
Abstract
Molecular breeding has a crucial role in improvement of crops. Conventional breeding techniques have failed to ameliorate food production. Next generation sequencing has established new concepts of molecular breeding. Exome sequencing has proven to be a significant tool for assessing natural evolution in plants, studying host pathogen interactions and betterment of crop production as exons assist in interpretation of allelic variation with respect to their phenotype. This review covers the platforms for exome sequencing, next generation sequencing technologies that have revolutionized exome sequencing and led toward development of third generation sequencing. Also discussed in this review are the uses of these sequencing technologies to improve wheat, rice and cotton yield and how these technologies are used in exploring the biodiversity of crops, providing better understanding of plant-host pathogen interaction and assessing the process of natural evolution in crops and it also covers how exome sequencing identifies the gene pool involved in symbiotic and other co-existential systems. Furthermore, we conclude how integration of other methodologies including whole genome sequencing, proteomics, transcriptomics and metabolomics with plant exomics covers the areas which are left untouched with exomics alone and in the end how these integration will transform the future of crops.
Abbreviations
| BAC | = | bacterial artificial chromosome |
| BGR | = | bacterial grain rot |
| CBOL | = | consortium for 860 the barcode of life |
| dNMPs | = | deoxyribosenucleoside monophosphates |
| ETI | = | effector-triggered immunity |
| HPRT | = | hypoxanthineguanine phosphoribosyl transferase |
| MMs | = | molecular markers |
| NGS | = | next generation sequencing |
| NITSR | = | nuclear internal transcribed spacer region |
| OPC | = | open promoter complex |
| QTL | = | quantitative trait locus |
| SMRT | = | single molecule real time |
| SNPs | = | single nucleotide poly-morphisms |
| SOLiD | = | sequencing by oligonucleotide ligation and detection |
| WES | = | whole exome sequencing |
| WGS | = | whole genome sequencing |
| WGS | = | whole genome shotgun |
Introduction
Crop improvement is hampered by the breeding strategies that have been a tradition and further improvement in food production is demanded in order to encounter the energy needs of world. Breeding strategies established on methods that utilize molecular markers (MMs) have undoubtedly contributed in the improvement of crop quality. Though like every new method that emerges alongside its limitations, these techniques additionally have limitations that mostly deal with identification of specific MMs for different phenotypic traits. NGS (next-generation sequencing) strategies have proven to sequence whole genomes easily by facilitating the new MM identification and have substituted the PCR or Restriction sequence based biomarker screening using exome sequencing. Whole Genome Sequencing (WGS) methodology although possesses the ability of identification of single nucleotide variations but in comparison with Whole Exome Sequencing (WES) are not preferred due to elevated price and time period required for identification of whole sequence. Furthermore, analysis of whole genome can be enormous and time consuming as WGS assay has been found to be 6 times more expensive than that of WES assay.Citation1
Plant breeding techniques require to concentrate on those traits that have greatest yielding potential under continuously changing climatic conditions. Previous practices in agriculture have terminated local conventional varieties and its wild relatives, which have resulted in loss of biodiversity. Thus, genotypic and phenotypic methods are to be improved by development of new technologies in order to accelerate plant breeding.Citation2 Progress has made in many aspects from crop improvementCitation3 to forest tree management.Citation4-5
Exome pertains to exons, which code for the protein region in whole genome of an organism. These sequences representing about a hundredth of whole genome includes all the sequences that code for proteins, which are responsible for phenotypic regulation although they do not give a complete picture of gene regulation.Citation6 Exome Sequencing has emerged as a vital genetic tool for revealing molecular basis of diseases and phenotypic traits.
