
Abstract
Diagnosis and treatment of epithelial ovarian cancer is challenging due to the poor understanding of the pathogenesis of the disease. Our aim was to investigate epigenetic mechanisms in ovarian tumorigenesis and, especially, whether tumors with different histological subtypes or hereditary background (Lynch syndrome) exhibit differential susceptibility to epigenetic inactivation of growth regulatory genes. Gene candidates for epigenetic regulation were identified from the literature and by expression profiling of ovarian and endometrial cancer cell lines treated with demethylating agents. Thirteen genes were chosen for methylation-specific multiplex ligation-dependent probe amplification assays on 104 (85 sporadic and 19 Lynch syndrome-associated) ovarian carcinomas. Increased methylation (i.e., hypermethylation) of variable degree was characteristic of ovarian carcinomas relative to the corresponding normal tissues, and hypermethylation was consistently more prominent in non-serous than serous tumors for individual genes and gene sets investigated. Lynch syndrome-associated clear cell carcinomas showed the highest frequencies of hypermethylation. Among endometrioid ovarian carcinomas, lower levels of promoter methylation of RSK4, SPARC, and HOXA9 were significantly associated with higher tumor grade; thus, the methylation patterns showed a shift to the direction of high-grade serous tumors. In conclusion, we provide evidence of a frequent epigenetic inactivation of RSK4, SPARC, PROM1, HOXA10, HOXA9, WT1-AS, SFRP2, SFRP5, OPCML, and MIR34B in the development of non-serous ovarian carcinomas of Lynch and sporadic origin, as compared to serous tumors. Our findings shed light on the role of epigenetic mechanisms in ovarian tumorigenesis and identify potential targets for translational applications.
Abbreviations
| Dm | = | methylation dosage ratio |
| miRNAs | = | microRNAs |
| MS-MLPA | = | methylation-specific multiplex ligation-dependent probe amplification |
| LS | = | Lynch syndrome |
| TSG | = | tumor suppressor gene |
| WT1 | = | Wilms tumor suppressor 1 sense |
| WT1-AS | = | Wilms tumor suppressor 1 antisense |
Introduction
Epithelial ovarian cancer is the most lethal gynecological malignancy due to late diagnosis of the disease.Citation1 Heredity is a major risk factor; at least 1 ovarian cancer in 10 is estimated to develop as the result of autosomal dominant predisposition with high penetrance.Citation2 Lynch syndrome (LS), which is associated with germline mutations in 1 of 4 DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2), is the third most common cause of inherited ovarian cancer after BRCA1 and BRCA2 mutations. The estimated lifetime risk of ovarian carcinoma in women with LS is up to 12%.Citation3,4
Epithelial ovarian cancer is classified into 5 major histotypes (serous, endometrioid, clear cell, mucinous, and undifferentiated), each histology displaying remarkable differences in their molecular pathogenesis and clinical outcome.Citation5 In addition to the type of differentiation, ovarian carcinomas can be classified based on the degree of differentiation, with grade 1 corresponding to low grade and grades 2 and 3 corresponding to high grade.Citation6 Histology and tumor grade stratify ovarian carcinomas into 2 broad categories, type I and type II, which correlate with molecular and clinical features. Type I tumors (low-grade serous, low-grade endometrioid, clear cell, and mucinous carcinomas) are thought to develop in a stepwise manner from borderline tumors or endometriosis, and have frequent mutations in KRAS/BRAF and Wnt signaling pathway genes, PIK3CA and ARID1A mutations especially in clear cell carcinoma), and occasional microsatellite instability (MSI).Citation7-9 in Clear cell and endometrioid ovarian cancers account for approximately 15–20% of epithelial ovarian cancers in Western countries and are the predominant types in LS.Citation8 Type II tumors (high-grade serous, high-grade endometrioid, malignant mixed mesodermal and undifferentiated tumors) may arise de novo or from precursor lesions remaining to be reliably identified, with fallopian tube as the likely origin for high-grade serous carcinoma.Citation8 Molecularly, type II tumors are characterized by TP53 alterations, positive WT1 expression (in high-grade serous carcinoma), and chromosomal instability.Citation7-9 High-grade serous cancers are the most common type of ovarian cancer overall (70%) and the predominant type in BRCA1/2-associated ovarian cancer.Citation8 The prognosis of high-grade serous ovarian carcinoma is generally poor, compared to intermediate prognosis for clear cell ovarian carcinoma, and favorable prognosis for endometrioid ovarian carcinoma.Citation8
Besides genetic changes, epigenetic events are important for cancer initiation and progression, and may provide molecular tools to detect and manage cancer. DNA methylation of promoter CpG islands is the best established epigenetic modification capable of silencing conventional tumor suppressor genes and tumor suppressive microRNAs (miRNAs) (hypermethylation) or activating oncogenes and oncogenic miRNAs (hypomethylation).Citation10 While yet to be incorporated in ovarian cancer classifications, genome-wide studies have identified DNA methylation signatures associated with histological subtype of epithelial ovarian cancerCitation11,12 and disease progression,Citation13 and ovarian cancer is viewed as a model of translational applications of epigenetic alterations.Citation14,15 Epigenetically silenced genes may offer new targets for therapeutic intervention, based on re-expression of tumor suppressor genes via demethylating drugs.Citation16
We took advantage of expression profiling of ovarian and endometrial cancer cell lines treated with demethylating agents to identify candidates for epigenetically silenced tumor suppressor genes in epithelial ovarian cancer. The extent to which epigenetic dysregulation of tumor suppressor genes is histology-specific in ovarian cancer is incompletely understood; moreover, it is unknown if the origin of ovarian cancer as hereditary vs. sporadic disease influences the epigenetic patterns. We find that inactivation of tumor suppressor genes is selective and depends on the histology and clinical category of ovarian cancer, as well as the individual genes in question, to be described in detail below.
