
Abstract
The polycomb proteins BMI-1 and EZH2 are highly overexpressed by Ewing sarcoma (ES), a tumor of stem cell origin that is driven by EWS-ETS fusion oncogenes, most commonly EWS-FLI1. In the current study we analyzed expression of transcription programs that are controlled by polycomb proteins during embryonic development to determine if they are abnormal in ES. Our results show that polycomb target gene expression in ES deviates from normal tissues and stem cells and that, as expected, most targets are relatively repressed. However, we also discovered a paradoxical up regulation of numerous polycomb targets and these were highly enriched for homeobox (HOX) genes. Comparison of HOX profiles between malignant and non-malignant tissues revealed a distinctive HOX profile in ES, which was characterized by overexpression of posterior HOXD genes. In addition, ectopic expression of EWS-FLI1 during stem cell differentiation led to aberrant up regulation of posterior HOXD genes. Mechanistically, this up regulation was associated with altered epigenetic regulation. Specifically, ES and EWS-FLI1+ stem cells displayed a relative loss of polycomb-dependent H3K27me3 and gain of trithorax-dependent H3K4me3 at the promoters of posterior HOXD genes and also at the HOXD11.12 polycomb response element. In addition, a striking correlation was evident between HOXD13 and other genes whose regulation is coordinately regulated during embryonic development by distal enhancer elements. Together, these studies demonstrate that epigenetic regulation of polycomb target genes, in particular HOXD genes, is altered in ES and that these changes are mediated downstream of EWS-FLI1.
Keywords:
Abbreviations
| ARMS | = | alveolar rhabdomyosarcoma |
| BM-MSC | = | adult bone marrow-derived mesenchymal stem cells |
| ChIP | = | chromatin immunoprecipitation |
| ChIP-seq | = | chromatin immunoprecipitation/high throughput sequencing |
| ERMS | = | embryonal rhabdomyosarcoma |
| ES | = | Ewing sarcoma |
| GCR | = | global control region |
| hESC | = | human embryonic stem cells |
| HOX | = | homeobox |
| MSC | = | mesenchymal stem cells |
| NC-MSC | = | neural crest stem cell-derived mesenchymal stem cells |
| NCSC | = | neural crest stem cells |
| OS | = | osteosarcoma |
| PCA | = | principal components analysis |
| PRE | = | polycomb response element |
| qPCR | = | quantitative polymerase chain reaction |
| RT-PCR | = | reverse transcriptase polymerase chain reaction |
Introduction
Hijacking of biologic processes that are central to normal embryonic development is an essential feature of human malignancy. Nowhere is this more apparent than in pediatric tumors, where disruptions to normal fetal development are implicated in initiating the process of malignant transformation by interfering with tissue differentiation.Citation1 Normal differentiation of stem and progenitor cells into terminally differentiated progeny is characterized by global changes in transcriptional programs that are, in turn controlled by epigenetic modifications to chromatin. In particular, the competing actions of the polycomb and trithorax group protein complexes modify chromatin and to support repression and activation of target genes, respectively.Citation2 Disruptions to the composition, regulation, and function of these epigenetic regulatory complexes contribute to the initiation and progression of numerous pediatric as well as adult malignancies.Citation2,3
Ewing sarcoma (ES) is a highly aggressive bone and soft tissue tumor that predominately affects adolescents and young adults.Citation4 Although the precise cellular ontogeny of ES is unknown, experimental models suggest that ES arises from stem and progenitor cells of mesenchymal and/or neural crest origin.Citation5-8 Significantly, ectopic expression of the ES-associated oncoprotein EWS-FLI1 in human stem cells induces up regulation of the polycomb proteins BMI-1 and EZH2, leading to maintenance of stemness and inhibition of cellular differentiation.Citation5,7,9 In addition, overexpression of BMI-1 and EZH2 in established ES cells are required to support their tumorigenicity.Citation5,9,10 Thus, a critical role for polycomb proteins in ES pathogenesis has been established. However, it is as yet unknown whether disruption of polycomb-modulated developmental programs contributes to tumorigenesis.
In the current study, we have investigated gene expression in primary ES tumors and a human stem cell-based model of ES tumor initiation to determine if EWS-FLI1-dependent overexpression of polycomb proteins is associated with altered regulation of polycomb-dependent developmental programs. As expected, our findings confirm that these differentiation-associated transcriptional programs are abnormal in ES and that their regulation is altered by EWS-FLI1 during stem cell differentiation. Unexpectedly, however, we discovered that many polycomb-regulated genes were not repressed but were instead up regulated in ES and these overexpressed genes were highly enriched for homeobox (HOX) genes. In particular, our studies showed that ES highly overexpresses posterior HOXD genes and that this up regulation is mechanistically linked to altered epigenetic regulation of the posterior HOXD locus. Together, these findings demonstrate a novel role for EWS-FLI1 in disrupting expression of polycomb target genes and, for the first time, implicate altered epigenetic regulation of HOX genes in ES pathogenesis.
Results
Expression of developmentally regulated polycomb target genes is abnormal in ES
BMI-1 and EZH2 are overexpressed by ES and both proteins contribute to tumor initiation and maintenance.Citation5,7,9,10 Polycomb proteins play critical roles in regulating gene transcription during stem cell differentiationCitation11 and, given the presumptive stem cell origin of ES, we hypothesized that overexpression of polycomb proteins in ES would be associated with altered expression of polycomb-dependent developmental programs. To test this, we analyzed polycomb target gene expression in 32 ES and 11 different normal adult tissues. Human NCSC, neural crest-derived MSC and adult bone marrow MSC were also analyzed given their prior characterization as candidate cells of ES origin.Citation5-9 Significantly, despite their diverse histologies, clustering analysis revealed that the pattern of polycomb target gene expression in adult tissues is remarkably similar with only testes and cerebellum deviating from the main cluster (). In contrast, considerable variability was evident among the ES tumors (). Nevertheless, despite inter-tumor heterogeneity, over half of the polycomb target genes were found to be significantly differentially expressed by tumors compared to adult tissues ( and Supplemental Table 1). Consistent with their presumptive cellular origins, the pattern of polycomb target gene expression in ES was found to be more similar to stem cells (); however, nearly 500 polycomb target genes were found to be significantly differentially expressed by the tumors (Supplemental Table 2).