Exome sequencing involves identification of genes (whole exome, genes responsible for a disease or class of genes) followed by specificity step (designing of primers or capturing chip) leading to capture of regions of interest (e.g. whole genome) and hence creation of small sequence fragments during a sequencing reaction, which can either be stored as library or used to determine phenotypic traits determination, identification of thousands of exome SNPs, and further computational and statistical use in identification of disease associated signals.Citation7
First Generation Sequencing Technologies (Conventional Sequencing Methods)
Gene sequencing is the core of molecular biology and is highly recommended for identification of molecular basis of diseases and phenotypic traits. DNA sequencing technology entered into a new era after DNA sequencing (chemical sequencing method) based on the chemical-alteration dependent cleavage methodCitation8 which soon became the method of choice that time. Dideoxy method of chain termination was common method that is still in practice, published by Sanger and CoulsonCitation9 which laid foundation of first generation high throughput sequencers. This method makes use of dideoxynuecleotide using it as chain termination due to absence of 3′–OH group resulting in blockage of chain elongation. Bands thus formed are visualized using nucleotides incorporated with radioactive phosphorus. This method dominated over Maxam and Gilbert method because of its high efficiency and low toxicity. One major breakthrough in DNA sequencing field was the introduction of automation which provided users reliability over results, excluded the chances of human error and provided cost effectiveness during procedure. Sanger's method kept its process of evolution with advancement with time. Smith et al.Citation10 introduced 4 labeled Dideoxynuecleotide (ddATP, ddGTP, ddCTP, ddTTP) with fluorescent dyes for each ddNTP in single reaction.Citation10 Amplicon incorporated ddNTP resolution by capillary electrophoresis and detection by LASER technology was one evolution that is more significant. Despite being time effective and method of choice of current automated sequencing, this method has limitations in spite of all the precision. Few of these include poor quality results initially at 20–50 bases, and has upper limit of 600–1000 bases because large size DNA fragments are not properly resolved by capillary electrophoresis which also causes non specific primer binding and inclusion of DNA secondary structures. Sequence contamination with Vector sequences occur when attempts were made to sequence longer DNA fragments for cloning shorter sequences before sequencing. This issue was alleviated when sequencing strategies integrating PCR cloned fragments were improved along with improvement in bioinformatics tools. Combined amplicationCitation11 technologies further revolutionised methods for the amplification of longer fragments of DNA without chances of being contaminated.Citation12 Hypoxanthineguanine phosphoribosyl transferase gene was sequenced using an end-pair approach for sequencing of whole HPRT gene through automated fluorescent DNA sequencer.Citation13
Template Generation for Sequencing
dsDNA generally is used as a starting material for all NGS experiments however source can either be genomic, reverse transcribed RNA, cDNA or DNA obtained by immunoprecipitation. This starting material is used to make sequence reaction templates thus converting it into sequencing library by fragment generation followed by size selection and adaptor ligation.Citation14 First 2 steps function in break down of DNA templates into short fragments whose size is wholly dependent on sequencing platform's specifications. Third step ligate adaptor molecule serving as primer for the fragments generated in first 2 steps. These 3 steps ideally generate a sequencing library that accurately shows DNA population of sample.Citation15 Library sequencing is performed either by single molecule templates (directly sequenced) or is amplified first and sequenced afterwards which are categorized as clonally amplified templates (indirectly sequenced). Template generation also functions to separate and immobilize DNA fragments for sequencing by attaching DNA to solid surfaces (usually beads). Each template is having millions of micro reactions carrying out parallel in spatial dimension allowing the downstream sequencing.Citation16
Clonally amplified vs. single molecule template
Single molecule reactions cannot be monitored by couple of sequencing platformsCitation17 therefore template amplification method is hence adopted to produce adequate signal for nucleotide detection by system.Citation18 Emulsion PCR or bridging amplification are commonly used strategies by different platforms. Notably all amplification steps since include DNA polymerase are prone to experimental errors as these reagents being not accurate can introduce mutations into populations that are clonally amplified. Where as sequencing of single molecule template rules out the requirement for amplification steps thereby sequence detection can be done using lesser material.Citation19 Amplification and Single molecule base sequence technologies have been declared as second and third generation sequencing technologies respectively.
Second Generation Sequencing Technologies for Exome Sequencing
With emergence of exome sequencing concept, large sequence demands are continuously increasing, which can not be fulfilled by first generation sequencing technologies and their high throughput automated sequencers because of their limitations.Citation6 New sequencing techniques are thus formulated and commercialized recently to provide users novel approaches that can be utilized in exome sequencing to study physical traits, molecular markers identification, larger exome library formulation and unlocking Mendelian disease.Citation20 These Ultra-high sequencing platforms that omit Sanger's Chain termination sequencing technology are classified as next or second generation sequencing platforms.Citation6 These methodologies are based on diverse chemistries and detection approaches. While some of the techniques involved share very few features, key characteristic is common to all. These NGS technologies work on the principle of immobilization (up to billions) DNA templates on a solid surface, which in most of cases are beads.Citation15 Roche/454 a Genome Sequencer, GenomeAnalyzer from Illumina/Solexa, SOLiD of Applied Biosystems, and Polonator from Dover Systems are some of the commercially available next-generation sequencers.
Pyrosequencing
It is one of the efficient techniques used to sequence larger exon fragments in faster run times. The principle of detecting a pyrophosphate was first explained in 1985 and was later refined and developed into a functional method for DNA sequencing by Ansorge.Citation21 Pyrosequencing uses the Roche/454 FLX and was launched commercially in 2004. This process explains nucleotide integration by DNA polymerase, followed by release of pyrophosphate causing the commencement of a series of downstream reactions. Luciferase at the end causes the release of luminance by converting luciferin into oxyluciferin and its intensity is relative to the number of nucleotides added in the DNA polymerase.Citation6 The agarose beads used in the process have oligonucleotides on their surfaces, corresponding to the 454-exclusive adaptors on the library of DNA fragments.Citation22 This causes the association of a single bead with a single fragment and these complexes are then separated and added to micelles. The colloid also contains reactants of PCR. The micelles are added to a thermal cycle which causes the production of millions DNA copies on the surface of every bead.