Results
Selection of epigenetic markers
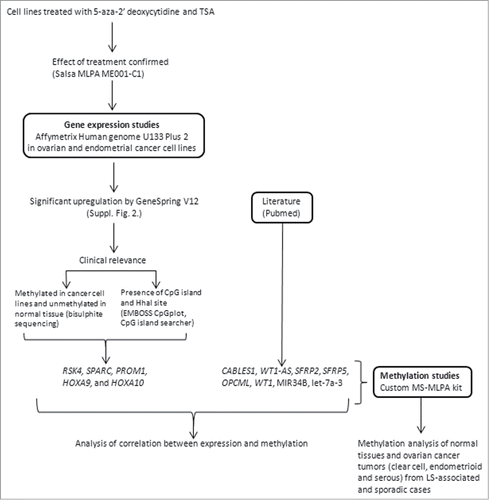
The study design is illustrated in . Ovarian and endometrial cancer cell lines (Suppl. Table 1, Suppl. Fig. 1) were treated with 5-aza-CdR and TSA, resulting in the identification of 1 to 5 thousand significantly up-regulated genes depending on the cell line. Genes specific to non-serous vs. serous cancers as well as genes shared by non-serous and serous cancers were detected (Suppl. Fig. 2; data to be published in detail separately). Since our aim was to address histology- and patient group-specificity of tumor suppressor gene inactivation by epigenetic mechanisms, genes with known function in pathways relevant to ovarian tumorigenesis (see below and ref.Citation17) and with available experimental and/or literature evidence of methylation-sensitivity were prioritized in marker selection. Accordingly, 5 genes from the expression microarrays (RSK4, SPARC, PROM1, HOXA9 and HOXA10) were combined with 8 genes from the literature (CABLES1, WT1-AS, WT1, SFRP2, SFRP5, OPCML, MIR34B, and let-7a-3) to compile an informative epigenetic marker panel for custom-made methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) assays on patient samples (Suppl. Table 2A, Suppl. Table 2B, Suppl. Fig. 3, Suppl. Fig. 4). Among genes identified through our expression profiling experiments, RSK4 showed statistically significant upregulation after treatment in the non-serous cancer cell lines ES2 (2-fold) and AN3CA (2 – 3-fold depending on the probe), HOXA9-HOXA10 in several non-serous (ES2 2-fold, HEC59 2-fold) and serous cell lines (CAOV3 2-fold, SKOV3 2 – 3-fold), SPARC in HEC59 (non-serous, 5 – 15-fold) and PROM1 in CAOV3 (serous, 3-fold).
Figure 1. Flowchart of this investigation.
Analysis of tumor and normal tissues for promoter methylation
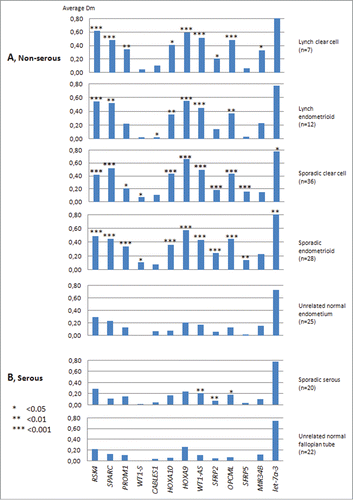
MS-MLPA was our method of choice as it allows for multiplex, quantitative analysis of methylation in archival formalin-fixed paraffin-embedded (FFPE) specimens without the need of bisulphite conversion (Suppl. Fig. 3B). The clinical series (Table 1) comprised ovarian cancers of the clear cell and endometrioid subtypes of sporadic (n = 65) and LS-associated cases (n = 19). Sporadic serous (n = 20) samples were also analyzed. Based on the assumed tissue of origin (see Introduction), normal (unrelated) endometrium was used as a reference for endometrioid and clear cell ovarian carcinomas, whereas specimens of normal (unrelated) fallopian tubes served as a reference for serous tumors. With the exception of WT1 and CABLES1 (with low degree of methylation in both tumor and normal tissues) and let-7a-3 (with high degree of methylation in tumor and normal tissues), the remaining genes displayed low levels of methylation in normal endometrium and fallopian tubes and increased methylation of variable degree in ovarian carcinomas (, Suppl. Table 3).
Figure 2. Average Dm values from MS-MLPA analyses on non-serous and serous ovarian carcinomas and the corresponding normal tissue references. Asterisks denote significantly elevated methylation in tumor vs. normal tissue by t-test for independent samples. The average Dm values of tumor DNAs may in fact be somewhat higher than those shown if possible “contamination” with normal cells is taken into account (see Materials and Methods).
