Biochem Biophys Res Commun 2012; 420:102-7; PMID:22405772; http://dx.doi.org/10.1016/j.bbrc.2012.02.122
- Bian S, Sun X, Bai A, Zhang C, Li L, Enjyoji K, Junger WG, Robson SC, Wu Y. P2´7 integrates PI3K/AKT and AMPK-PRAS40-mTOR signaling pathways to mediate tumor cell death. PLoS One 2013; 8:e60184; PMID:23565201; http://dx.doi.org/10.1371/journal.pone.0060184
- Di Virgilio F. Dr. Jekyll/Mr. Hyde: the dual role of extracellular ATP. J Auton Nerv Syst 2000; 81:59-63; PMID:10869701; http://dx.doi.org/10.1016/S0165-1838(00)00114-4
- Amoroso F, Falzoni S, Adinolfi E, Ferrari D, Di Virgilio F. The P2´7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis 2012; 3:e370; PMID:22898868; http://dx.doi.org/10.1038/cddis.2012.105
- Martinello T, Baldoin MC, Morbiato L, Paganin M, Tarricone E, Schiavo G, Bianchini E, Sandonà D, Betto R. Extracellular ATP signaling during differentiation of C2C12 skeletal muscle cells: role in proliferation. Mol Cell Biochem 2011; 351:183-96; PMID:21308481
- Araya R, Riquelme MA, Brandan E, Saez JC. The formation of skeletal muscle myotubes requires functional membrane receptors activated by extracellular ATP. Brain Res Brain Res Rev 2004; 47:174-88; PMID:15572171; http://dx.doi.org/10.1016/j.brainresrev.2004.06.003
- Rawat R, Cohen TV, Ampong B, Francia D, Henriques-Pons A, Hoffman EP, Nagaraju K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am J Pathol 2010; 176:2891-900; PMID:20413686; http://dx.doi.org/10.2353/ajpath.2010.090058
- Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol 2010; 21:683-90; PMID:20223289; http://dx.doi.org/10.1016/j.semcdb.2010.03.002
- Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 2013; 36:7-16; PMID:23708729; http://dx.doi.org/10.1007/s10059-013-0140-8
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507-15; PMID:19945408; http://dx.doi.org/10.1016/j.cmet.2009.10.008
- Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, Yan Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 2013; 27:4184-93; PMID:23825228; http://dx.doi.org/10.1096/fj.13-228486
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481:511-5; PMID:22258505; http://dx.doi.org/10.1038/nature10758
- Grumati P, Bonaldo P. Autophagy in Skeletal Muscle Homeostasis and in MuscularDystrophies. Cells 2012; 1:325-45; PMID:24710479; http://dx.doi.org/10.3390/cells1030325
- Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech 2013; 6:25-39; PMID:23268536; http://dx.doi.org/10.1242/dmm.010389
- Xu C, Liu J, Hsu LC, Luo Y, Xiang R, Chuang TH. Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J 2011; 25:2700-10; PMID:21543763; http://dx.doi.org/10.1096/fj.10-167676
- Gehrig SM, van der Poel C, Sayer TA, Schertzer JD, Henstridge DC, Church JE, Lamon S, Russell AP, Davies KE, Febbraio MA, et al. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 2012; 484:394-8; PMID:22495301; http://dx.doi.org/10.1038/nature10980
- Carmignac V, Svensson M, Körner Z, Elowsson L, Matsumura C, Gawlik KI, Allamand V, Durbeej M. Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum Mol Genet 2011; 20:4891-902; PMID:21920942; http://dx.doi.org/10.1093/hmg/ddr427
- Coutinho-Silva R, Persechini PM, Bisaggio RD, Perfettini JL, Neto AC, Kanellopoulos JM, Motta-Ly I, Dautry-Varsat A, Ojcius DM. P2Z/P2´7 receptor-dependent apoptosis of dendritic cells. Am J Physiol 1999; 276:C1139-47; PMID:10329963