After amplification, the molecules are sequenced all together. Firstly, beads are fitted into a solid-phase sequencing substrate (a Pico Titter Plate), which comprises of 1.6 million wells and has the ability to contain a single bead along with sequencing reagents (polymerase, luciferase, ATP sulfurylase). Each of the dNTPs is cycled over the wells by microfluidics allowing the nucleotide incorporation which causes the release of a pyrophosphate molecule. This molecule functions as the substrate for the luminance reaction. A CCD (Charge Coupled Device) camera is placed in front of the PPT to record the emission of light. The final data of luminance is recorded in the form of a flow diagram, which also corresponds to a chromatogram displaying the order of A, T, G and C residues of a sequencing template.Citation33
About 450,000 wells with detectable sequencing templates are presentCitation23 About 2.5 nucleotides during a normal run of 8 hours are integrated. Since the process allows for the flow of a single nucleotide phosphatase at a time, chances of substitution are highly unlikely.Citation24
The Roche system allows for comparable read lengths and a higher sequencing capacity as compared to Sanger's method. Genome of James Watson sequenced in 2 months at a cost of US$1 million by massively parallel pyrosequencing technique as compared to the 11 y and US$3 million HGP speaks for the efficiency of this procedure.Citation24 An upgraded version of Roche sequencer was released in 2009, with improved methods of library preparation and data management.Citation25
Sequencing by oligonucleotide ligation and detection
This exome sequencing approach uses the SOLiD (Sequencing by Oligonucleotide Ligation and Detection) sequencer, which was acquired by Applied Biosystems in the year 2006.Citation25 The technology was developed in the lab of George Church and the research was published in 2005 along with coli's resequenced genome.Citation26 The process initiated by construction of DNA libraries includes the first step of obtaining DNA fragments which are modified further by ligation of 25 bp adaptors (P1 along with P2) on the 3′ as well as 5′ ends.Citation21 This is followed by attachment of tailored fragments with beads and then the resulting fragment-beads complex is added to PCR containing a mixture of water, oil emulsion and amplification reagents.
The amplified libraries are then annexed to the sequencer flow cells where hybridization of a primer with the adaptor takes place. Next, oligonucleotide octamers are inserted followed by the addition of ligation mixture. The octamers constitute of a ligation site (base 1), a cleavage site (base 5) and 4 incandescent dyes (coupled with the last base). Any one of the 4 labels can differentiate fourth and fifth base pair. After fluorescence is detected, sequence of the doublet is determined and then the octamer is cleaved past the fifth base. The fragment is again passed through hybridization and ligation cycles and the sequences of bases 9th and 10th are determined followed by the 14th and 15th bases.Citation21 The templates might be sequenced by employing another primer, which is a base shorter than the previous one, in order to determine the sequence of the residual bases. Approximately 80% of the beads possess the ability to generate signals.Citation27
Accuracy of the process that allows the examination of each base twice is further improved by employment of a specialized system. The read length of the sequencer was primarily 35 bp per bead but recent advances in SOLiD technology have enhanced the read length to about 85 bp with a data output of about 3–6 Gbp.Citation27 A single run takes about 6–10 d.
Reversible terminator-based sequencing
This technology is utilized by the Illumina/Solexa Genome analyzer, which was conceived by Shankar and David by Solexa in 2006 and then purchased by Illumina in 2007. Central rationale behind its conception was to sequence DNA fragments affixed with microspheres but later the focus shifted to sequencing of clonally amplified DNA. The Illumina Genome Analyzer works on the theory of Sequencing-by-Synthesis (SBS) and employs a flow cell (also known as single-molecule array) which consists of 8 channels of microfabricated glass having oligonucleotide anchors on its surface and provides the platform for bridge amplification.
First step is the preparation of DNA fragment for ligation by splitting it into numerous base pairs, which are later, end-refurbished to fashion phosphorylated 5′ blunt ends. Anon, an Adenine base pair is inserted to the 3′ side by polymerase action of Klenow fragment. This allows ligation of DNA to oligonucleotide anchors, which contain a T overhang thus increasing the ligation competence.Citation26 Immediate hybridization of the DNA template occurs with anchors after it has been inserted into flow cell under conditions of minimal dilutions. The single-stranded DNA is augmented by Bridge amplification, which causes the in situ production of abundant copies of DNA (approx 5 × 10^6 clusters with each cluster containing 1000 clonally amplified molecules). Before the sequencing occurs, the clusters undergo denaturation and cleavage, which leaves forward single-stranded DNA fragments. The sequencing is instigated by affixing a primer with the adaptor sequences and later, there occurs the addition of polymerase and 4 fluorescently labeled, 3′-OH blocked nucleotides (ddATP, ddGTP, ddCTP, ddTTP). Excess reagents are washed off and a scan buffer is added. The optical interrogation with CCD takes place and recorded. Imaging of each lane takes place in 3 segments of 100-tile each.Citation22 After the image is documented, the 3′ ends are unblocked and the labels are cleaved and washed. The strands are incorporated with fresh nucleotides and the cycle is repeated.
