
Abstract
The molecular basis of male infertility is poorly understood, the majority of cases remaining unsolved. The association of aberrant sperm DNA methylation patterns and compromised semen parameters suggests that disturbances in male germline epigenetic reprogramming contribute to this problem. So far there are only few data on the epigenetic heterogeneity of sperm within a given sample and how to select the best sperm for successful infertility treatment. Limiting dilution bisulfite sequencing of small pools of sperm from fertile donors did not reveal significant differences in the occurrence of abnormal methylation imprints between sperm with and without morphological abnormalities. Intracytoplasmic morphologically selected sperm injection was not associated with an improved epigenetic quality, compared to standard intracytoplasmatic sperm injection. Deep bisulfite sequencing (DBS) of 2 imprinted and 2 pluripotency genes in sperm from men attending a fertility center showed that in both samples with normozoospermia and oligoasthenoteratozoospermia (OAT) the vast majority of sperm alleles was normally (de)methylated and the percentage of epimutations (allele methylation errors) was generally low (<1%). However, DBS allowed one to identify and quantify these rare epimutations with high accuracy. Sperm samples not leading to a pregnancy, in particular in the OAT group, had significantly more epimutations in the paternally methylated GTL2 gene than samples leading to a live birth. All 13 normozoospermic and 13 OAT samples leading to a child had <1% GTL2 epimutations, whereas one (7%) of 14 normozoospermic and 7 (50%) of 14 OAT samples without pregnancy displayed 1–14% GTL2 epimutations.
Abbreviations
| ART | = | assisted reproductive technology |
| DBS | = | deep bisulfite sequencing |
| ICSI | = | intracytoplasmatic sperm injection |
| IMSI | = | intracytoplasmic morphologically selected sperm injection |
| LD | = | limiting dilution |
| OAT | = | oligoasthenoteratozoospermia |
| PCR | = | polymerase chain reaction |
| SNP | = | single nucleotide polymorphism |
Introduction
The ever-increasing shift in parental age is associated with a growing number of couples with fertility problems seeking assisted reproductive technologies (ARTs). Although overall 5–10% of all men are infertile, currently known genetic causes account only for a small proportion of them.Citation1,2 In male germ cells, the number of de novo mutations increases with paternal age and the number of spermatogonial cell divisions.Citation3 Since copying of the epigenetic information (in particular of DNA methylation patterns) during cell division is much more error-prone than replication of the DNA sequence itself,Citation4,5 it is plausible that epimutations are also accumulating in aging male germ cells.
A genome-wide demethylation and remethylation wave occurs in the mammalian germline. Around the time when the primordial germ cells reach the genital ridge, the methylation patterns of previous generations are erased. New parent-specific methylation patterns are set during male and female germ-cell differentiation.Citation6,7 In the male germline, remethylation is initiated after prenatal mitotic arrest and completed postnatally during pachytene.Citation8,9 This implies that in IVF/ICSI sperm all paternal methylation imprints must be established correctly. A second demethylation and remethylation wave occurs after fertilization to restore totipotency in embryonal cells.Citation10,11 Only 100–200 imprinted genes (out of >22,000 genes) escape this postzygotic reprogramming and maintain their germline methylation patterns and parent-specific activities.Citation12
Accumulating experimental evidence links epigenetic modifications affecting DNA methylation and histone-protamine configuration in sperm to male infertility.Citation13,14 Aberrant sperm DNA methylation patterns, mainly in imprinted genes have been associated with poor semen parameters and male infertilityCitation15-18 as well as with abortionsCitation19 and ART outcome.Citation20 Genome-wide analyses with methylation arrays suggest broad methylation changes in both imprinted and non-imprinted genes in sperm from infertile and older men, but none with large effect size.Citation21-24 Most candidate gene and genome-wide approaches have measured the average methylation level of genes/loci in samples consisting of millions of sperm. The observed changes in mean methylation, e.g., of an imprinted gene, could be due to an increased rate of single CpG methylation errors at random positions of each or most sperm or to allele methylation errors. Since it is usually the density of methylated CpGs rather than individual CpGs that turns a gene "on" or "off," only alleles with the majority of CpGs being aberrantly (de)methylated (allele methylation errors) are considered as epimutations, whereas single CpG errors are most likely without functional consequences.Citation25,26 Plasmid bisulfite sequencing of a limited number of sperm DNA alleles suggests the existence of true epimutations of both paternally (e.g., H19) and maternally methylated alleles (e.g., MEST) in sperm of infertile men.Citation15,27 Here we employed different techniques for in depth methylation analysis of individual DNA molecules in different classes of sperm.
Results
Methylation analysis of single or a few sperm
Sperm for intracytoplasmatic sperm injection (ICSI) are usually selected under a standard microscope (magnification ∼400×) on the basis of their motility and morphology. Intracytoplasmic morphologically selected sperm injection (IMSI) requires a much higher magnification (∼6000×) for better morphological assessment.Citation28 We employed limiting dilution (LD) bisulfite pyrosequencingCitation26 to analyze single-allele methylation of one paternally methylated (GTL2) and 2 maternally methylated (LIT1 and PEG3) imprinted genes in 4 different classes of sperm from fertile donors: IMSI+ are the "best" sperm selected under a high-powered microscope. They display normal morphology and do not contain any vacuoles. IMSI- sperm have large vacuoles. IMSI-/- are sperm with vacuoles and morphological abnormalities. ICSI sperm appear to be normal at lower magnification but still may contain vacuoles. and present the number of alleles without CpG methylation errors (all analyzed CpGs show the correct sperm methylation imprint), with single CpG methylation errors (one, rarely 2 CpGs are aberrant), and >50% CpG methylation errors (epimutations). All together, we recovered 382 individual alleles in the IMSI+, 120 in the IMSI−, 73 in the IMSI-/-, and 168 in the ICSI group. Neither the number of single CpG nor the number of allele methylation errors (>50% aberrant CpGs) differed significantly between groups. It is interesting to note that only 1 of 104 (1%) analyzed LIT1 alleles (in all 4 groups) and 1 of 303 (0.3%) PEG3 alleles showed single CpG errors, compared to 62 of 336 (18.5%) GTL2 alleles. Similarly, none (0%) of the 104 LIT1 alleles and only 3 of 303 (1%) PEG3 alleles were classified as epimutations, whereas 13 of 336 (3.9%) GTL2 alleles were aberrantly demethylated. Single CpG errors (Fisher's exact test; P < 0.0001) and epimutations (P < 0.05) were significantly more frequent in GTL2 than in LIT1 and PEG3.