Figure 1. Abnormal expression of polycomb target genes in ES. (A). Affymetrix whole genome expression profiling was performed on 32 ES and 11 normal adult tissues (each in triplicate). Principal components analysis (PCA) of 1306 polycomb target genes shows that ES deviate widely from normal tissues. (B). PCA of polycomb target gene expression in ES, stem cells and adult tissues shows that ES are more similar to their putative cells of origin than to adult tissues. BM-MSC: adult bone marrow-derived mesenchymal stem cells (MSC), NCSC: human embryonic stem cell-derived neural crest stem cells, NC-MSC: NCSC-derived MSC. (C). DAVID was used to functionally categorize polycomb target genes that were identified as being overexpressed in ES. Enrichment was determined relative to the polycomb target gene subset and the top 10 most enriched categories are shown (y axis=p-value for level of enrichment; N=number of genes/category). (D). PCA of 37 HOX genes segregates ES from normal tissues and stem cells. Labels as in B. (E). PCA plot of HOX gene expression in pediatric solid tumors (ES, neuroblastoma, embryonal rhabdomyosarcoma (ERMS), alveolar rhabdomyosarcoma (ARMS), osteosarcoma (OS)) and adult tissues shows the unique nature of the ES HOX profile. (F). PCA plot of HOX gene expression in 124 ENCODE cell lines – includes both malignant and non-malignant cell types. Cancer cells cluster with non-cancer cells.
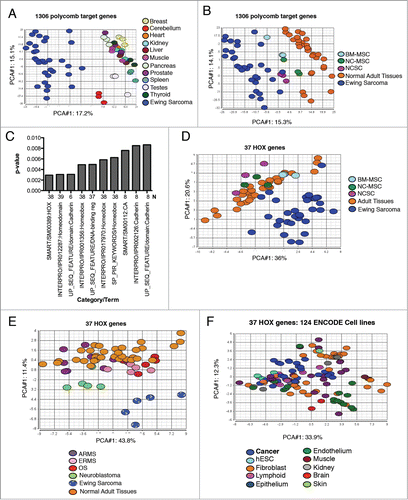
In stem cells, polycomb proteins repress developmental transcription programs.Citation11 Given the undifferentiated phenotype of ES, their high levels of polycomb protein expression, and their presumptive stem cell origins, we anticipated that polycomb-regulated transcription programs would be relatively repressed in the tumors. It was therefore unexpected when our analysis showed that a large proportion of the differentially expressed polycomb target genes were overexpressed in ES (Supplemental Tables 1 and 2). Interestingly, biologic categorization of these overexpressed genes revealed that homeobox genes, in particular HOX genes, were highly over represented (). We therefore next interrogated the data to specifically define HOX gene expression patterns in the various cell and tissue types. There are 39 different HOX genes in the human genome, located in 4 paralogous clusters – HOXA, HOXB, HOXC, and HOXD – and together they function as major determinants of embryonic patterning and development (reviewed in Ref. 12). Data for 37 of these 39 HOX genes were available in the Affymetrix data and, remarkably, principal components analysis (PCA) of just these 37 transcripts was able to segregate nearly all ES from both adult tissues and stem cells (). Given the pediatric age distribution of the profiled tumors we hypothesized that the ES HOX profile might represent a general feature of pediatric solid tumors. To address this, we interrogated array data from an independent set of pediatric tumors, which included ES, osteosarcoma, embryonal and alveolar rhabdomyosarcoma, and neuroblastoma (data kindly provided by Dr. Tim Triche). As shown, the HOX gene profiles of the other pediatric tumors did not recapitulate that of ES, indicating the unique nature of the ES HOX phenotype (). Likewise, the 37 HOX genes were unable to discriminate malignant from non-malignant cells in a panel of 124 ENCODE cell linesCitation13 (). Thus, these results show that the pattern of HOX gene expression in ES deviates significantly from both differentiated tissues and undifferentiated stem cells and is also distinct from other malignant tumors.
ES overexpresses posterior HOXD genes
Having established that HOX gene expression profiles in ES were unique when compared to normal tissues as well as other tumors, we next sought to determine which genes specifically contributed to the ES HOX phenotype. Comparison of ES, adult tissues, and stem cells revealed that 24 of 37 HOX genes were differentially expressed (FDR < 0.05) among the 3 groups and genes in all 4 clusters were represented (). Anterior HOXA genes were downregulated in ES while posterior HOXB, HOXC and HOXD genes were generally upregulated, with a notable and striking differential up regulation of HOXB13, HOXD10, HOXD11 and HOXD13 (). This pattern of HOX gene expression was confirmed by RT-PCR of cell lines and, although several HOXB and HOXC genes were detected in different cancer types, high-level expression of HOXD10, HOXD11 and HOXD13 was restricted to ES cells (Supplemental ).
Figure 2. HOX genes are abnormally expressed in ES. (A). 24 of 37 HOX genes were differentially expressed in ES, stem cells and adult tissues (ANOVA FDR < 0.05). Note profound up regulation of HOXB13, HOXD10, HOXD11, and HOXD13 in ES relative to other cell types. (B). High levels of posterior HOXD genes HOXD10, HOXD11, and HOXD13 were detected in primary ES irrespective of their anatomic site of origin – expression was equally high in tumors outside of the pelvis and extremities. (C). High posterior HOXD gene expression was evident in both bone and soft tissue ES. (D). Low posterior HOXD gene expression was evident in 2 EWS-ETS fusion negative tumors and 4 fusion positive tumors. (E). Affymetrix data show that expression of posterior HOXD genes is higher in primary ES tumors than osteosarcoma (OS) tumors.
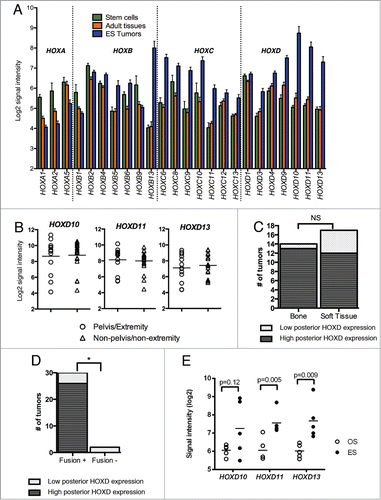
In pediatric ES, tumors most commonly arise in the pelvis and appendicular skeleton, anatomic sites that express posterior HOXD genes during development.Citation14 We therefore hypothesized that the relative overexpression of posterior HOXD genes in ES might be a mark of their anatomic sites of origin. In contrast to our hypothesis, we found that the expression of posterior HOXD genes was equally high in tumors that were derived from non-pelvis/non-extremity sites indicating that the anatomic site of origin was not the only determinant of HOXD gene expression (). Significantly, however, we noted on this analysis that there were 6 tumors that did not express high levels of HOXD10, HOXD11 and HOXD13 (). Closer analysis of the pathologic features of these 6 tumors revealed them to be of both bone and soft tissue origin, but with a trend to being over-represented among soft tissue tumors (). In addition, 2 of the HOXD-low tumors did not amplify either EWS-FLI1 or EWS-ERG, the 2 most common ES-associated fusion genes, at the time of clinical diagnosis (). In contrast, all of the posterior HOXD overexpressing tumors expressed 1 of the 2 fusions. Notably, posterior HOXD expression was also higher in ES than osteosarcoma, demonstrating that tissue site was not the primary determinant of expression (). Thus, overexpression of posterior HOXD genes is prevalent in ES and these studies of primary tumors suggest that both the histologic origin of the tumor and EWS-ETS fusions to contribute to its unique HOX gene profile.