- Schachter J, Motta AP, de Souza Zamorano A, da Silva-Souza HA, Guimarães MZ, Persechini PM. ATP-induced P2´7-associated uptake of large molecules involves distinct mechanisms for cations and anions in macrophages. J Cell Sci 2008; 121:3261-70; PMID:18782864; http://dx.doi.org/10.1242/jcs.029991
- Cankurtaran-Sayar S, Sayar K, Ugur M. P2´7 receptor activates multiple selective dye-permeation pathways in RAW 264.7 and human embryonic kidney 293 cells. Mol Pharmacol 2009; 76:1323-32; PMID:19749088; http://dx.doi.org/10.1124/mol.109.059923
- Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct 2002; 27:421-9; PMID:12576635; http://dx.doi.org/10.1247/csf.27.421
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/10.4161/auto.4600
- Ogier-Denis E, Pattingre S, El Benna J, Codogno P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J Biol Chem 2000; 275:39090-5; PMID:10993892; http://dx.doi.org/10.1074/jbc.M006198200
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson Å. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287:19094-104; PMID:22505714; http://dx.doi.org/10.1074/jbc.M111.322933
- Onopiuk M, Brutkowski W, Wierzbicka K, Wojciechowska S, Szczepanowska J, Fronk J, Lochmüller H, Górecki DC, Zabłocki K. Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem Biophys Res Commun 2009; 386:463-6; PMID:19527684; http://dx.doi.org/10.1016/j.bbrc.2009.06.053
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84-91; PMID:16874052; http://dx.doi.org/10.4161/auto.1.2.1697
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/10.1101/gad.1599207
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Liu Y, Shoji-Kawata S, Sumpter RM, Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 2013; 110:20364-71; PMID:24277826; http://dx.doi.org/10.1073/pnas.1319661110
- Schwacke JH, Hill EG, Krug EL, Comte-Walters S, Schey KL. iQuantitator: a tool for protein expression inference using iTRAQ. BMC Bioinformatics 2009; 10:342; PMID:19835628; http://dx.doi.org/10.1186/1471-2105-10-342
- Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43:572-85; PMID:21855797; http://dx.doi.org/10.1016/j.molcel.2011.06.018
- Kim M, Jiang LH, Wilson HL, North RA, Surprenant A. Proteomic and functional evidence for a P2´7 receptor signaling complex. EMBO J 2001; 20:6347-58; PMID:11707406; http://dx.doi.org/10.1093/emboj/20.22.6347
- Haanes KA, Schwab A, Novak I. The P2´7 receptor supports both life and death in fibrogenic pancreatic stellate cells. PloS One 2012; 7:e51164; PMID:23284663; http://dx.doi.org/10.1371/journal.pone.0051164
- Hanley PJ, Kronlage M, Kirschning C, del Rey A, Di Virgilio F, Leipziger J, Chessell IP, Sargin S, Filippov MA, Lindemann O, et al. Transient P2´7 receptor activation triggers macrophage death independent of Toll-like receptors 2 and 4, caspase-1, and pannexin-1 proteins. J Biol Chem 2012; 287:10650-63; PMID:22235111; http://dx.doi.org/10.1074/jbc.M111.332676
- Ryten M, Dunn PM, Neary JT, Burnstock G. ATP regulates the differentiation of mammalian skeletal muscle by activation of a P2´5 receptor on satellite cells. J Cell Biol 2002; 158:345-55; PMID:12135987; http://dx.doi.org/10.1083/jcb.200202025
- Guha S, Baltazar GC, Coffey EE, Tu LA, Lim JC, Beckel JM, Patel S, Eysteinsson T, Lu W, O'Brien-Jenkins A, et al. Lysosomal alkalinization, lipid oxidation, and reduced phagosome clearance triggered by activation of the P2´7 receptor. FASEB J 2013; 27:4500-9; PMID:23964074; http://dx.doi.org/10.1096/fj.13-236166
- Yan Z, Li S, Liang Z, Tomic M, Stojilkovic SS. The P2´7 receptor channel pore dilates under physiological ion conditions. J Gen Physiol 2008; 132:563-73; PMID:18852304; http://dx.doi.org/10.1085/jgp.200810059