Table 1. Methylation status of imprinted genes in human sperm
Figure 1. Methylation patterns of 743 individual DNA molecules (336 for the paternally methylated GTL2, 104 for the maternally methylated LIT1, and 303 for the maternally methylated PEG3 gene) in different groups of sperm from fertile donors. IMSI+ are the "best" sperm selected at high magnification, IMSI- sperm contain vacuoles, IMSI-/- sperm show abnormal morphology and vacuoles, and ICSI sperm appear to be normal at standard magnification. Each line represents the methylation pattern of an individual DNA molecule. The number at the left-hand side indicates how many independent alleles of this type were recovered. Open circles represent unmethylated and filled circles methylated CpG sites. In normal alleles, all CpGs show the correct sperm methylation imprint. Alleles with single CpG errors contain one or at most 2 aberrant CpG sites. Abnormal alleles have more than 50% aberrantly (de)methylated CpG sites, indicative of an epimutation.

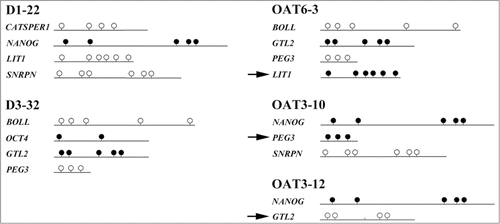
In addition to LD analysis of small pools of morphologically selected sperm, we developed a multiplex assay for methylation analysis of 2 spermatogenesis (BOLL and CATSPER1) genes, which are unmethylated in sperm, 2 pluripotency (NANOG and OCT4) genes, which are methylated in sperm, and 4 (paternally methylated GTL2 and maternally methylated LIT1, PEG3, and SNRPN) imprinted genes. Single sperm were selected under low magnification (ICSI criteria) from samples of 3 fertile men with normal semen parameters and 8 infertile men with OAT syndrome, respectively. shows the methylation patterns of 5 individual sperm, 2 without and 3 with epimutations. All together, we recovered 94 alleles from 61 sperm of infertile men, 8 (8.5%) of which showed single CpG errors and 11 (12%) of which showed >50% CpG errors (). Most epimutations (8 of 11) were found in imprinted genes. Eighty alleles were recovered from 40 sperm of fertile men, 2 (2.5%) of which showed single CpG errors and 5 (6%) epimutations, all in imprinted genes (). Overall, 11 of 61 (18%) OAT sperm and 5 of 35 (14%) of sperm from fertile men displayed epimutations. Although men with OAT syndrome showed a higher rate of epimutations and alleles with single CpG errors, these differences were not statistically significant.
Table 2. Methylation analysis of 8 genes in single sperm
Figure 2. Methylation analysis of single sperm. Alleles recovered from the same sperm are grouped together. Sperm 22 of fertile donor 1 (D1–22) and sperm 32 from donor 3 (D3–32) display normal methylation patterns in all 4 recovered alleles. Sperm 3 of OAT patient 6 (OAT6–3) displays an abnormally methylated LIT1 allele, sperm 10 and 12 of OAT patient 3 (OAT3–10 and −12) display an abnormally methylated PEG3 and an abnormally demethylated GTL2 allele, respectively. Sperm epimutations are indicated by arrows.
Deep bisulfite sequencing (DBS)
Methylation analysis of small pools of cells or even individual cells is relatively time-consuming and only a limited number of individual DNA molecules (several dozen to several hundred) can be sequenced. Next generation sequencing of bisulfite-treated DNA samples can generate methylation data of several hundred to several thousand individual alleles each of different target genes and multiple samples in a single experiment. We used DBSCitation29 for methylation analysis of 2 pluripotency (NANOG and OCT4) and 2 (paternally methylated GTL2 and maternally methylated PEG3) imprinted genes in 54 sperm samples from men (attending a fertility center) with normal semen parameters (N) and OAT syndrome (O). In both groups, approximately half of the samples had led to a live birth after ICSI (N+ and O+) and the other half had not (N- and O-). On average, each sample yielded 1164 (range 101–4455) reads for GTL2, 1473 (297–6759) for PEG3, 581 (50–1537) for OCT4, and 976 (77–2697) for NANOG (). The mean methylation (number of methylated CpGs divided by the total number of CpGs in all analyzed alleles) was 94.6 ± 2.7% for GTL2, 1.5 ± 0.3% for PEG3, 92.3 ± 0.4% for OCT4, and 96.4 ± 0.2% for NANOG. Since DBS analyzes a larger number of CpGs (8 for GTL2, 19 for PEG3, 5 for OCT4, and 8 for NANOG) than LD, sperm alleles with no or a single CpG methylation error were considered as normal and alleles with >50% aberrant CpGs as abnormal (epimutations). Mixed alleles contained at least 2 but <50% CpG sites with the wrong (oocyte) methylation imprint.