Expression of developmental transcription programs is disrupted by EWS-FLI1
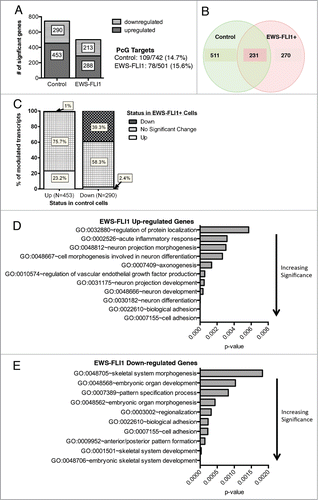
The activation and repression of HOX genes and other embryonic transcriptional programs is dependent on signals from the microenvironment that serve to initiate and inhibit differentiation programs in a developmentally and contextually appropriate fashion. Although it is known that EWS-FLI1 acutely alters gene expression in undifferentiated stem cells,Citation7,9,15 it has not yet been established whether and how expression of the oncogene impacts on transcriptional programs during stem cell differentiation. To address this, we performed transcriptional profiling of control and EWS-FLI1-transduced NCSC before and after exposure to differentiation conditions as previously described.Citation7 We have previously shown that these cells possess both neural and mesenchymal differentiation potential and that, unlike control cells, EWS-FLI1+ cells maintain high levels of both BMI-1 and EZH2 after transfer to differentiation conditions.Citation7 Whole genome expression profiles of both control vector- and EWS-FLI1-transduced cells showed that exposure to differentiation conditions resulted in significant changes to gene expression and that polycomb targets featured prominently among the altered genes ( & Supplemental Table 3). However, less than half of the altered genes were coordinately modulated in both cell populations (). Gene ontology characterization of the divergent transcripts revealed that the presence of EWS-FLI1 resulted in preferential activation of neuronal programs () and repression of mesenchymal differentiation programs (). This finding is in keeping with studies of established ES cells, which showed that EWS-FLI1 maintains tumor cells in an undifferentiated stem cell-like state and prevents mesenchymal differentiation.Citation8 Interestingly, adhesion-related gene programs were among those both up-and down-modulated in the presence of EWS-FLI, reinforcing the recently identified role for EWS-FLI1 in modulation of cell adhesion.Citation16,17
Figure 3. EWS-FLI1 disrupts normal regulation of developmental transcriptional programs during stem cell differentiation. (A). Gene expression profiling was performed in triplicate on EGFP-(control) and EWS-FLI1-EGFP (EWS-FLI1)-transduced NCSC before and after exposure to differentiation conditions for 6 weeks. Histograms show the number of genes that changed over time in each condition following exposure to differentiation media (FDR<0.05 and Fold change >1.5). Polycomb target genes represented ∼15% of the genes in each gene set. (B). Venn diagram shows minimal overlap between differentially modulated genes in the control and EWS-FLI1+ cells. (C). Histograms of gene expression profiling data outlined in A-B show that the presence of EWS-FLI1 results in differential modulation of both up and downregulated genes with less than half of significantly altered genes in control cells being similarly altered in EWS-FLI1+ cells. (D). Gene ontologic analysis of divergently regulated genes shows that genes involved in neural differentiation and cell adhesion were over-represented among genes that were aberrantly upregulated in the presence of EWS-FLI1. (E). Skeletal system morphogenesis and development genes were aberrantly downregulated in EWS-FLI1+ cells (x axis=enrichment score P-value calculated relative to all HuEx trancripts).
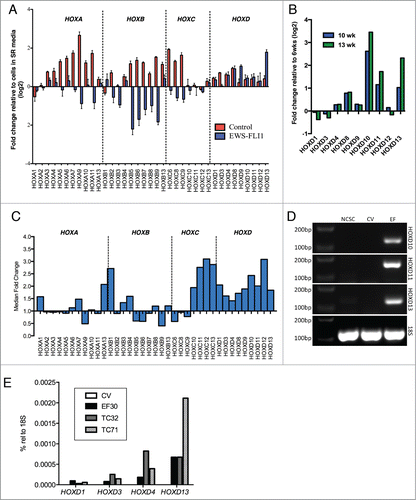
Next, we specifically assessed whether EWS-FLI1 impacted on the expression of HOX genes. Changes in HOX expression were evident across all 4 clusters in control cells and levels of 13 genes in the HOXA, HOXB, and HOXC clusters increased by 2-fold or greater after 6 weeks in differentiation conditions (HOXA5, HOXA6, HOXA7, HOXA9, HOXA10, HOXA11, HOXB6, HOXB7, HOXB9, HOXB13, HOXC6, HOXC8, HOXC9). No HOX genes were downregulated. In contrast, EWS-FLI1+ cells showed a very different pattern of HOX gene regulation (). In particular, HOXD9 and HOXD13 were induced more than 2-fold while several other genes were downregulated (HOXA9, HOXB5, HOXB6, HOXB7, HOXB9). Profiling of EWS-FLI1+ cells after an additional 4 to 7 weeks in differentiation conditions showed continued evolution of HOX gene expression, with notable up regulation of the 3 posterior HOXD genes HOXD10, HOXD11 and HOXD13 (). Replicate experiments with independently isolated and transduced NCSC also showed altered HOX gene expression patterns in EWS-FLI1+ cells, including up regulation of both posterior HOXC and posterior HOXD genes (). Persistent and selective overexpression of the posterior HOXD genes in EWS-FLI1+ cells was confirmed by RT-PCR () and, like established ES cells, posterior genes were more highly expressed than anterior genes in the same cluster (). Thus, regulation of HOX genes is altered by EWS-FLI1 in differentiating stem cells; in particular the expression of posterior HOXD genes is up regulated.