Hypermethylation frequencies in non-serous versus serous ovarian carcinomas
As evident from and Suppl. Table 3, some genes showed relatively high levels of methylation in normal tissues already (e.g., RSK4 and let-7a-3 with methylation dosage ratio (Dm) values in normal tissues clearly above the detection threshold for methylation of Dm = 0.20, see Materials and Methods). Additionally, some genes showed significant differences between the normal reference tissues (e.g. SPARC with average Dm = 0.23 for normal endometrium vs. Dm = 0.13 for normal fallopian tubes, P < 0.000). For meaningful comparisons across markers and patient groups, we adopted the concept of hypermethylation to describe increased methylation in tumor relative to normal DNA, and determined gene-specific thresholds based on Dm values in the respective normal reference tissues (Materials and Methods, Suppl. Table 4). The percentages of tumors with hypermethylation are given in . Non-serous vs. serous comparisons were restricted to sporadic ovarian cancers since there was no serous group among the LS-associated ovarian carcinomas. SPARC (77% vs. 15%, P < 0.001), HOXA10 (72% vs. 30%, P < 0.05), HOXA9 (88% vs. 35%, P < 0.001), WT1-AS (81% vs. 30%, P < 0.001), and OPCML (80% vs. 30%, P < 0.001) showed significantly higher frequencies of tumors with hypermethylation in the combined non-serous (endometrioid + clear cell) vs. serous group (the indicated P values include correction for multiple testing).
Table 1. Clinicopathological data of sporadic and Lynch-associated ovarian carcinoma
Table 2. Hypermethylation frequencies in different patient groups
Hypermethylation in LS-associated vs. sporadic ovarian cancer
Among all ovarian cancer groups investigated, LS-associated clear cell tumors showed the highest frequencies of hypermethylation (). RSK4 was hypermethylated in 7/7 (100%) LS-clear cell carcinomas vs. 21/36 (58%) in sporadic clear cell carcinomas (borderline significant with P = 0.077 by Fisher's exact test). The corresponding frequencies were 5/7 (71%) vs. 10/36 (28%) for PROM1 (P = 0.040) and 5/7 (71%) vs. 8/36 (22%) (P = 0.019) for MIR34B. None of the above differences, however, remained significant after correction for multiple testing. Hypermethylation frequencies in LS-associated endometrioid ovarian carcinomas were comparable to sporadic endometrioid carcinomas.
Hypermethylation with 13 “ovarian-cancer-related” genes vs. 24 “general” tumor suppressor genes (TSGs)
The number of hypermethylated genes out of 13 was determined for each individual tumor and the average values calculated for each ovarian cancer group. The average values from the highest to the lowest were 8.3 for LS-clear cell, 6.7 for sporadic clear cell, 6.4 for LS-endometrioid, 6.4 for sporadic endometrioid, and 2.7 for sporadic serous ovarian carcinomas. The values provided further support to the observations noted above, namely, higher frequencies of hypermethylation in non-serous vs. serous ovarian tumors (P < 0.0001 by t- test for independent samples) and higher frequencies of hypermethylation in clear cell carcinomas from LS-associated vs. sporadic cases (P = 0.052). We have previouslyCitation18 determined the TSG methylator phenotype for the tumors using 24 TSGs that are commonly methylated in various cancers (). The average fraction of hypermethylated genes was higher for the set of 13 vs. 24 genes in all patient groups (). In a sample-specific comparison of the proportions of hypermethylated genes, the present 13 “ovarian cancer-related” genes showed a positive correlation with the 24 “general” TSGs in each individual patient group, with the P-value reaching statistical significance for sporadic clear cell ovarian carcinoma (P = 0.031 by Spearman correlation analysis).
Clinical correlations
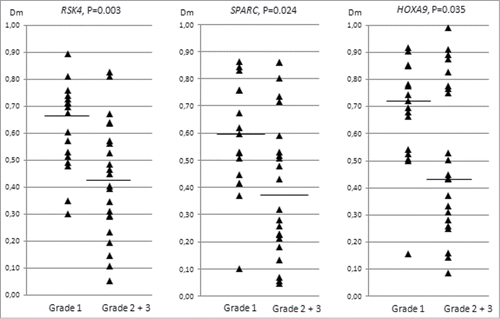
CpG island methylation of the 13 genes of interest showed no association with the clinical stage of ovarian cancer. Grade analysis was restricted to endometrioid ovarian carcinomas since clear cell carcinomas are not graded and all serous carcinomas were of high grade (). Interestingly, Dm values for RSK4, SPARC, and HOXA9 decreased with increasing grade (grade 1 compared with grades 2 and 3 combined) (). Thus, the methylation pattern of high-grade endometrioid ovarian carcinomas showed a trend toward lower methylation levels characteristic of high-grade serous tumors.
Figure 3. Distribution of methylation dosage ratios (Dm values) for RSK4, SPARC, and HOXA9 in endometrioid ovarian carcinomas (sporadic and Lynch-associated combined) stratified by grade (low refers to grade 1 and high to grades 2 and 3). The horizontal line denotes the median and each triangle represents the Dm value of individual data point. Significance values by t-test for independent samples are shown.
Correlation of methylation with expression in cancer cell lines
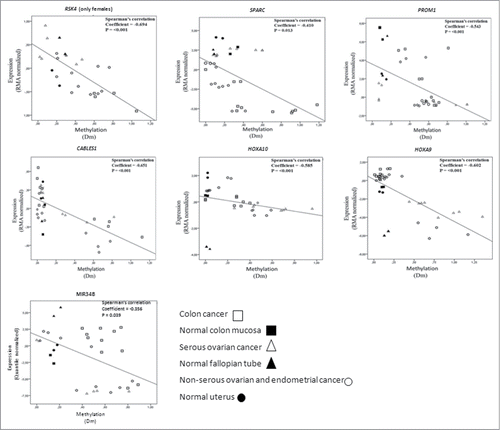
DNA methylation (Dm values) showed significant inverse correlation with expression for RSK4, SPARC, PROM1, CABLES1, HOXA10, HOXA9, and MIR34B in the cancer cell lines and normal tissues investigated (). The observed patterns as a whole suggested that loss of expression was likely linked to promoter hypermethylation of these genes. Future studies are warranted to confirm the cell line data in patient specimens. While no RNA was available from the present archival samples for expression studies, the available literature does provide evidence of inverse correlation between methylation and expression for the genes in question in clinical specimens of ovarian and endometrial cancer.Citation19-22
Figure 4. Correlation analysis of expression (Y-axis, RMA normalized values for protein coding genes and quantile normalized values for MIR34B from arrays) and methylation (X-axis, Dm values from MS-MLPA). The analysis includes cancer cell lines and normal tissue references for which high molecular weight DNA and RNA were available. Data points for normal tissues predominantly clustered in the left top quadrants, compatible with low methylation and high expression. Cancer cells with high degree of methylation often showed low expression (hence, were located in the right bottom quadrants), whereas cancer cells with low methylation showed high or low expression depending on the intrinsic properties of the genes and tissue types in question. While methylation for all these genes significantly correlated with transcriptional repression overall, subsets of specimens occasionally showed transcriptional regulation apparently unrelated to methylation (see, e.g., MIR34B).