Table 3. Number and methylation patterns of sperm alleles analyzed by deep bisulfite sequencing
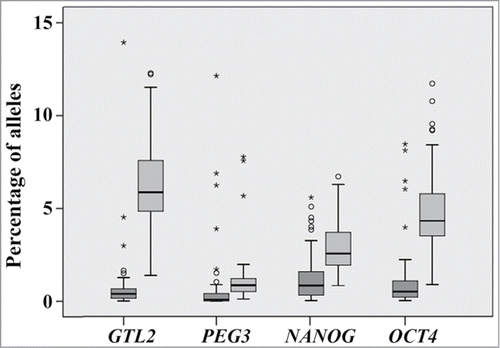
Across all genes and samples, the majority (>85%) of alleles displayed normal methylation patterns (). The percentage of mixed alleles differed significantly (ANOVA with Bonferroni post hoc test; P < 0.001) between the 4 studied genes, being the highest (6.5%) for GTL2 and the lowest (1.2%) for PEG3 (). The percentage of abnormal alleles did not differ significantly between genes. For most DBS measurements (4 genes x 54 samples), the percentage of abnormal alleles was lower than 1%, in one fifth of the cases even 0% (). In 22 (10%) of the 224 measurements the percentage of abnormal alleles ranged from 1–3%, in 15 (7%) cases from 3–10%, and in 2 (1%) cases was >10%.
Figure 3. Box plots showing the percentages of alleles with abnormal (dark gray) and mixed (light gray) methylation patterns for GTL2, PEG3, NANOG, and OCT4 in 54 sperm samples. The median is represented by a horizontal line. The bottom of the box indicates the 25th percentile, the top the 75th percentile. Outliers are shown as circles and extreme outliers as stars.
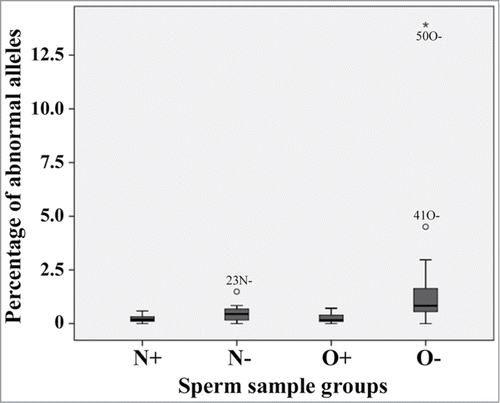
After correction for multiple testing, pluripotency genes did not show significant differences () between the 4 studied groups (N+, N-, O+, and O-). In contrast, the percentage of abnormally demethylated GTL2 alleles was significantly (P < 0.0001) higher in sperm samples (N- and O-) with negative ART outcome than in those (N+ and O+) leading to a child after ICSI. Box plots show a higher variation in the percentage of GTL2 epimutations in sperm samples with negative ART outcome, especially in the O- group than in sperm samples leading to live-birth (). Sperm samples from men with OAT syndrome and negative ART outcome (O-) had significantly more GTL2 epimutations than all other groups (P < 0.0001, compared to both N+ and O+; P = 0.02 for N-). To a lesser extent, the percentage of abnormally methylated PEG3 alleles also appeared to differ between groups (). Interestingly, we could not find any difference in methylation of the 4 studied genes between normozoospermic and OAT samples leading to a child (N+ versus O+).
Table 4. Between-group differences in the percentage of normal, mixed and abnormal alleles
Figure 4. Box plots showing the percentages of abnormal GTL2 alleles (>50% unmethylated CpGs) in sperm samples of men with normal semen parameters with (N+) and without child (N−), respectively, and men with OAT syndrome with (O+) and without child (O−), respectively. The median is represented by a horizontal line. The bottom of the box indicates the 25th percentile, the top the 75th percentile. Outliers are shown as circles and extreme outliers as stars.
In addition, we used binary logistic regression models to test the predictive values of percentages of normal, mixed and/or abnormal alleles of the 4 studied genes for ART outcome (live-birth of a child vs. no pregnancy). For NANOG, OCT4 and PEG3, the predictive values were low, ranging from 52% to 58%. A model based on the percentages of abnormal and normal GTL2 alleles (excluding intermediate alleles as covariate) had the highest predictive value (72%).
A single nucleotide polymorphism (SNP rs71594112) in the GTL2 amplicon allowed us to distinguish the 2 parental alleles. Of the 54 analyzed sperm samples 34 were homozygous TT, 4 homozygous CC, and 16 heterozygous TC. When using a more stringent filter setting (than for standard methylation analysis of low complexity bisulfite-converted DNA) to avoid SNP-mistyping, 9 of the 16 heterozygous samples still displayed multiple GTL2 alleles with epimutations. In five of these informative samples (17N-, 18N-, 20N-, 39O+, and 49O-) the GTL2 epimutations were confined to a single parental allele, whereas in 4 cases (26N-, 40O+, 45O-, and 47O-) both the T and the C allele were affected. Since we did not have parental DNAs, the parental origin of the T and C allele, respectively, in a given sample could not be determined.
Discussion
IMSI has been developed to pick out the "best" sperm of samples from men with compromised semen parameters, using a 10- to 15-times higher magnification than ICSI to assess sperm morphology. Vacuoles can be present in sperm of fertile and infertile men. So far, there is no conclusive evidence whether or not these vacuoles are associated with functional abnormalities such as DNA damage, chromatin decondensation, immaturity, and/or fertilization problems.Citation30-32 Some studies reported higher pregnancy rates and lower abortion rates using IMSI.Citation28,33-35 However, a recent meta-analysis did not find evidence for an improved ART outcome using IMSI, compared to standard ICSI.Citation36 This is consistent with our observation that the epimutation rate (of 3 imprinted genes) in the "best" sperm (IMSI+), sperm with vacuoles (IMSI-) and morphological abnormalities (IMSI-/-) is comparable to that of ICSI sperm. At the single sperm level the presence of vacuoles or morphological abnormalities did not correlate with epigenetic abnormalities. Low levels of epimutations (2–4%) were found in all 4 classes of sperm tested. In addition to LD bisulfite sequencing which studies methylation of individual DNA molecules from small pools of sperm, we have developed a multiplex PCR assay for methylation analysis of up to 8 genes in single sperm. When we compared individual sperm from men with OAT and fertile controls, we found a higher rate of epimutations in the OAT sperm, but the majority of sperm in both groups showed normal methylation patterns for the studied genes.