Figure 4 (See previous page). EWS-FLI1 alters expression of HOX genes during stem cell differentiation. (A). Expression of 37 HOX genes was assessed using Affymetrix whole genome expression profiling before and after exposure to differentiation conditions in control and EWS-FLI1+ cells. Histograms represent the mean fold change in expression (log2) after 6 weeks in differentiation conditions relative to the same cells in self-renewal (SR) media (± SEM for triplicate samples in each condition). As shown, EWS-FLI1 altered the regulation of HOX genes and induced marked up regulation of HOXD13. (B). Affymetrix whole genome expression profiling demonstrates continued up regulation of posterior HOXD genes in EWS-FLI1+ cells following continued exposure to differentiation conditions. (C). Up regulation of posterior HOXD genes was reproducibly detected using Affymetrix whole genome expression profiling in EWS-FLI1-transduced NCSC in 2 additional independent experiments. Notably, posterior HOXC genes were also up regulated in these experiments. Histograms represent the median fold change of 2 experiments in EWS-FLI1+ cells compared to control cells after 13 weeks in differentiation conditions. (D). RT-PCR confirmed up regulation of HOXD10, HOXD11 and HOXD13 in EWS-FLI1+ cells. None of these genes was detected in control vector (CV) transduced cells or undifferentiated neural crest stem cells (NCSC). (E). Taqman quantitative RT-PCR analysis of EWS-FLI1+ (EF) and control vector (CV) transduced NCSC and ES cell lines TC71 and TC32 cells. No HOXD genes were detected in CV cells. In contrast, EWS-FLI1+ cells expressed low levels of anterior HOXD genes and high levels of HOXD13, consistent with expression patterns in the ES cell lines.
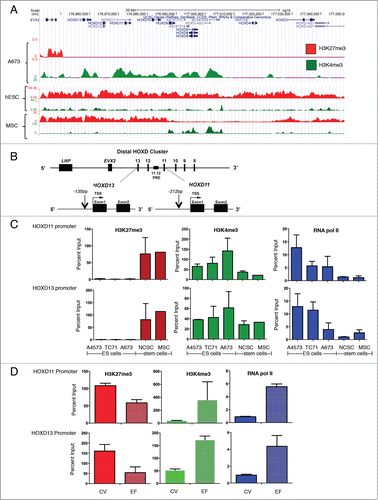
Overexpression of posterior HOXD genes in ES is associated with absence of polycomb-dependent silencing
During embryonic patterning and limb development polycomb-dependent repression of posterior HOXD genes is lost and the loci become activated.Citation14 Thus, we hypothesized that overexpression of posterior HOXD genes in ES cells would be associated with absence of polycomb-dependent repression. To test this, we interrogated genome-wide H3K27me3 and H3K4me3 ChIP-seq data along the HOX gene clusters (E. Tomazou, manuscript in preparation). This analysis revealed a striking absence of H3K27me3 across the HOXD cluster in A673 ES cells (). In contrast, robust H3K4me3 enrichment was detected (). Interestingly, this pattern of absent H3K27me3 and enriched H3K4me3 was also evident at the promoters of HOXB and HOXC genes while the reverse pattern was detected across the HOXA cluster (Supplemental ). Using publicly available ChIP-seq data, we next assessed the pattern of these histone modifications in human embryonic stem cells (hESC) and adipose-derived MSC.Citation18,19 Both marks were detected at HOXD promoters in hESC (). In MSC, the posterior HOXD region was heavily marked with the repressive H3K27me3 modification and also showed intermittent peaks of H3K4me3 enrichment (). This analysis suggested that histone modifications at HOX promoters in ES deviate from normal stem cells. To validate these findings we performed targeted ChIP-qPCR studies of the posterior HOXD region in additional ES cell lines and stem cells (). These studies confirmed a complete absence of H3K27me3 at both the HOXD11 and HOX13 promoters in 3 different ES cell lines (). Conversely, abundant H3K27me3 was detected at the HOXD11 and HOXD13 promoters in both NCSC and BM-MSC (). Evidence of active transcription at the loci in ES cells was supported by H3K4me3 enrichment and RNA polymerase II occupancy (). Thus, despite an abundance of polycomb proteins in ES cells, the promoters of posterior HOXD genes are not targeted for polycomb-dependent repression. Next we performed ChIP-qPCR studies on the control and EWS-FLI1+ differentiating NCSC. In accordance with the observed overexpression of posterior HOXD genes in EWS-FLI1+ cells, we detected a relative loss of H3K27me3 at the HOXD11 and HOXD13 promoters in these cells (). In contrast, H3K4me3 and RNA polymerase II were relatively enriched (). Thus, EWS-FLI1 alters the chromatin state of posterior HOXD gene promoters to support gene activation.
Figure 5 (See previous page). The polycomb-dependent repressive mark H3K27me3 is absent from the posterior HOXD cluster in ES. (A). ChIP-seq studies of A673 ES cells showed a complete absence of the repressive H3K27me3 mark throughout the posterior HOXD cluster. In contrast, enrichment with the H3K4me3 activating mark was evident across the region. Tracks shown are representative of 2 independent experiments for each condition. Publicly available ChIP-seq data were interrogated for H3K27me3 and H3K4me3 binding in hESC and adipose-derived MSC (see methods). Both marks were detected at the locus in both stem cell types. (B). PCR primers were designed to amplify the promoter regions of HOXD11 and HOXD13, close to their transcription start sites, and ChIP performed to interrogate H3K27me3 and H3K4me3 marks at these precise regions. (C). H3K27me3 was absent from the HOXD11 and HOXD13 promoters in 3 different ES cell lines (N = 2 each). Conversely, the mark was detected at the same promoters in human neural crest (NCSC, N = 2) and mesenchymal (MSC, N = 1) stem cells. H3K4me3 and RNA polymerase II were both detected at both promoters and were more highly enriched in ES cells than in either NCSC or MSC (N = 2 each). Histograms and error bars represent mean ± SEM. (D). ChIP-PCR was performed on control (CV) and EWS-FLI1 (EF)-transduced cells after more than 13 weeks in differentiation conditions. Studies of histone modifications at the HOXD11 and HOXD13 promoters showed a relative loss of H3K27me3 and gain of H3K4me3 in EF cells. Increased binding of RNA polymerase II was also evident in EF cells. Histograms and error bars represent mean ± SEM for 2 independent experiments.