- Pauly M, Daussin F, Burelle Y, Li T, Godin R, Fauconnier J, Koechlin-Ramonatxo C, Hugon G, Lacampagne A, Coisy-Quivy M, et al. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am J Pathol 2012; 181:583-92; PMID:22683340; http://dx.doi.org/10.1016/j.ajpath.2012.04.004
- Johnson BD, Schumacher RJ, Ross ED, Toft DO. Hop modulates Hsp70/Hsp90 interactions in protein folding. J Biol Chem 1998; 273:3679-86; PMID:9452498; http://dx.doi.org/10.1074/jbc.273.6.3679
- Adinolfi E, Kim M, Young MT, Di Virgilio F, Surprenant A. Tyrosine phosphorylation of HSP90 within the P2´7 receptor complex negatively regulates P2´7 receptors. J Biol Chem 2003; 278:37344-51; PMID:12869560; http://dx.doi.org/10.1074/jbc.M301508200
- Smart ML, Gu B, Panchal RG, Wiley J, Cromer B, Williams DA, Petrou S. P2´7 receptor cell surface expression and cytolytic pore formation are regulated by a distal C-terminal region. J Biol Chem 2003; 278:8853-60; PMID:12496266; http://dx.doi.org/10.1074/jbc.M211094200
- Gu BJ, Rathsam C, Stokes L, McGeachie AB, Wiley JS. Extracellular ATP dissociates nonmuscle myosin from P2X(7) complex: this dissociation regulates P2X(7) pore formation. Am J Physiol Cell Physiol 2009; 297:C430-9; PMID:19494237; http://dx.doi.org/10.1152/ajpcell.00079.2009
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 2007; 17:839-49; PMID:17893711; http://dx.doi.org/10.1038/cr.2007.78
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 2000; 10:524-30; PMID:11121744; http://dx.doi.org/10.1016/S0962-8924(00)01852-3
- Sandri M, Coletto L, Grumati P, Bonaldo P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci 2013; 126:5325-33; PMID:24293330; http://dx.doi.org/10.1242/jcs.114041
- Nakae Y, Stoward PJ, Kashiyama T, Shono M, Akagi A, Matsuzaki T, Nonaka I. Early onset of lipofuscin accumulation in dystrophin-deficient skeletal muscles of DMD patients and mdx mice. J Mol Histol 2004; 35:489-99; PMID:15571326; http://dx.doi.org/10.1023/B:HIJO.0000045947.83628.a7
- Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina DL, Settembre C, Gavina M, Raia V, Ballabio A, Maiuri L. Cystic fibrosis: a disorder with defective autophagy. Autophagy 2011; 7:104-6; PMID:21048426; http://dx.doi.org/10.4161/auto.7.1.13987
- Dokladny K, Zuhl MN, Mandell M, Bhattacharya D, Schneider S, Deretic V, Moseley PL. Regulatory coordination between two major intracellular homeostatic systems: heat shock response and autophagy. J Biol Chem 2013; 288:14959-72; PMID:23576438; http://dx.doi.org/10.1074/jbc.M113.462408
- Das S, Seth RK, Kumar A, Kadiiska MB, Michelotti G, Diehl AM, Chatterjee S. Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2013; 305:G950-63; PMID:24157968; http://dx.doi.org/10.1152/ajpgi.00235.2013
- Rubinstein AD, Kimchi A. Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci 2012; 125:5259-68; PMID:23377657; http://dx.doi.org/10.1242/jcs.115865
- Smith J, Goldsmith C, Ward A, LeDieu R. IGF-II ameliorates the dystrophic phenotype and coordinately down-regulates programmed cell death. Cell Death Differ 2000; 7:1109-18; PMID:11139285; http://dx.doi.org/10.1038/sj.cdd.4400738
- Clarke PG, Puyal J. Autophagic cell death exists. Autophagy 2012; 8:867-9; PMID:22652592; http://dx.doi.org/10.4161/auto.20380
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004-10; PMID:18971948; http://dx.doi.org/10.1038/nrm2529
- Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011; 7:457-65; PMID:21150268; http://dx.doi.org/10.4161/auto.7.5.14226