Because both LD and single sperm methylation analysis are time-consuming and expensive, only a limited number of samples and/or alleles can be analyzed, making it difficult to reach statistical significance. In contrast, DBS can generate methylation information on a large number of individual DNA molecules (from different target genes and different samples) in a single experiment. Unlike bisulfite pyrosequencing, which determines the average methylation level of a large number of DNA molecules at single CpG resolution,Citation26 DBS can distinguish between normally and abnormally methylated sperm alleles and, thus, detect rare epimutations in a much larger pool of normal alleles ("needles in a haystack"). In this context, it is important to emphasize that for imprinted genes the maternal alleles from somatic cells would mimic epimutations in sperm. Thus, even a very minor contamination of a given sperm sample with somatic cells interferes with correct data interpretation. For the quantification of sperm epimutations, meticulous care has to be taken to avoid contamination with somatic DNA, i.e., from lymphocytes and epithelial cells in the ejaculate. To this end, the swim-up sperm fraction was purified further by density gradient centrifugation and the resulting sperm purity was then checked by inverted light microscopy.
In all (normozoospermic and OAT) samples the vast majority of alleles of all 4 studied genes showed normal methylation patterns and in most (>80%) cases the percentage of epimutations was very low (<1%). Interestingly, the percentage of mixed alleles with multiple single CpG methylation errors differed significantly between genes (1.2% for PEG3, 2.9% for NANOG, 4.8% for OCT4, and 6.5% for GTL2). Since the same bisulfite-converted sperm DNAs were used for all measurements, this is unlikely due to a technical artifact, i.e., different rates of bisulfite conversion errors. Moreover, LD bisulfite sequencing also showed a significantly increased number of CpG methylation errors in GTL2, compared to LIT1 and PEG3. Thus, some genes may be more prone than others to stochastic methylation errors in sperm, e.g., by less stringent maintenance of the methylation imprints established in the male germline.
When looking for possible epigenetic signatures for sperm quality, the 2 pluripotency genes did not correlate with semen parameters (normozoospermia versus OAT) or ART outcome (live-birth vs. no pregnancy). In contrast, the percentage of GTL2 epimutations was significantly lower in sperm samples producing a child than in those that did not. The association of GTL2 epimutations with negative ART outcome was highly significant (P < 0.0001) for OAT sperm samples (O+ versus O-); a similar trend (P = 0.03) was observed for normozoospermic samples (N+ vs. N-). GTL2 is the only studied gene with a paternal methylation imprint that must be maintained after fertilization. PEG3 is demethylated on the paternal allele in both sperm and embryo, whereas the methylated NANOG and OCT4 sperm alleles become actively demethylated after fertilization.Citation10,11 It is tempting to speculate that transmission of correct paternal methylation imprints into the embryo are more crucial for further development and, therefore, a better predictor of sperm quality and ART success than genes that are either demethylated in sperm or become demethylated after fertilization. However, we need information on more sperm genes to test this hypothesis.
In most analyzed samples, sperm epimutations were found at low frequency in all 4 studied genes. The epimutations in different genes most likely arose in different precursor cells, resulting in extensive epigenetic mosaicism in the germline of a given male. There is no evidence that epimutations in different genes accumulate in the same germ cell and its clonal descendants. First, multiplex methylation analysis of single sperm () usually revealed only a single epimutated allele and normal methylation patterns in the additional recovered alleles. Second, the percentage of epimutations determined by DBS usually varied between the 4 studied genes in a given sperm sample. For example, sample 13 (N+) showed 0% GTL2, 0.1% PEG3, 0.2% NANOG, and 6% OCT4 allele methylation errors; sample 48 (O-) 3% GTL2, 12% PEG3, and 1.3% NANOG epimutations. Our results are consistent with the view that in most cases of male infertility with an epigenetic component there may be only a relatively modest increase in the number of epimutations at the individual gene level, but if multiple genes are affected, this may result in a large proportion of sperm with compromised epigenome. A conceptually related DBS studyCitation37 demonstrated much higher epimutation rates in 7 OAT sperm samples, affecting 4 imprinted genes more or less equally. This may be explained by technical differences or, more likely, by preselection of sperm samples with known gross epigenetic abnormalities. The 27 OAT samples in our study were only selected on the basis of semen parameters and ART outcome and, therefore may be more representative of the "average" infertile man attending a fertility center.
In approximately half of the sperm samples that were heterozygous for a GTL2 SNP, epimutations were confined to one of the 2 parental alleles, consistent with a single hit in a precursor cell. In the other half, both alleles were affected, suggesting (at least) 2 hits, one in the paternal and one in the maternal allele in different precursor cells. Sperm epimutations can result from incomplete erasure of the parental methylation patterns in primordial germ cells, failures in the establishment of paternal methylation imprints after erasure (from prenatal mitotic arrest to postnatal pachytene stage), or failures in the maintenance of established methylation patterns (from pachytene to mature sperm). At least for the paternally methylated GTL2 gene, epimutations (abnormal demethylation) can affect both parental alleles.