Normal developmental regulation of the posterior HOXD cluster is disrupted by EWS-FLI1
The epigenetic regulation of posterior HOXD genes during embryonic development is controlled by a complex interplay between both local and distal regulatory elements, including small intrinsic sequences within the cluster itself Citation20,21 as well as long-range enhancers.Citation22,23 In particular, 3-dimensional folding of chromatin permits long-range interactions between distal enhancer regions, including a global control region (GCR) and gene promoters, resulting in gene activation.Citation24 This GCR coordinately regulates expression of all posterior HOXD genes, as well as EVX2 and LNP (KIAA1715), adjacent genes in the region (see ).Citation25,26 Given that HOXD10, HOXD11, and HOXD13 are all overexpressed by ES, we reasoned that GCR-dependent control of the HOXD cluster might be deregulated. In support of this we discovered a striking direct correlation among expression of all 3 genes in the ES tumors (). Moreover, this correlation extended to include EVX2 and, to a lesser extent, LNP () lending further support for GCR-dependent regulation. In contrast, anterior HOXD genes, which are outside the control of the GCR, exhibited no correlation with HOXD13 (). Similarly, there was no correlation between HOXB13, another highly expressed HOX gene, and the posterior HOXD genes demonstrating that an alternate mechanism of regulation drives HOXB13 up regulation in these tumors (). RT-PCR analysis of ES cell lines () as well as differentiating stem cells () detected expression of EVX2 in the EWS-FLI1+, posterior HOXD+ cells further supporting a mechanistic link between EWS-FLI1 and the posterior HOXD GCR.
Figure 6. Altered epigenetic regulation of the posterior HOXD cluster in ES. (A). A highly statistically significant correlation exists in ES tumors between expression of HOXD13 and HOXD9, HOXD10, and HOXD11, as well as EVX2 and LNP, adjacent genes upstream of HOXD13. All of these genes are under the control of a distal global control region (GCR) during embryonic development. Pearson correlations (r-value) determined using Graphpad Prism Software. (B). Expression of anterior HOXD genes, HOXD3 and HOXD1, which are not under GCR control, is not correlated with HOXD13 in ES tumors. Likewise, no correlation exists between HOXD13 and HOXB13, an equally overexpressed HOX gene that is outside the control of the HOXD GCR. (C). RT-PCR of TC-71 and A673 confirms parallel expression of EVX2, HOXD11, and HOXD13 in ES cells. (D). EVX2, HOXD11, and HOXD13 are detected by RT-PCR of RNA samples obtained from EWS-FLI1+ (EF) but not control vector (CV) transduced neural crest cells. Results shown are representative of 3 independent RNA samples for each condition. (E). EWS-FLI1 ChIP using a FLI1 antibody showed that the fusion protein binds to the HOXD11.12 PRE in A673 cells. Confirmation of EWS-FLI1 binding specificity was demonstrated by binding of the protein at an established EWS-FLI1 target gene promoter, ATAD2,Citation31 with minimal binding outside the ATAD2 promoter (neg control). Results shown are mean ± SD of 3 independent experiments. (F). ChIP-PCR studies of the HOXD11.12 PRE showed absence of H3K27me3 and enrichment of H3K4me3 in ES cells. H3K27me3 was enriched in stem cells. (G). EWS-FLI1+ cells showed reduced levels of H3K27me3 marks and enhanced H3K4me3 modification compared to empty vector control cells (CV). Cells were grown and analyzed as in .
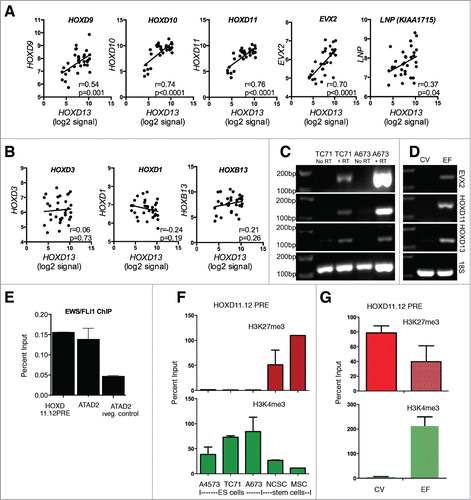
Polycomb response elements (PRE) are well characterized in Drosophila and recruit polycomb proteins to target loci.Citation27 By contrast, the mechanism of polycomb recruitment to target genes in mammalian cells is poorly understood.Citation20,28-30 Nevertheless, a well-characterized mammalian PRE exists between HOXD11 and HOXD12 and recruitment of polycomb proteins to this locus contributes to repression of posterior HOXD gene expression during MSC differentiationCitation20 (see ). Although no EWS-FLI1 binding sites have been identified at HOX gene promoters, binding of the fusion protein to the HOXD11.12 PRE was suggested by recently reported ChIP-seq data.Citation31 We therefore performed ChIP-qPCR studies and confirmed that EWS-FLI1 does indeed bind directly to the HOXD11.12 PRE (). In addition, we observed a complete absence of H3K27me3 and a corresponding enrichment of H3K4me3 at the PRE in ES cell lines (). Finally, diminished H3K27me3 and increased H3K4me3 marks were detected at the HOXD11.12 PRE in EWS-FLI1+ differentiating stem cells (). Thus, in addition to altering GCR-dependent regulation of posterior HOXD gene expression, these data support the hypothesis that EWS-FLI1 could disrupt epigenetic control of the posterior HOXD locus by directly binding to the HOXD11.12 PRE.
Discussion
It is well established that overexpression of polycomb proteins in ES cells contributes to maintenance of stemness and tumorigenicity and that this is mediated in large part by polycomb-dependent repression of genes that instruct differentiation and development.Citation9,10,32 In the current study, we have shown that despite high levels of BMI-1 and EZH2, expression of many developmentally regulated polycomb target genes is paradoxically increased in ES. In particular, our studies of primary tumors and cell lines revealed a marked up regulation of posterior HOXD genes, as well as overexpression of several genes in the HOXB and HOXC clusters. Several lines of evidence suggest that this aberrant HOX expression profile is mediated downstream of EWS-FLI1. Notably, neither of the 2 clinically annotated ES tumors that lacked an EWS-ETS fusion exhibited high levels of posterior HOXD genes. Recent genomic sequencing of Ewing sarcoma tumors showed that some clinically annotated Ewing tumors that lacked an EWS-ETS fusion had gene expression profiles that were distinct from Ewing sarcoma,Citation33 raising the possibility that these tumors were incorrectly classified as Ewing sarcoma at the time of diagnosis. Nevertheless, the failure of the 2 fusion negative cases to express high levels of posterior HOXD genes lends further support to our hypothesis that the presence of the fusion protein is a key contributor to the abnormal HOX expression profile of Ewing sarcomas. Moreover, we further demonstrated that EWS-FLI1 disrupted normal regulation of HOX genes in a human stem cell model of tumor initiation. In line with our findings, overexpression of EWS-FLI1 in pediatric mesenchymal stem cells resulted in significant up regulation of HOXD10 and HOXD13, with concomitant repression of several HOXA genes.Citation15 Furthermore, 2 comprehensive analyses of EWS-FLI1 knockdown in ES cell lines identified HOXC13, HOXD4, and HOXD13 as induced targets of EWS-FLI1.Citation34,35 Thus, these studies together reveal that EWS-FLI1 deregulates HOX gene expression and suggest that, like MLL-fusion driven leukemogenesis,Citation36 disordered HOX gene expression might contribute to EWS-ETS-driven sarcomagenesis and maintenance.