- Young CN, Sinadinos A, Gorecki D. P2X receptor signaling in skeletal muscle health and disease. WIREs Memb Transp Signal 2013; 2:265-274. doi: 10.1002/wmts.96
- De Palma C, Morisi F, Cheli S, Pambianco S, Cappello V, Vezzoli M, Rovere-Querini P, Moggio M, Ripolone M, Francolini M, et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis 2012; 3:e418; PMID:23152054; http://dx.doi.org/10.1038/cddis.2012.159
- Spitali P, Grumati P, Hiller M, Chrisam M, Aartsma-Rus A, Bonaldo P. Autophagy is impaired in the tibialis anterior of dystrophin null mice. PLoS Curr 2013; 5; PMID:24292657
- Eghtesad S, Jhunjhunwala S, Little SR, Clemens PR. Rapamycin ameliorates dystrophic phenotype in mdx mouse skeletal muscle. Mol Med 2011; 17:917-24; PMID:21607286; http://dx.doi.org/10.2119/molmed.2010.00256
- Gurpur PB, Liu J, Burkin DJ, Kaufman SJ. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol 2009; 174:999-1008; PMID:19179609; http://dx.doi.org/10.2353/ajpath.2009.080537
- Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A. A block of autophagy in lysosomal storage disorders. Hum Mol Genet 2008; 17:119-29; PMID:17913701; http://dx.doi.org/10.1093/hmg/ddm289
- Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 2010; 16:1313-20; PMID:21037586; http://dx.doi.org/10.1038/nm.2247
- Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 2011; 286:9587-97; PMID:21228274; http://dx.doi.org/10.1074/jbc.M110.202911
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J 2011; 30:4701-11; PMID:22068051; http://dx.doi.org/10.1038/emboj.2011.398
- Peral de Castro C, Jones SA, Ní Cheallaigh C, Hearnden CA, Williams L, Winter J, Lavelle EC, Mills KH, Harris J. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J Immunol 2012; 189:4144-53; PMID:22972933; http://dx.doi.org/10.4049/jimmunol.1201946
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12:222-30; PMID:21151103; http://dx.doi.org/10.1038/ni.1980
- Arulkumaran N, Unwin RJ, Tam FW. A potential therapeutic role for P2´7 receptor (P2´7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs 2011; 20:897-915; PMID:21510825; http://dx.doi.org/10.1517/13543784.2011.578068
- Sorge RE, Trang T, Dorfman R, Smith SB, Beggs S, Ritchie J, Austin JS, Zaykin DV, Vander Meulen H, Costigan M, et al. Genetically determined P2´7 receptor pore formation regulates variability in chronic pain sensitivity. Nat Med 2012; 18:595-9; PMID:22447075; http://dx.doi.org/10.1038/nm.2710
- Heier CR, Damsker JM, Yu Q, Dillingham BC, Huynh T, Van der Meulen JH, Sali A, Miller BK, Phadke A, Scheffer L, et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med 2013; 5:1569-85; PMID:24014378; http://dx.doi.org/10.1002/emmm.201302621
- Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem 2001; 276:125-32; PMID:11016935; http://dx.doi.org/10.1074/jbc.M006781200
- Amalfitano A, Chamberlain JS. The mdx-amplification-resistant mutation system assay, a simple and rapid polymerase chain reaction-based detection of the mdx allele. Muscle Nerve 1996; 19:1549-53; PMID:8941268; http://dx.doi.org/10.1002/(SICI)1097-4598(199612)19:12%3c1549::AID-MUS4%3e3.0.CO;2-A
- Yeung D, Zablocki K, Lien CF, Jiang T, Arkle S, Brutkowski W, Brown J, Lochmuller H, Simon J, Barnard EA, et al. Increased susceptibility to ATP via alteration of P2X receptor function in dystrophic mdx mouse muscle cells. FASEB J 2006; 20:610-20; PMID:16581969; http://dx.doi.org/10.1096/fj.05-4022com
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with imageJ. Biophoton Int 2004; 11:36-42.