Overall, DBS is a high-throughput technique, which allows one to identify and precisely quantify the number of epimutations which amount to just a few percent (in most cases <1%) of all DNA target molecules in a given sperm sample. Our results show that in both normozoospermic and OAT samples the vast majority of alleles display normal methylation patterns (at least for the 4 studied genes). For our best marker gene of ART outcome, GTL2, the average percentage of abnormally demethylated alleles was 0.2% in normozoospermic (N+) and 0.3% in OAT (O+) samples leading to a child, compared to 0.5% in normozoospermic (N-) and 1.8% in OAT (O-) samples not leading to a pregnancy. Although these between-group differences are highly significant (P < 0.0001), most conventional techniques for methylation analysis, including bisulfite plasmid sequencing, LD analysis of small sperm pools, and bisulfite pyrosequencing of sperm DNA, are unable to detect such small increments in epimutations in individual genes. Massive parallel sequencing of thousands of individual DNA target molecules in a biological sample can overcome this problem. From a clinical point of view, it is helpful to define thresholds for the prediction of ART outcome. When looking at our data set (), an empirical threshold of 1% GTL2 epimutations seems to be practicable. None (0%) of the 26 samples (N+ and O+) leading to a child, but 8 (29%) of 28 samples (N- and O-) with negative ART outcome had ≥1% GTL2 epimutations. Combined with classical semen parameters, the predictive power is even better: 7 (50%) of 14 OAT samples not leading to a child (O-) displayed an increased GTL2 epimutation rate.
Materials and Methods
Sperm samples
The study was approved by the Ethics Committee at the Medical Faculty of Wuerzburg University and informed consent was obtained from all participating subjects. Sperm samples for DBS were collected from 54 couples undergoing infertility treatment at the Fertility Center Wiesbaden and pseudonymized before methylation analysis. Twenty-seven samples were from men with repeatedly normal semen parameters and 27 from men with OAT syndrome. Following ICSI, 13 samples of each group led to a live birth and 14 did not lead to a pregnancy. Semen parameters were measured according to the WHO guidelines.Citation38 OAT was assumed in males with sperm concentration fewer than 15 × 106/ml (oligozoospermia), fewer than 32% spermatozoa with progressive motility (asthenozoospermia), and fewer than 4% spermatozoa with normal morphology (teratozoospermia).
The left-over swim-up sperm fraction (excess material) was frozen at −80°C until further use. After thawing, sperm cells were purified by PureSperm 40/80 (Nidacon, Mölndal, Sweden) gradient centrifugation. Purified sperm were incubated for 2 h at 56°C (on a thermomixer) with 100 mM TrisCl, 10 mM EDTA, 500 mM NaCl, 1% SDS, 2% β-mercaptoethanol, and 100 μl proteinase K (>600 mAU/ml). Additional 20 μl proteinase K were added and incubated for another 2 h at 56°C. DNA was isolated with the DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) following the recommendations of the manufacturer. DNA concentration was measured using a NanoDrop 2000c spectrophotometer (PeqLab, Erlangen, Germany). Bisulfite conversion of sperm DNA was performed using the EZ DNA Methylation kit (Zymo Research Corporation, Irvine, CA, USA).
For LD bisulfite pyrosequencing,Citation26 pools of 10 sperm each (in 10 μl PBS) were selected from sperm samples of 3 healthy donors with normal semen parameters and proven fertility. A high-powered microscope (magnification 6000×) for IMSI was used to pick out sperm based on morphological criteria. IMSI+ sperm were motile, had normal morphology and did not contain vacuoles,Citation39 IMSI- sperm had large vacuoles, and IMSI-/- showed morphological (head and/or neck) abnormalities as well as vacuoles. ICSI sperm were picked out at lower magnification (400×) according to standard criteria (normal motility and morphology but small inclusions in sperm head cannot be excluded). For single sperm analysis, 80 swim-up sperm from 3 fertile donors with normal semen parameters and 144 sperm from 8 infertile men with OAT syndrome were collected in 224 individual tubes.
Deep bisulfite sequencing
The PyroMark Assay Design 2.0 software (Biotage, Uppsala, Sweden) was used for primer design (Supplemental Table 1). The GTL2 target sequence contained a T/C-SNP (rs71594112; chr.14:100,809,346 bp) with a minor allele frequency of 0.13. Cis-regulatory regions of 2 pluripotency (NANOG and OCT4) genes, one paternally methylated (GTL2) and one maternally methylated (PEG3) imprinted gene were amplified in a first round polymerase chain reaction (PCR) from bisulfite-treated sperm DNA. The 25 μl reaction mixture consisted of 2.5 μl 10× PCR buffer with MgCl2 (Roche Diagnostics, Mannheim, Germany), 0.5 μl (10 mM dNTPs) PCR Grade Nucleotide Mix (Roche Diagnostics), 0.2 μl (5 U/μl) FastStart Taq DNA Polymerase (Roche Diagnostics), 1 μl (10 pmol/μl) of forward and reverse primer (Metabion, München-Planegg, Germany) as well as 1 μl bisulfite-converted sperm DNA. For GTL2, 2.5 μl 10× PCR buffer without MgCl2 and 4 μl MgCl2 (25 mM) were used. PCR was carried out with an initial denaturation step at 95°C for 5 min, 40 (for NANOG and PEG3) and 50 cycles (OCT4 and GTL2), respectively, of 95°C for 30 s (denaturation), primer-specific annealing temperature (56°C for NANOG, 60°C for GTL2 and PEG3, and 62°C for OCT4) for 30 s, and 72°C for 45 s (elongation), and a final extension step at 72°C for 10 min. Sample-specific multiplex identifiers (MIDs), 454 Titanium A and B sequences, and key (TCAG) sequences were added in a second round PCR. After initial denaturation at 95°C for 10 min, 45 cycles of 95°C for 20 s, and 72°C for 45 s (annealing and elongation) with a final elongation step at 72°C for 7 min were performed.