Discussion
This study was undertaken to investigate the ability of epigenetic markers to stratify epithelial ovarian carcinomas according to histological subtype and hereditary background (Lynch syndrome). In regard to genes ascertained through expression profiling of cancer cell lines, RSK4 encodes a putative suppressor of RAS/ERK signaling,Citation23 the product of SPARC modulates cell-matrix interactions,Citation24 PROM1/CD133 codes for a cancer stem cell marker,Citation20 and the homeobox genes HOXA9 and HOXA10 control differentiation of the Müllerian ducts into the fallopian tubes, uterus, and cervix.Citation25 As for genes ascertained through the literature, Wilms tumor suppressor 1 sense (WT1) and antisense (WT1-AS) expression are markers of serous ovarian cancer,Citation7 CABLES1 encodes a cyclin-dependent kinase,Citation26 SFRP2 and SFRP5 code for secreted antagonists of Wnt signaling,Citation27 and the product of OPCML/OBCAM is a cell adhesion molecule initially identified as a tumor suppressor for epithelial ovarian cancer.Citation28 Finally, the panel of protein-coding genes was supplemented with 2 ovarian cancer-associated miRNAs: MIR34B, a tumor suppressorCitation22 and let-7a-3, a putative oncogene.Citation29
One of the major findings of this study was a high frequency of hypermethylation in both LS-associated and sporadic non-serous types as compared with sporadic serous ovarian cancer (). We observed low frequencies of hypermethylation for serous ovarian cancer even for genes with high rates of methylation previously reported for the serous subtype, including SPARC,Citation30 HOXA10,Citation31 HOXA9,Citation32 OPCML,Citation33 and MIR34B.Citation22 While the CpG islands investigated were generally the same, different methods for methylation analyses, definitions for hypermethylation, and tissues used for reference may in part explain the observed discrepancies.
The expression of a number of genes from our gene panel has been associated with specific histology of ovarian cancer in the literature. For example, WT1 is expressed in serous, but not in endometrioid, clear cell, or mucinous types.Citation7 Moreover, WT1 sense and antisense mRNAs show a similar expression pattern relative to each other in each tissue.Citation34 These findings are in agreement with our methylation data showing higher frequencies of inactivation by hypermethylation (of mainly WT1-AS) in non-serous vs. serous tumors (). The HOXA gene cluster coordinates the patterning of the Müllerian system, with HOXA9 normally expressed in fallopian tubes, HOXA10 in endometrium, and HOXA11 in endocervix. Ectopic expression of HOXA9 is postulated to give rise to serous ovarian carcinoma, that of HOXA10 to endometrioid ovarian carcinoma, and that of HOXA11 to mucinous ovarian carcinoma.Citation25 In reality, regulation of ovarian cancer development by the HOX system appears complex and the above delineated patterns were poorly reflected by our observations of equally low methylation of HOXA9 and HOXA10 in fallopian tubes vs. endometrium and HOXA9 and HOXA10 both showing significantly more frequent inactivation by hypermethylation in non-serous than serous ovarian cancers. Our finding of lower methylation – a serous-like shift – in HOXA9 associated with high-grade endometrioid ovarian carcinomas () however fits the hypothesis of ectopic expression of HOXA9 accompanying the development of serous ovarian carcinoma.