Expression of posterior HOXD genes is normally restricted to posterior tissues during early primary axis development and patterning and to the limb buds during secondary axis development.Citation14,22 Like all HOX genes, HOXD gene promoters exist in a poised, bivalent state in stem cells, marked by both the polycomb-dependent H3K27me3 and the trithorax-dependent H3K4me3 histone modifications.Citation37 During the appropriate spatio-temporal developmental windows the H3K27me3 mark is lost, resulting in a univalent H3K4me3-marked state and gene activation.Citation14,21,37 Conversely, outside of these windows the genes are transcriptionally repressed and robust H3K27me3 enrichment is evident at the promoters of posterior HOXD genes in terminally differentiated cells.Citation21,37,38 Our ChIP-seq studies of the A673 ES cell line revealed that the HOXB, HOXC and HOXD clusters are generally devoid of H3K27me3 marks. This is consistent with published data from another ES cell line, EWS502,Citation39 and was further validated by directed ChIP-qPCR studies of the posterior HOXD locus. Thus, the epigenetic regulation of HOX genes in ES cells deviates from both normal differentiated cells as well as normal stem cells and is likely to be more closely related to an as yet undefined progenitor state that has been coopted by the EWS-ETS fusion.
Epigenetic regulation of posterior HOXD expression during development is mediated in cis and in trans by diverse epigenetic mechanisms and an orchestrated interplay between both internal and external regulatory elements contribute to gene regulation in discrete areas of time and space.Citation22 Our findings suggest that EWS-ETS fusions might interfere with both of these elements. First, we observed a striking direct correlation between expression of all posterior HOXD genes as well as adjacent genes EVX2 and LNP in primary tumors. These genes are all coordinately regulated and expressed in a landscape fashion during development and this landscape regulation is mediated by a distal GCR as well as other distal enhancer elements.Citation24,25 In addition, EWS-FLI1-transduced stem cells displayed synchronized up regulation of EVX2, HOXD10, HOXD11 and HOXD13, indicating that the presence of the oncogenic fusion impacted on GCR function. Our discovery that EWS-FLI1 binds to the internal HOXD11.12 PRE also raises the possibility that the fusion protein might also inhibit epigenetic repression of the locus by directly interfering with polycomb recruitment. Recruitment of polycomb complexes to the posterior HOXD region during terminal differentiation of MSC is dependent on binding of the YY-1 transcription factor to the HOXD11.12 PRE.Citation20 In addition, recruitment of polycomb to HOXB and HOXC clusters also depends, at least in part, on YY-1 binding.Citation20,30 These findings raise the intriguing possibility that EWS-FLI1 could interfere with YY-1 binding and polycomb recruitment more generally. In support of this hypothesis, YY1 binding sites were found to be highly enriched among EWS-FLI1 binding sites in genome wide ChIP-seq studies of ES cells (Kauer et al., unpublished and Ref.Citation31).
Based on these data we propose that expression of EWS-FLI1 in an early embryonic progenitor cell leads to maintenance of an active chromatin state at multiple HOX promoters, resulting in sustained overexpression of HOX genes outside of appropriate developmental windows. This model is particularly relevant for the posterior HOXD genes and for ES tumors that arise in the pelvis and appendicular skeleton, the most common sites of pediatric disease. These tissues arise from embryonic structures that transiently express posterior HOXD genes during development. However, our data also suggest that EWS-ETS fusions can aberrantly induce expression of HOX genes in inappropriate cellular contexts. We base this on our discovery that posterior HOXD genes were also overexpressed in tumors from anatomic sites that do not normally express high levels of posterior HOXD genes during development. These observations also offer potential insights into the difficulties that have been encountered with the creation of transgenic ES models. The most successful model so far was generated by inducing EWS-FLI1, using a lineage-specific Prx1-Cre, in a p53-null background.Citation40 Although the Prx1-Cre/EWS-FLI1 mice did not by themselves generate tumors, they demonstrated varying degrees of developmental abnormalities, ranging from mild defects in mesenchymal differentiation to complete disruption of limb bud development and embryonic lethality.Citation40 These effects support the notion that EWS-FLI1 can have profound deleterious effects on HOX regulation in vivo and suggest that the generation of genetically modified mouse models of ES is impeded by oncogene-induced disruptions to HOX-dependent embryonic patterning and tissue development.
In summary, these studies demonstrate that ES tumors display a unique HOX profile that is distinct from stem cells, differentiated cells, and other cancer cells. In particular, ES overexpress numerous HOX genes including posterior HOXD genes and this aberrant up regulation of the posterior HOXD cluster is mediated downstream of EWS-FLI1 and is associated with loss of the polycomb-dependent H3K27me3 mark and gain of the trithorax-dependent H3K4me3 mark. We hypothesize that a key function of EWS-FLI1 in tumorigenesis is to maintain the epigenetic memory of activated HOX programs, either by aberrantly inducing and/or by promoting maintenance of active chromatin states outside of normal spatio-temporally-defined developmental windows.
Materials and Methods
Cell lines and stem cells
ES cell lines were kindly provided by Dr. Heinrich Kovar and the Children's Oncology Group cell bank (cogcell.org). Identities were confirmed by short tandem repeat profiling. Neuroblastoma, osteosarcoma and HeLa cell lines were obtained from the American Type Culture Collection (ATCC). HEK293FT cells were obtained from Invitrogen. All cell lines were cultured according to COG- or ATCC-recommended protocols. Human embryonic stem cells (hESC) were purchased from Wicell (WA-09) (Madison, Wisconsin) and hESC-derived neural crest stem cells (NCSC) derived as previously described.Citation7,41 Adult bone marrow-derived MSC (BM-MSC) were cultured as previously described.Citation7
Stem cell differentiation
NCSC were transduced with EWS-FLI1-EGFP or EGFP lentiviral vectors and transduced cells were selected by FACS-sorting as previously described.Citation7 Cells were maintained in self-renewal media prior to transfer to differentiation conditions for between 6 and 13 weeks. Differentiation conditions were as describedCitation41 and included replacement of self-renewal media with 5% FBS-containing media. RNA was collected for analysis by Affymetrix arrays and RT-PCR at different times before and cells were transferred to differentiation condition.