Prior to DBS second-round amplification products were purified using Agencourt AMPure XP Beads (Beckman Coulter, Krefeld, Germany) and quantified using the NanoDrop 2000c spectrophotometer. After pooling, dilution and emulsion PCR (emPCR), amplification products were sequenced on a Roche/454 GS Junior system, following the protocol described in the Roche emPCR Amplification Method and Sequencing Method Manual. Sequencing runs were processed using a special filter setting (bisRecommendation filter) for low complexity bisulfite-converted DNA that is depleted of C (sense) or G (antisense strand) nucleotides. Briefly, the default "filterOnlyAmplicons" that is applied when using the “full processing for amplicons” option in the Roche Genome Sequencer application was modified to increase the yield of reads from GC-poor bisulfite-treated DNA.Citation29 Sequence reads were then analyzed further using the Amplikyzer program.Citation40 For SNP-typing the more stringent filter setting of the Roche Genome Sequencer application was used (instead of the bisRecommendation filter).
LD bisulfite pyrosequencing
DNA was extracted from pools of 10 sperm each and bisulfite-treated with the EZ DNA methylation direct kit (Zymo Research Corporation), which is particularly suited for small amounts of DNA. Bisulfite-treated DNA was eluted in 10 μl of elution buffer, diluted to a final volume of 200 μl and evenly distributed into 20 wells of a 96-well plate. Additionally, 4 negative controls were added for each PCR reaction. Multiplex (GTL2, LIT1 and PEG3) PCR was performed in 25 μl reactions, each well containing 2.5 μl 10× PCR buffer with MgCl2, 0.5 μl (10 mM dNTPs) PCR Grade Nucleotide Mix, 0.2 μl (5 U/μl) FastStart Taq DNA Polymerase, and 0.33 μl (33 μM) forward and reverse outer primers (Metabion) (Supplemental Table 1). PCR was carried out with an initial denaturation step at 95°C for 5 min, 35 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 45 s, and a final extension step at 72°C for 5 min. Nested singleplex PCRs for each of the studied genes were performed using 1 μl of the first-round multiplex PCR product as a template. The 25 μl reaction volume consisted of 2.5 μl 10× PCR buffer with MgCl2, 0.5 μl PCR Grade Nucleotide Mix, 0.2 μl FastStart Taq DNA Polymerase, and 1 μl (10 pmol/μl) forward and reverse inner primers. Cycling conditions were as follows: 95°C for 5 min, 32 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 45 s, and a final extension of 7 min at 72°C. For LIT1, 40 cyles with an annealing temperature of 54°C were performed. The nested PCRs (4 μl each) were run on 2% agarose gels to visualize reactions yielding a product representing a single DNA molecule (allele) in the starting sample. Because bisulfite-treated DNA is heavily degraded, the number of wells containing an amplifiable DNA template was always markedly lower than the number of DNA molecules in the starting sample.
Pyrosequencing was performed on a Pyromark Q96MD system with the PyroGold SQA reagent kit (Qiagen). The Pyromark Q-CpG software was used to quantify methylation of individual CpGs. Sequestration of individual DNA molecules from the starting sample in separate PCR reactions reduces the methylation status of the targeted sequence to a binary state: an analyzed CpG site is either methylated (100%) or not (0%). However, because the actual measurements also depend on the sequence context and other factors, methylation values of <20% are typical for unmethylated and >80% for methylated sites. Lollipop diagrams were generated using BiQ Analyzer.Citation41
Single sperm analysis
Following bisulfite treatment with the EZ DNA methylation direct kit, the DNA of a single sperm was eluted in 10 μl. Multiplex (BOLL, CATSPER1, OCT4, NANOG, GTL2, LIT1, PEG3, and SNRPN) PCR of single sperm DNA was performed in 25 μl reactions containing 2.5 μl 10× PCR buffer without MgCl2, 4 μl (25 mM) MgCl2, 0.5 μl (10 mM dNTPs) PCR Grade Nucleotide Mix, 0.2 μl (5 U/μl) FastStart Taq DNA Polymerase, and 0.33 μl (33 μM) of each forward and reverse outer primer (Supplemental Table 1). PCR was carried out with an initial denaturation at 95°C for 5 min, 35 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 45 s, and a final extension step at 72°C for 5 min. Nested PCRs were generally performed using 2.5 μl 10× PCR buffer with MgCl2, 0.5 μl PCR Grade Nucleotide Mix, 0.2 μl FastStart Taq DNA Polymerase, 1 μl (10 pmol/μl) forward and reverse inner primer, and 1 μl multiplex PCR product. For BOLL, 2.5 μl 10× PCR Rxn Buffer without MgCl2 (Invitrogen, Darmstadt, Germany), 0.75 μl (50 mM) MgCl2, 0.5 μl PCR Grade Nucleotide Mix, and 0.2 μl (5 U/μl) Platinum Taq DNA Polymerase (Invitrogen) were used. Cycling conditions were initial denaturation at 95°C for 5 min, 32 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 45 s, and a final extension at 72°C for 5 min. For BOLL, 45 cycles and an annealing temperature of 58°C; for CATSPER1, 40 cycles and 60°C; for LIT1, 35 cycles and 57°C were used.
Statistical analyses
All descriptive and inferential statistical analyses were performed with IBM SPSS version 22.0.0.0 (http://www.spss.com). Depending on data distribution, the Mann-Whitney test or ANOVA with Bonferroni post hoc test was applied for group comparisons. P values were adjusted for multiple testing by the Bonferroni method. P < 0.05 was considered as significant. To estimate the predictive value of DBS data (), a binary logistic regression analysis was performed using "live-birth" versus "no pregnancy" as outcome variable and the percentages of normal, mixed and abnormal alleles of the 4 studied genes as covariates.