Comparison of Lynch-associated ovarian carcinomas with their sporadic counterparts revealed less striking differences than the non-serous vs. serous comparison described above. Clear cell carcinomas from LS patients showed the highest frequencies of hypermethylation among all subgroups of ovarian carcinoma we have investigated to date, irrespective of the gene set (13 vs. 24 genes) used. In particular, MIR34B was hypermethylated in 71% of LS-associated vs. 22% of sporadic clear cell carcinomas (). Studies have shown that MIR34B is induced by p53 and upon overexpression mediates apoptosis or growth arrest in response to cellular stress, whereas reduction of miR-34 attenuates these functions.Citation35 While p53 expression is abnormal in a majority of serous and a considerable proportion of endometrioid and clear cell ovarian carcinomas from sporadic cases, LS-associated ovarian carcinomas (endometrioid and clear cell) lack p53 aberrations.Citation18 Our finding of a more frequent inactivation of MIR34B by hypermethylation in LS-associated than sporadic clear cell carcinomas, together with the absence of abnormal p53 in LS-associated as opposed to sporadic clear cell tumors, supports the idea that MIR34B inactivation and abnormal p53 in part function independently to achieve the same tumorigenic purpose. Indeed, p53-independent regulation for MIR34B was recently reported.Citation36
Our study illustrates well the known advantages and disadvantages of the “expression-based” strategy to identify epigenetic markers.Citation37 The method allows for large-scale screening of differentially expressed genes, and re-expression of silenced genes after treatment can be taken as evidence of functional importance of methylation. An important disadvantage is that treatments with demethylating agents can only be performed on cell lines, whereas primary tumors remain to be investigated for methylation and expression by different methods. Several lines of evidence support functional significance of the methylation findings we describe. First, methylation was inversely correlated with mRNA expression in cell lines (). Second, promoter methylation of the 13 genes of interest was part of a more generalized TSG methylator phenotype. Third, our cell line studies and available literature suggest that these genes are likely to be involved (via epigenetic or other mechanisms) in the pathogenesis of several other common cancers beyond ovarian cancer (e.g., colorectal cancer). Fourth, lower levels of promoter methylation of RSK4, SPARC, and HOXA9 () may identify a more aggressive (high-grade) subgroup among endometrioid ovarian cancers that are generally considered to be associated with a favorable prognosis (see Introduction). These 3 genes have been reported to have both oncogenic and tumor suppressor properties, and their increased expression (the predicted result of reduced promoter methylation) promotes tumor growth.Citation38-40
Among the 13 ovarian cancer-related genes investigated, WT1-S, WT1-AS, SFRP2, OPCML, SFRP5, and let-7a-3 did not show significant inverse correlation between methylation and expression in our cell lines (), raising a question about the functional significance of promoter methylation observed in these genes in the different patient groups (, ). It is possible that the methylation changes reflected a generalized CpG island methylator (CIMP) phenotype where a majority of methylation events may represent “passenger” methylation.Citation41 On the other hand, for at least some of the genes listed above, other investigations provide evidence of significant inverse correlation between methylation and expression in ovarian cancer.Citation28,42
In conclusion, our data demonstrate that aberrant DNA methylation of RSK4, SPARC, PROM1, HOXA10, HOXA9, WT1-AS, SFRP2, SFRP5, OPCML, and MIR34B is a frequent and selective event in ovarian tumorigenesis (, ). Our findings increase the understanding of the significance of epigenetic mechanisms in ovarian tumorigenesis and identify possible targets to be evaluated for diagnostic and/or therapeutic purposes.
Materials and Methods
Patient material
The study material consisted of normal and tumor samples of all available LS-associated ovarian carcinomas in Finland, combined with sporadic cases of corresponding histological types ().Citation18 DNA was extracted from formalin-fixed paraffin-embedded (FFPE) samples from representative tumor sections shown to contain > 60% tumor epithelium. The average tumor percentages for the different patient groups were 80% for Lynch associated, 85% for sporadic clear cell, 77% for sporadic endometrioid and 76% for sporadic serous ovarian carcinomas. The Institutional Review Boards of the Departments of Surgery (466/E6/01) and the Obstetrics and Gynecology (040/95) of the Helsinki University Central Hospital (Helsinki, Finland) and that of the Jyväskylä Central Hospital (Jyväskylä, Finland) (Dnro 5/2007) as well as the National Authority for Medicolegal Affairs (Dnro 1272/04/044/07) approved this study.
Bisulphite Modification and Sequencing
Cancer cell line (Suppl. Table 1) and normal samples (600 ng of DNA) were bisulphite converted using EZ DNA Methylation-DirectTM Kit (Zymo Research, Orange, CA, USA). Bisulphite modified DNA was amplified with methylation-unbiased primers (Suppl. Table 2A) and sequenced either directly or after cloning (Suppl. Fig. 3). For the latter purpose, amplification products were cloned into a pCR2.1 TOPO vector by using the TOPO TA Cloning System (Invitrogen, Carlsbad, CA, USA). All resulting white colonies were used for the purification of DNA for sequencing. Gene promoter regions were identified by the EMBOSS CpGplot1 and CpG island searcher2 programs, and promoter information from the literature was taken into account when appropriate (Suppl. Fig. 4).
1http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot
2http://ccat.hcs.usc.edu/cpgislands2
Custom methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) for methylation analysis
The methylation profiles of CpG sites within the CpG-islands in genes under investigation were detected by bisulphite sequencing in cancer cell lines and normal tissues. Those CpG dinucleotides that were part of the methylation-sensitive enzyme HhaI restriction site (GCGC) were chosen for the probe-design for custom-made MS-MLPA (Suppl. Table 2B). When the CpG dinucleotide is methylated, HhaI enzyme cannot recognize the site and a PCR product together with a signal peak will be generated. To complete MS-MLPA assay, Salsa MLPA kit P-300-A1 human DNA reference-2 (Lot 0408, Amsterdam, the Netherlands) was added to the custom MS-MLPA probe mix. MS-MLPA was carried out following the manufacturer´s instructions (http://www.mrc-holland.com) using 100 to 200 ng of DNA. The PCR products were separated by capillary electrophoresis (ABI 3730 Automatic DNA Sequencer, Applied Biosystems, Carlsbad, CA, USA) and analyzed by GeneMapper4.0 genotyping software (Applied Biosystems).
The methylation dosage ratio (Dm) was calculated as described.Citation43 Dm varies from 0 to 1.0, which corresponds to the percentage of methylated DNA. Based on comparison with bisulphite sequencing results, Dm values of ≥ 0.20 were considered to reliably indicate methylation. Thresholds for hypermethylation that distinguish tumor from normal DNA in patient samples were specified for each gene on the basis of methylation values in normal DNAs of the tissue of origin (26 specimens of unrelated normal endometrium and 22 unrelated normal fallopian tubes) as described.Citation18
Tumor suppressor gene (TSG) methylator phenotype
The TSG methylator phenotype was established for the tumors previouslyCitation18 and utilized the SALSA MS-MLPA ME001-C1 Tumor suppressor-1 kit (MRC-Holland, Amsterdam, the Netherlands) for 24 commonly methylated TSGs.