Gene expression
Total RNA that had been isolated from NCSC following transfer to differentiation conditions as above was processed and hybridized to Affymetrix HuEx 1.0 arrays according to Affymetrix protocols. HuEx cel files from normal human tissues were downloaded from the Affymetrix Netaffx website (www.netaffx.com). Source HuEx cel files for undifferentiated NCSC, NCSC-derived MSC (NC-MSC), BM-MSC and genetically modified control and EWS-FLI1-transduced cells were previously published (GEO accession: GSE21511).Citation7 HuEx cel files from de-identified tumor biopsies were obtained from the Children's Oncology Group (COG) and Children's Hospital Los Angeles (CHLA). All tumor data were provided in compliance with the Health Insurance Portability and Accountability Act (HIPAA). HuEx cel files for 124 human cell lines that were profiled for the ENCODE project were downloaded from GEO (GSE19090).Citation13 Data for core probeset regions from the cel files were quantile normalized using robust multi-chip averaging in the Partek Genomics Suite software platform (Partek, St. Louis, Mo). Transcript level data were derived from normalized exon data using median summarization and all further analyses performed on transcript-summarized data. Developmentally regulated polycomb target genes were defined as genes whose promoters were identified as being bound by polycomb in hESC.Citation11 Official symbols for these genes were matched to Affymetrix transcript IDs resulting in the designation of 1306 core summarized HuEx transcripts as polycomb target genes. ANOVA was used to detect differential expression of polycomb targets between different cell/tissue types and transcripts with a false detection rate (FDR) <0.05 were deemed significant irrespective of fold change. Gene ontology analysis was performed using DAVID bioinformatics resources.Citation42 HOX gene expression was validated by reverse-transcriptase PCR using conditions and primers as detailed in the Supplemental Methods.
Chromatin immunoprecipitation
ChIP-seq of A673 cells was performed using the iDeal ChIP-seq kit (Diagenode). Publicly available ChIP-seq data for hESC were from ENCODECitation19 (GEO series GSE29611, samples GSM733748 and GSM733657) and human MSC from the Roadmap Epigenomics ProjectCitation18 (GEO series GSE19465, samples GSM621454 and GSM621422). The UCSC Genome Browser (http://genome.ucsc.edu) was used for data visualization and the MSC data were converted from the provided Wig format to BigWig prior to visualization. ChIP-PCR was performed using standard procedures as detailed, along with antibodies and primer sequences, in the Supplemental Methods.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental_Table_3.pdf
Download PDF (269.2 KB)Supplemental_Table_2.pdf
Download PDF (228.6 KB)Supplemental_Table_1.pdf
Download PDF (218.7 KB)Supplemental_Methods.pdf
Download PDF (124.4 KB)Supplemental_Figures.pdf
Download PDF (614.7 KB)Acknowledgments
The authors would like to thank Jenny Tran, Victor Chen, and Dr. Beth Starnes for technical help as well as other member of the Lawlor and Kovar labs for intellectual input. We gratefully acknowledge the support of the COG Tumor Biorepository, the CHLA Genome Core, Dr. Tim Triche for tumor array data and cell lines, as well as Dr. Pat Reynolds and the COG Cell Bank for STR profiling and cell lines.
Funding
Grant support for this work was provided by R01-CA134604, SU2C-AACR-IRG1309, and the University of Michigan's Cancer Center Support Grant (P30 CA046592) by the use of the following Cancer Center Core(s): Flow Cytometry, Vector, and DNA Sequencing. LS was supported by NIH/NCI T32 009676.This study was supported in part by the European framework program 7 grant 259348 (“ASSET”) and the Austrian Science Fund grant M1448-B13. Philanthropic support from the Russell G. Adderley Endowment and the Evan Shapiro Fund to Combat Pediatric Cancer is gratefully acknowledged.
References
- Maris JM, Denny CT. Focus on embryonal malignancies. Cancer Cell 2002; 2:447-50; PMID:12498713; http://dx.doi.org/10.1016/S1535-6108(02)00206-4
- Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer 2010; 10:669-82; PMID:20865010; http://dx.doi.org/10.1038/nrc2931
- Lawlor ER, Thiele CJ. Epigenetic changes in pediatric solid tumors: promising new targets. Clin Cancer Res 2012; 18:2768-79; PMID:22589485; http://dx.doi.org/10.1158/1078-0432.CCR-11-1921
- Balamuth NJ, Womer RB. Ewing's sarcoma. Lancet Oncol 2010; 11:184-92; PMID:20152770; http://dx.doi.org/10.1016/S1470-2045(09)70286-4
- Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A 2009; 106:5324-9; PMID:19289832; http://dx.doi.org/10.1073/pnas.0810759106
- Riggi N, Suva ML, Stamenkovic I. Ewing's sarcoma origin: from duel to duality. Expert Rev Anticancer Ther 2009; 9:1025-30; PMID:19671021; http://dx.doi.org/10.1586/era.09.81
- von Levetzow C, Jiang X, Gwye Y, von Levetzow G, Hung L, Cooper A, Hsu JH, Lawlor ER. Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS One 2011; 6:e19305; PMID:21559395; http://dx.doi.org/10.1371/journal.pone.0019305
- Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007; 11:421-9; PMID:17482132; http://dx.doi.org/10.1016/j.ccr.2007.02.027
- Riggi N, Suva ML, Suva D, Cironi L, Provero P, Tercier S, Joseph JM, Stehle JC, Baumer K, Kindler V, et al. EWS-FLI-1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res 2008; 68:2176-85; PMID:18381423; http://dx.doi.org/10.1158/0008-5472.CAN-07-1761
- Douglas D, Hsu JH, Hung L, Cooper A, Abdueva D, van Doorninck J, Peng G, Shimada H, Triche TJ, Lawlor ER. BMI-1 promotes Ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res 2008; 68:6507-15; PMID:18701473; http://dx.doi.org/10.1158/0008-5472.CAN-07-6152
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006; 125:301-13; PMID:16630818; http://dx.doi.org/10.1016/j.cell.2006.02.043
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 2006; 6:846-56; PMID:17060944; http://dx.doi.org/10.1038/nrc1991
- Consortium EP. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 2011; 9:e1001046; PMID:21526222; http://dx.doi.org/10.1371/journal.pbio.1001046