Outlook
A predictive test based on GTL2 alone (using a 1% threshold) has a high specificity (no false positives in this pilot study) but relatively low sensitivity (50% for the OAT and 30% for all samples). This may still be inadequate for a stand-alone test in a clinical setting. However, it is feasible to identify additional epigenetic signatures for ART outcome, e.g., by genome-wide methylation analyses or to combine classical and epigenetic semen parameters, improving sensitivity. Considering that multiple sperm samples and marker genes can be analyzed in a single next generation sequencing run, epigenetic signatures can provide an additional layer of information for assessing sperm quality before infertility treatment. With an improved epigenetic marker panel, DBS may become a powerful tool for differential diagnosis of male infertility and the prediction of ART outcome.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Table_S1.docx
Download MS Word (25.8 KB)Funding
This study was supported by the German Research Foundation (grant no. HA 1374/13‑1).
References
- Singh K, Jaiswal D. Human male infertility: a complex multifactorial phenotype. Reprod Sci 2011; 18:418-25; PMID:21421900; http://dx.doi.org/10.1177/1933719111398148
- Tüttelmann F, Werny F, Cooper TG, Kliesch S, Simoni M, Nieschlag E. Clinical experience with azoospermia: aetiology and chances for spermatozoa detection upon biopsy. Int J Androl 2011; 34:291-8; PMID:20609027; http://dx.doi.org/10.1111/j.1365-2605.2010.01087.x
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature 2012; 488:471-5; PMID:22914163; http://dx.doi.org/10.1038/nature11396
- Bennett-Baker PE, Wilkowski J, Burke DT. Age-associated activation of epigenetically repressed genes in the mouse. Genetics 2003; 165:2055-62; PMID:14704185
- Goyal R, Reinhardt R, Jeltsch A. Accuracy of DNA methylation pattern preservation by the Dnmt1 methyltransferase. Nucleic Acids Res 2006; 34:1182-88; PMID:16500889; http://dx.doi.org/10.1093/nar/gkl002
- Hajkova P, Erhardt S, Lane N, Haaf T, El-Maarri O, Reik W, Walter J, Surani MA. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev 2002; 117:15-23; PMID:12204247; http://dx.doi.org/10.1016/S0925-4773(02)00181-8
- Allegrucci C, Thurston A, Lucas E, Young L. Epigenetics and the germline. Reproduction 2005; 129:137-49; PMID:15695608; http://dx.doi.org/10.1530/rep.1.00360
- Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol 2007; 307:368-79; PMID:17559830; http://dx.doi.org/10.1016/j.ydbio.2007.05.002
- Boyano MD, Andollo N, Zalduendo MM, Aréchaga J. Imprinting of mammalian male gametes is gene specific and does not occur at a single stage of differentiation. Int J Dev Biol 2008; 52:1105-11; PMID:18956343; http://dx.doi.org/10.1387/ijdb.072284mb
- Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science 2001; 293:1089-93; PMID:11498579; http://dx.doi.org/10.1126/science.1063443
- Haaf T. Methylation dynamics in the early mammalian embryo: implications of genome reprogramming defects for development. Curr Top Microbiol Immunol 2006; 310:13-22; PMID:16909904
- Kelsey G. Genomic imprinting - roles and regulation in development. Endocr Dev 2007; 12:99-112; PMID:17923773
- Carrell DT. Epigenetics of the male gamete. Fertil Steril 2012; 97:267-74; PMID:22289286; http://dx.doi.org/10.1016/j.fertnstert.2011.12.036
- Boissonnas CC, Jouannet P, Jammes H. Epigenetic disorders and male subfertility. Fertil Steril 2013; 99:624-31; PMID:23714437; http://dx.doi.org/10.1016/j.fertnstert.2013.01.124
- Marques CJ, Costa P, Vaz B, Carvalho F, Fernandes S, Barros A, Sousa M. Abnormal methylation of imprinted genes in human sperm is associated with oligozoospermia. Mol Hum Reprod 2008; 14:67-74; PMID:18178607; http://dx.doi.org/10.1093/molehr/gam093
- Hammoud SS, Purwar J, Pflueger C, Cairns BR, Carrell DT. Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil Steril 2010; 94:1728-33; PMID:19880108; http://dx.doi.org/10.1016/j.fertnstert.2009.09.010
- Poplinski A, Tüttelmann F, Kanber D, Horsthemke B, Gromoll J. Idiopathic male infertility is strongly associated with aberrant methylation of MEST and IGF2/H19 ICR1. Int J Androl 2010; 33:642-9; PMID:19878521
- Sato A, Hiura H, Okae H, Miyauchi N, Abe Y, Utsunomiya T, Yaegashi N, Arima T. Assessing loss of imprint methylation in sperm from subfertile men using novel methylation polymerase chain reaction Luminex analysis. Fertil Steril 2011; 95:129-34; PMID:20655520; http://dx.doi.org/10.1016/j.fertnstert.2010.06.076
- Kobayashi H, Hiura H, John RM, Sato A, Otsu E, Kobayashi N, Suzuki R, Suzuki F, Hayashi C, Utsunomiya T, et al. DNA methylation errors at imprinted loci after assisted conception originate in the parental sperm. Eur J Hum Genet 2009; 17:1582-91; PMID:19471309; http://dx.doi.org/10.1038/ejhg.2009.68
- El Hajj N, Zechner U, Schneider E, Tresch A, Gromoll J, Hahn T, Schorsch M, Haaf T. Methylation status of imprinted genes and repetitive elements in sperm DNA from infertile males. Sex Dev 2011; 5:60-9; PMID:21293114; http://dx.doi.org/10.1159/000323806