Cell culturing and epigenetic drug treatments
Ovarian cancer (CAOV3, SKOV3, and ES2), colon cancer (RKO, HCT15 and HCT116) and endometrial cancer cell lines (HEC59 and AN3CA) were cultured according to the supplier's protocol (ATCC, Rockville, MD, USA). Drug treatments were done according to Derks et al.Citation44 cells were treated with 1 μM of global genomic DNA demethylating agent 5-aza-2’deoxycytidine (Sigma, A3656) and 300 nM of trichostatin A (Sigma, T1952) for 96 h and 18 h, respectively. All treatments were performed in duplicates. DNA was isolated using standard protocols and total RNA extracted with miRNeasy mini kit (Qiagen, Valencia, CA, USA). The efficiency of the drug treatments was confirmed by methylation (SALSA MS-MLPA ME001-C1 Tumor suppressor -1 kit, MRC-Holland, Amsterdam, the Netherlands) analyses of selected tumor suppressor genes.
Genome-wide gene expression analysis
mRNA gene expression was analyzed using Affymetrix Human Genome U133 Plus 2.0 GeneChip® microarrays (Affymetrix, Santa Clara, CA), containing over 54,000 probe sets covering 47,000 transcripts. Samples of RNA from the cell lines shown in Suppl. Table 1 (treated and untreated) and respective normal tissues (purchased from Amsbio, Abingdon, UK or fresh-frozen tissues obtained from local hospitals) were amplified, labeled and hybridized as described.Citation45 Array image was analyzed using the GeneChip operating software (GCOS; Affymetrix, Santa Clara, CA) and comparison analysis was done according to the manufacturer's instructions. After image acquisition, raw fluorescent signal (cel. file) from Affymetrix GeneChip Operating Software (GCOS) was used for analysis.
Agilent's human miRNA microarrays (8 × 15 K from Agilent Technologies, G4470B), containing 723 human and 76 human viral miRNAs sourced from the Sanger miRBase v. 10.1, were used to analyze miRNA gene expression. Signal intensities of fluorescence were calculated by Agilent's Feature Extraction software version 10.7.3.1.
Analysis of microarray data
GeneSpring GX software, version 12 (Agilent Technologies) was used for microarray data analysis. RMA normalization was used for mRNA data and quantile normalization for miRNA data. Statistically significant differentially expressed genes identified by moderated t-Test combined with the Benjamini and Hochberg correction for multiple testing and using filters based on P-value cut-off 0.05 and fold change cut-off +/-1.5.
Statistical analysis
Statistical analyses of gene expression data were performed as described above. Statistical analysis of other data was performed with the software SPSS 20.0 (SPSS, Chicago, IL, USA). For methylation and expression correlation analysis, Pearson product-moment correlation coefficient (r) or Spearman rank correlation coefficient (rho) was used (depending on whether or not the data were normally distributed, as evaluated by Shapiro-Wilk test). Significance for the differences between groups was determined using Student's t test (for independent samples) or Fisher's exact test as appropriate. P values < 0.05 (2-tailed) were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Tables.docx
Download MS Word (48.2 KB)Supplemental_Figures.pptx
Download MS Power Point (2.1 MB)Acknowledgments
We thank the patients and clinicians contributing to this study. Saila Saarinen is thanked for technical assistance and Satu Valo for help in statistical analyses.
Funding
This study was supported by the Academy of Finland, The Finnish Cancer Organizations, the Sigrid Juselius Foundation, the Integrative Life Science Doctoral Program ILS (to A Niskakoski), Päivikki and Sakari Sohlberg Foundation, Biocentrum Helsinki, and the European Research Council (FP7-ERC-232635).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Jelovac D, Armstrong DK. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J Clin 2011; 61:183-203; PMID:21521830; http://dx.doi.org/10.3322/caac.20113.
- Lynch HT, Casey MJ, Snyder CL, Bewtra C, Lynch JF, Butts M, Godwin AK. Hereditary ovarian carcinoma: Heterogeneity, molecular genetics, pathology, and management. Mol Oncol 2009; 3:97-137; PMID:19383374; http://dx.doi.org/10.1016/j.molonc.2009.02.004
- Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomaki P, Mecklin JP, Jarvinen HJ. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999; 81:214-8; PMID:10188721; http://dx.doi.org/10.1002/(SICI)1097-0215(19990412)81:2%3c214::AID-IJC8%3e3.0.CO;2-L.