- Williamson I, Eskeland R, Lettice LA, Hill AE, Boyle S, Grimes GR, Hill RE, Bickmore WA. Anterior-posterior differences in HoxD chromatin topology in limb development. Development 2012; 139:3157-67; PMID:22872084; http://dx.doi.org/10.1242/dev.081174
- Riggi N, Suva ML, De Vito C, Provero P, Stehle JC, Baumer K, Cironi L, Janiszewska M, Petricevic T, Suva D, et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev 2010; 24:916-32; PMID:20382729; http://dx.doi.org/10.1101/gad.1899710
- Chaturvedi A, Hoffman LM, Welm AL, Lessnick SL, Beckerle MC. The EWS/FLI oncogene drives changes in cellular morphology, adhesion, and migration in Ewing sarcoma. Genes Cancer 2012; 3:102-16; PMID:23050043; http://dx.doi.org/10.1177/1947601912457024
- Chaturvedi A, Hoffman LM, Jensen CC, Lin YC, Grossmann AH, Randall RL, Lessnick SL, Welm AL, Beckerle MC. Molecular dissection of the mechanism by which EWS/FLI expression compromises actin cytoskeletal integrity and cell adhesion in Ewing sarcoma. Mol Biol Cell 2014; 25:2695-709; PMID:25057021; http://dx.doi.org/10.1091/mbc.E14-01-0007
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol 2010; 28:1045-8; PMID:20944595; http://dx.doi.org/10.1038/nbt1010-1045
- Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/10.1038/nature11247
- Woo CJ, Kharchenko PV, Daheron L, Park PJ, Kingston RE. A region of the human HOXD cluster that confers polycomb-group responsiveness. Cell 2010; 140:99-110; PMID:20085705; http://dx.doi.org/10.1016/j.cell.2009.12.022
- Schorderet P, Lonfat N, Darbellay F, Tschopp P, Gitto S, Soshnikova N, Duboule D. A genetic approach to the recruitment of PRC2 at the HoxD locus. PLoS Genet 2013; 9:e1003951; PMID:24244202; http://dx.doi.org/10.1371/journal.pgen.1003951
- Andrey G, Duboule D. SnapShot: Hox gene regulation. Cell 2014; 156:856- e1; PMID:24529385; http://dx.doi.org/10.1016/j.cell.2014.01.060
- Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, Leleu M, Trono D, Spitz F, Duboule D. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 2013; 340:1234167; PMID:23744951; http://dx.doi.org/10.1126/science.1234167
- Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, Duboule D. A regulatory archipelago controls Hox genes transcription in digits. Cell 2011; 147:1132-45; PMID:22118467; http://dx.doi.org/10.1016/j.cell.2011.10.023
- Spitz F, Gonzalez F, Duboule D. A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell 2003; 113:405-17; PMID:12732147; http://dx.doi.org/10.1016/S0092-8674(03)00310-6
- Tschopp P, Duboule D. A regulatory 'landscape effect' over the HoxD cluster. Dev Biol 2011; 351:288-96; PMID:21195707; http://dx.doi.org/10.1016/j.ydbio.2010.12.034
- Kassis JA, Brown JL. Polycomb group response elements in Drosophila and vertebrates. Adv Genet 2013; 81:83-118; PMID:23419717; http://dx.doi.org/10.1016/B978-0-12-407677-8.00003-8
- Bengani H, Mendiratta S, Maini J, Vasanthi D, Sultana H, Ghasemi M, Ahluwalia J, Ramachandran S, Mishra RK, Brahmachari V. Identification and validation of a putative polycomb responsive element in the human genome. PLoS One 2013; 8:e67217; PMID:23805300; http://dx.doi.org/10.1371/journal.pone.0067217
- Sing A, Pannell D, Karaiskakis A, Sturgeon K, Djabali M, Ellis J, Lipshitz HD, Cordes SP. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell 2009; 138:885-97; PMID:19737517; http://dx.doi.org/10.1016/j.cell.2009.08.020
- Woo CJ, Kharchenko PV, Daheron L, Park PJ, Kingston RE. Variable requirements for DNA-binding proteins at polycomb-dependent repressive regions in human HOX clusters. Mol Cell Biol 2013; 33:3274-85; PMID:23775117; http://dx.doi.org/10.1128/MCB.00275-13
- Bilke S, Schwentner R, Yang F, Kauer M, Jug G, Walker RL, Davis S, Zhu YJ, Pineda M, Meltzer PS, et al. Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res 2013; 23:1797-809; PMID:23940108; http://dx.doi.org/10.1101/gr.151340.112
- Burdach S, Plehm S, Unland R, Dirksen U, Borkhardt A, Staege MS, Muller-Tidow C, Richter GH. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle 2009; 8:1991-6; PMID:19502792; http://dx.doi.org/10.4161/cc.8.13.8929
- Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, et al. The genomic landscape of the Ewing sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 2014; 10:e1004475; PMID:25010205; http://dx.doi.org/10.1371/journal.pgen.1004475
- Kauer M, Ban J, Kofler R, Walker B, Davis S, Meltzer P, Kovar H. A molecular function map of Ewing's sarcoma. PLoS One 2009; 4:e5415; PMID:19404404; http://dx.doi.org/10.1371/journal.pone.0005415
- Stoll G, Surdez D, Tirode F, Laud K, Barillot E, Zinovyev A, Delattre O. Systems biology of Ewing sarcoma: a network model of EWS-FLI1 effect on proliferation and apoptosis. Nucleic Acids Res 2013; 41:8853-71; PMID:23935076; http://dx.doi.org/10.1093/nar/gkt678
- Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Ann Rev Pathol 2012; 7:283-301; PMID:22017583; http://dx.doi.org/10.1146/annurev-pathol-011811-132434
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006; 125:315-26; PMID:16630819; http://dx.doi.org/10.1016/j.cell.2006.02.041
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007; 129:1311-23; PMID:17604720; http://dx.doi.org/10.1016/j.cell.2007.05.022
- Patel M, Simon JM, Iglesia MD, Wu SB, McFadden AW, Lieb JD, Davis IJ. Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Res 2012; 22:259-70; PMID:22086061; http://dx.doi.org/10.1101/gr.125666.111
- Lin PP, Pandey MK, Jin F, Xiong S, Deavers M, Parant JM, Lozano G. EWS-FLI1 induces developmental abnormalities and accelerates sarcoma formation in a transgenic mouse model. Cancer Res 2008; 68:8968-75; PMID:18974141; http://dx.doi.org/10.1158/0008-5472.CAN-08-0573
- Jiang X, Gwye Y, McKeown SJ, Bronner-Fraser M, Lutzko C, Lawlor ER. Isolation and characterization of neural crest stem cells derived from in vitro-differentiated human embryonic stem cells. Stem Cells Dev 2009; 18:1059-70; PMID:19099373; http://dx.doi.org/10.1089/scd.2008.0362
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; http://dx.doi.org/10.1038/nprot.2008.211