- Aston KI, Punj V, Liu L, Carrell DT. Genome-wide sperm deoxyribonucleic acid methylation is altered in some men with abnormal chromatin packaging or poor in vitro fertilization embryogenesis. Fertil Steril 2011; 97:285-92; PMID:22154369; http://dx.doi.org/10.1016/j.fertnstert.2011.11.008
- Pacheco SE, Houseman EA, Christensen BC, Marsit CJ, Kelsey KT, Sigman M, Boekelheide K. Integrative DNA methylation and gene expression analyses identify DNA packaging and epigenetic regulatory genes associated with low motility sperm. PLoS One 2011; 6:e20280; PMID:21674046; http://dx.doi.org/10.1371/journal.pone.0020280
- Schütte B, El Hajj N, Kuhtz J, Nanda I, Gromoll J, Hahn T, Dittrich M, Schorsch M, Müller T, Haaf T. Broad DNA methylation changes of spermatogenesis, inflammation and immune response-related genes in a subgroup of sperm samples for assisted reproduction. Andrology 2013; 1:822-9; PMID:23996961; http://dx.doi.org/10.1111/j.2047-2927.2013.00122.x
- Jenkins TG, Aston KI, Pflueger C, Cairns BR, Carrell DT. Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet 2014; 10:e1004458; PMID:25010591; http://dx.doi.org/10.1371/journal.pgen.1004458
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457-66; PMID:17334365; http://dx.doi.org/10.1038/ng1990
- El Hajj N, Trapphoff T, Linke M, May A, Hansmann T, Kuhtz J, Reifenberg K, Heinzmann J, Niemann H, Daser A, et al. Limiting dilution bisulfite (pyro)sequencing reveals parent-specific methylation patterns in single early mouse embryos and bovine oocytes. Epigenetics 2011; 6:1176-88; PMID:21937882; http://dx.doi.org/10.4161/epi.6.10.17202
- Minor A, Chow V, Ma S. Aberrant DNA methylation at imprinted genes in testicular sperm retrieved from men with obstructive azoospermia and undergoing vasectomy reversal. Reproduction 2011; 141:749-57; PMID:21389080; http://dx.doi.org/10.1530/REP-11-0008
- Bartoov B, Berkovitz A, Eltes F. Selection of spermatozoa with normal nuclei to improve the pregnancy rate with intracytoplasmic sperm injection. N Engl J Med 2001; 345:1067-8; PMID:11586970; http://dx.doi.org/10.1056/NEJM200110043451416
- Beygo J, Ammerpohl O, Gritzan D, Heitmann M, Rademacher K, Richter J, Caliebe A, Siebert R, Horsthemke B, Buiting K. Deep bisulfite sequencing of aberrantly methylated loci in a patient with multiple methylation defects. PLoS One 2013; 8:e76953; PMID:24130816; http://dx.doi.org/10.1371/journal.pone.0076953
- Watanabe S, Tanaka A, Fujii S, Mizunuma H, Fukui A, Fukuhara R, Nakamura R, Yamada K, Tanaka I, Awata S, et al. An investigation of the potential effect of vacuoles in human sperm on DNA damage using a chromosome assay and the TUNEL assay. Hum Reprod 2011; 26:978-86; PMID:21362682; http://dx.doi.org/10.1093/humrep/der047
- Franco JG Jr, Mauri AL, Petersen CG, Massaro FC, Silva LF, Felipe V, Cavagna M, Pontes A, Baruffi RL, Oliveira JB, et al. Large nuclear vacuoles are indicative of abnormal chromatin packaging in human spermatozoa. Int J Androl 2012; 35:46-51; PMID:21535011; http://dx.doi.org/10.1111/j.1365-2605.2011.01154.x
- Perdrix A, Rives N. Motile sperm organelle morphology examination (MSOME) and sperm head vacuoles: state of the ART in 2013. Hum Reprod Update 2013; 19:527-41; PMID:23825157; http://dx.doi.org/10.1093/humupd/dmt021
- Bartoov B, Berkovitz A, Eltes F, Kogosovsky A, Yagoda A, Lederman H, Artzi S, Gross M, Barak Y. Pregnancy rates are higher with intracytoplasmic morphologically selected sperm injection than with conventional intracytoplasmic injection. Fertil Steril 2003; 80:1413-39; PMID:14667877; http://dx.doi.org/10.1016/j.fertnstert.2003.05.016
- Berkovitz A, Eltes F, Yaari S, Katz N, Barr I, Fishman A, Bartoov B. The morphological normalcy of the sperm nucleus and pregnancy rate of intracytoplasmic injection with morphologically selected sperm. Hum Reprod 2005; 20 185-90; PMID:15471930; http://dx.doi.org/10.1093/humrep/deh545
- Antinori M, Licata E, Dani G, Cerusico F, Versaci C, D'Angelo D, Antinori S. Intracytoplasmic morphologically selected sperm injection: a prospective randomized trial. Reprod Biomed Online 2008; 16:835-41; PMID:18549694; http://dx.doi.org/10.1016/S1472-6483(10)60150-2
- Teixeira DM, Barbosa MA, Ferriani RA, Navarro PA, Raine-Fenning N, Nastri CO, Martins WP. Regular (ICSI) vs. ultra-high magnification (IMSI) sperm selection for assisted reproduction. Cochrane Database Syst Rev 2013; 7:CD010167; PMID:23884963; http://dx.doi.org/10.1002/14651858.CD010167
- Laurentino S, Beygo J, Nordhoff V, Kliesch S, Wistuba J, Borgmann J, Buiting K, Horsthemke B, Gromoll J. Epigenetic germline mosaicism in infertile men. Hum Mol Genet 2014; Epub ahead of print: PMID:25336341
- World Health Organization. WHO Laboratory Manual for the Examination and Processing of Human Semen. 5th edition. WHO press 2010
- Ubaldi F, Rienzi L. Morphological selection of gametes. Placenta 2008; 29 Suppl B:115-20; PMID:18762336; http://dx.doi.org/10.1016/j.placenta.2008.08.009
- Rahmann S, Beygo J, Kanber D, Martin M, Horsthemke B, Buiting K. Amplikyzer: automated methylation analysis of amplicons from bisulfite flowgram sequencing. PeerJ PrePrints 2013; 1:e122v2
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 2005; 21:4067-8; PMID:16141249; http://dx.doi.org/10.1093/bioinformatics/bti652