- Engel C, Loeffler M, Steinke V, Rahner N, Holinski-Feder E, Dietmaier W, Schackert HK, Goergens H, von Knebel Doeberitz M, Goecke TO, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol 2012; 30:4409-4415; PMID:23091106; http://dx.doi.org/10.1200/JCO.2012.43.2278
- Shih I, Kurman RJ. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am J Pathol 2004; 164:1511-8; PMID:15111296; http://dx.doi.org/10.1016/S0002-9440(10)63708-X
- Cho KR, Shih I. Ovarian cancer. Annu Rev Pathol 2009; 4:287-313; PMID:18842102; http://dx.doi.org/10.1146/annurev.pathol.4.110807.092246
- Soslow RA. Histologic subtypes of ovarian carcinoma: An overview. Int J Gynecol Pathol 2008; 27:161-74; PMID:18317227
- Prat J. Ovarian carcinomas: Five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch 2012; 460:237-249; PMID:22322322; http://dx.doi.org/10.1007/s00428-012-1203-5
- Kuhn E, Kurman RJ, Shih IM. Ovarian cancer is an imported disease: Fact or fiction? Curr Obstet Gynecol Rep 2012; 1:1-9; PMID:22506137; http://dx.doi.org/10.1007/s13669-011-0004-1
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis 2010; 31:27-36; PMID:19752007; http://dx.doi.org/10.1093/carcin/bgp220
- Kolbe DL, DeLoia JA, Porter-Gill P, Strange M, Petrykowska HM, Guirguis A, Krivak TC, Brody LC, Elnitski L. Differential analysis of ovarian and endometrial cancers identifies a methylator phenotype. PLoS One 2012; 7:e32941; PMID:22403726; http://dx.doi.org/10.1371/journal.pone.0032941
- Houshdaran S, Hawley S, Palmer C, Campan M, Olsen MN, Ventura AP, Knudsen BS, Drescher CW, Urban ND, Brown PO, et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One 2010; 5:e9359; PMID:20179752; http://dx.doi.org/10.1371/journal.pone.0009359
- Watts GS, Futscher BW, Holtan N, Degeest K, Domann FE, Rose SL. DNA methylation changes in ovarian cancer are cumulative with disease progression and identify tumor stage. BMC Med Genomics 2008; 1:47-8794-1-47; PMID:18237448; http://dx.doi.org/10.1186/1755-8794-1-47
- Maradeo ME, Cairns P. Translational application of epigenetic alterations: Ovarian cancer as a model. FEBS Lett 2011; 585:2112-20; PMID:21402071; http://dx.doi.org/10.1016/j.febslet.2011.03.016
- Widschwendter M, Jones A, Teschendorff AE. Epigenetics makes its mark on women-specific cancers–an opportunity to redefine oncological approaches? Gynecol Oncol 2013; 128:134-43; PMID:23036354; http://dx.doi.org/10.1016/j.ygyno.2012.09.027
- Balch C, Fang F, Matei DE, Huang TH, Nephew KP. Minireview: Epigenetic changes in ovarian cancer. Endocrinology 2009; 150:4003-11; PMID:19574400; http://dx.doi.org/10.1210/en.2009-0404
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474:609-15; PMID:21720365; http://dx.doi.org/10.1038/nature10166
- Niskakoski A, Kaur S, Renkonen-Sinisalo L, Lassus H, Jarvinen HJ, Mecklin JP, Butzow R, Peltomaki P. Distinct molecular profiles in lynch syndrome-associated and sporadic ovarian carcinomas. Int J Cancer 2013; 133:2596-608; PMID:23716351
- Dewdney SB, Rimel BJ, Thaker PH, Thompson DM, Jr, Schmidt A, Huettner P, Mutch DG, Gao F, Goodfellow PJ. Aberrant methylation of the X-linked ribosomal S6 kinase RPS6KA6 (RSK4) in endometrial cancers. Clin Cancer Res 2011; 17:2120-9; PMID:21372219; http://dx.doi.org/10.1158/1078-0432.CCR-10-2668
- Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, Huang Z, Bentley RC, Mori S, Fujii S, et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009; 28:209-18; PMID:18836486; http://dx.doi.org/10.1038/onc.2008.374
- Wu Y, Halverson G, Basir Z, Strawn E, Yan P, Guo SW. Aberrant methylation at HOXA10 may be responsible for its aberrant expression in the endometrium of patients with endometriosis. Am J Obstet Gynecol 2005; 193:371-80; PMID:16098858; http://dx.doi.org/10.1016/j.ajog.2005.01.034
- Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res 2010; 16:1119-28; PMID:20145172; http://dx.doi.org/10.1158/1078-0432.CCR-09-2642
- Myers AP, Corson LB, Rossant J, Baker JC. Characterization of mouse Rsk4 as an inhibitor of fibroblast growth factor-RAS-extracellular signal-regulated kinase signaling. Mol Cell Biol 2004; 24:4255-66; PMID:15121846; http://dx.doi.org/10.1128/MCB.24.10.4255-4266.2004
- Bornstein P, Sage EH. Matricellular proteins: Extracellular modulators of cell function. Curr Opin Cell Biol 2002; 14:608-16; PMID:12231357; http://dx.doi.org/10.1016/S0955-0674(02)00361-7
- Cheng W, Liu J, Yoshida H, Rosen D, Naora H. Lineage infidelity of epithelial ovarian cancers is controlled by HOX genes that specify regional identity in the reproductive tract. Nat Med 2005; 11:531-7; PMID:15821746; http://dx.doi.org/10.1038/nm1230
- Sakamoto H, Friel AM, Wood AW, Guo L, Ilic A, Seiden MV, Chung DC, Lynch MP, Serikawa T, Munro E, et al. Mechanisms of cables 1 gene inactivation in human ovarian cancer development. Cancer Biol Ther 2008; 7:180-8; PMID:18059193; http://dx.doi.org/10.4161/cbt.7.2.5253
- Kawano Y, Kypta R. Secreted antagonists of the wnt signalling pathway. J Cell Sci 2003; 116:2627-34; PMID:12775774; http://dx.doi.org/10.1242/jcs.00623
- Sellar GC, Watt KP, Rabiasz GJ, Stronach EA, Li L, Miller EP, Massie CE, Miller J, Contreras-Moreira B, Scott D, et al. OPCML at 11q25 is epigenetically inactivated and has tumor-suppressor function in epithelial ovarian cancer. Nat Genet 2003; 34:337-43; PMID:12819783; http://dx.doi.org/10.1038/ng1183
- Lu L, Katsaros D, de la Longrais IA, Sochirca O, Yu H. Hypermethylation of let-7a-3 in epithelial ovarian cancer is associated with low insulin-like growth factor-II expression and favorable prognosis. Cancer Res 2007; 67:10117-22; PMID:17974952; http://dx.doi.org/10.1158/0008-5472.CAN-07-2544
- Socha MJ, Said N, Dai Y, Kwong J, Ramalingam P, Trieu V, Desai N, Mok SC, Motamed K. Aberrant promoter methylation of SPARC in ovarian cancer. Neoplasia 2009; 11:126-35; PMID:19177197
- Fiegl H, Windbichler G, Mueller-Holzner E, Goebel G, Lechner M, Jacobs IJ, Widschwendter M. HOXA11 DNA methylation–a novel prognostic biomarker in ovarian cancer. Int J Cancer 2008; 123:725-9; PMID:18478570; http://dx.doi.org/10.1002/ijc.23563
- Montavon C, Gloss BS, Warton K, Barton CA, Statham AL, Scurry JP, Tabor B, Nguyen TV, Qu W, Samimi G, et al. Prognostic and diagnostic significance of DNA methylation patterns in high grade serous ovarian cancer. Gynecol Oncol 2012; 124:582-8; PMID:22115852; http://dx.doi.org/10.1016/j.ygyno.2011.11.026
- Chen H, Ye F, Zhang J, Lu W, Cheng Q, Xie X. Loss of OPCML expression and the correlation with CpG island methylation and LOH in ovarian serous carcinoma. Eur J Gynaecol Oncol 2007; 28:464-7; PMID:18179137
- Kaneuchi M, Sasaki M, Tanaka Y, Shiina H, Yamada H, Yamamoto R, Sakuragi N, Enokida H, Verma M, Dahiya R. WT1 and WT1-AS genes are inactivated by promoter methylation in ovarian clear cell adenocarcinoma. Cancer 2005; 104:1924-30; PMID:16134181; http://dx.doi.org/10.1002/cncr.21397
- He X, He L, Hannon GJ. The guardian's little helper: microRNAs in the p53 tumor suppressor network. Cancer Res 2007; 67:11099-101; PMID:18056431; http://dx.doi.org/10.1158/0008-5472.CAN-07-2672
- Agostini M, Knight RA. miR-34: From bench to bedside. Oncotarget 2014; 5:872-81; PMID:24657911
- Gal-Yam EN, Saito Y, Egger G, Jones PA. Cancer epigenetics: Modifications, screening, and therapy. Annu Rev Med 2008; 59:267-80; PMID:17937590; http://dx.doi.org/10.1146/annurev.med.59.061606.095816
- Sun Y, Cao S, Yang M, Wu S, Wang Z, Lin X, Song X, Liao DJ. Basic anatomy and tumor biology of the RPS6KA6 gene that encodes the p90 ribosomal S6 kinase-4. Oncogene 2013; 32:1794-810; PMID:22614021; 10.1038/onc.2012.200.
- Chen J, Wang M, Xi B, Xue J, He D, Zhang J, Zhao Y. SPARC is a key regulator of proliferation, apoptosis and invasion in human ovarian cancer. PLoS One 2012; 7:e42413; PMID:22879971; http://dx.doi.org/10.1371/journal.pone.0042413
- Ko SY, Barengo N, Ladanyi A, Lee JS, Marini F, Lengyel E, Naora H. HOXA9 promotes ovarian cancer growth by stimulating cancer-associated fibroblasts. J Clin Invest 2012; 122:3603-17; PMID:22945634; http://dx.doi.org/10.1172/JCI62229
- Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet 2010; 70:277-308; PMID:20920752; http://dx.doi.org/10.1016/B978-0-12-380866-0.60010-1
- Kaneuchi M, Sasaki M, Tanaka Y, Shiina H, Yamada H, Yamamoto R, Sakuragi N, Enokida H, Verma M, Dahiya R. WT1 and WT1-AS genes are inactivated by promoter methylation in ovarian clear cell adenocarcinoma. Cancer 2005; 104:1924-1930; PMID:16134181; http://dx.doi.org/10.1002/cncr.21397
- Gylling AH, Nieminen TT, Abdel-Rahman WM, Nuorva K, Juhola M, Joensuu EI, Jarvinen HJ, Mecklin JP, Aarnio M, Peltomaki PT. Differential cancer predisposition in lynch syndrome: Insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis 2008; 29:1351-9; PMID:18550572; http://dx.doi.org/10.1093/carcin/bgn133
- Derks S, Bosch LJ, Niessen HE, Moerkerk PT, van den Bosch SM, Carvalho B, Mongera S, Voncken JW, Meijer GA, de Bruine AP, et al. Promoter CpG island hypermethylation- and H3K9me3 and H3K27me3-mediated epigenetic silencing targets the deleted in colon cancer (DCC) gene in colorectal carcinogenesis without affecting neighboring genes on chromosomal region 18q21. Carcinogenesis 2009; 30:1041-8; PMID:19329758; 10.1093/carcin/bgp073.
- Nymark P, Lindholm PM, Korpela MV, Lahti L, Ruosaari S, Kaski S, Hollmen J, Anttila S, Kinnula VL, Knuutila S. Gene expression profiles in asbestos-exposed epithelial and mesothelial lung cell lines. BMC Genomics 2007; 8:62; PMID:17331233; http://dx.doi.org/10.1186/1471-2164-8-62