Approximately 1 billion base pairs are sequenced in one analytical run (about 2–3 days). A technical difficulty of the Illumina is that the base-call accuracy decreases with an increase in read length. This event occurs due to ‘Dephasing Noise′ which happens when the nucleotides are either over or under incorporated or the unblocking of 3′ end is defective. The software of Illumina is being modified to alleviate these analytical aberrations. The GA 2 is an upgraded version that posses the ability to sequence paired-end libraries. The throughput is increased to approximately 200 Gbp per run.
Ion-semiconductor-based non-optical sequencing
In 2011, Life Technologies marketed the Ion Torrent Personal Genome Machine (PGM), which works by employing advanced semi-conductor technologies and sequences DNA by utilizing ion-sensitive transistors. This system of exome sequencing is in a league of its own when compared with other techniques of NGS. This is because it does not require fluorescence or chemiluminence to work and therefore does not employ the use of optics (e.g., CCD camera).Citation28 Substituting fluorescence detection by electric signals has numerous advantages. For one, it does not require tailored nucleotides or costly optical detection instruments. Apart from that, the difficulty that arises from processing output images is also eliminated.Citation24 The raw materials include natural nucleotides and DNA polymerase and each polymerization is characterized by pH changes.
Methodology includes the preparation of a template library by adaptor ligation to DNA templates under optimal conditions. A template is then fastened to a bead, which is followed by amplification using emulsion PCR. The clonally amplified templates are loaded on an ion-chip, which is basically a massively parallel, semi-conductor sensor that is receptive to ion activity and contains pH meters that are connected to copious wells, which are the location of DNA polymerization. Deoxynucleotide Triphosphates (dNTPs) are flowed over the beads in a predetermined pattern and ligation occurs.Citation29 A single proton is released for every nucleotide integrated causing a decrease in the pH of the circumambient solution. The altering pH is detected by the ion-sensor. The PGM software processes the values and refines the reads by removing defects like signal or phase loss. Homopolymeric regions might create nonlinear electric impulses.
Each flow cycle lasts for about an hour and sequences about 100–200 nucleotides per run. Its efficiency can be measured by ability to prepare 8 samples in parallel in 6 hours.
DNA nano-ball sequencing
Complete Genomics carries out DNA sequencing through unchained ligation by means of cPAL (combinatorial probe-anchor) technology. DNA is amplified in the form of nanoballs and not by the emulsion PCR methodology. Firstly, the DNA to be sequenced is fragmented to about 500 bases additionally; 4 adaptors are added into each of the fragments using repetitive cutting utilizing restriction enzymes along with ligation resulting in circular DNA fragments.Citation24 DNA nanoballs (DNB) originate after the fragments are amplified into coils. Each DNB consists of repetitive copies of the original circles and is approximately 200 nm in diameter.Citation6 The DNBs are then adhered to a silicon chip. This approach of unchained ligation sequencing employs anchor probes (locate the adaptor sites) and probes (used for detection and consist of nanomeres along with fluorescent dye). Both of these probes are hybridized and ligated in a way that they decipher one base in every cycle. In the first step of sequencing, the anchor is appended to one of the 4 adaptors of the fragment and 4 probes (each containing an optically discrete pigment) are used to examine position of the base contiguous to the adaptor. Dye is only connected to a definite position on the probe whereas the other sites are occupied by degenerated nucleotides. Fluorescence is detected using a high-resolution CCD camera and the fragment is sequenced 5-base at a time. After the luminance is observed, the anchor-probe complex is detached and the system resets.
Since the order of each base is determined independently, the sequencing quality is significant.Citation24 The efficiency is further augmented by commissioning a highly accurate DNA polymerase, which eliminates any chance of flawed rolling amplification. Furthermore, miscalculations are minimized since this method does not call for probe ligation to end before the successive round.Citation6 A miscalculation rate of 1 faulty variant per one hundred Kb was reported by the company. The sequencer is not available commercially.Citation30
Third Generation Sequencing Technologies for Exome Sequencing
Single molecule real time (SMRT) sequencing
Pacific Biosciences introduced this sequencing that utilizes Zero Mode Waveguides (ZMW) and deals with single DNA molecule, developed by Craighead and Webb of Cornell University. SMRT technology is based on a nano scale structure (ZMW) to observe real time DNA polymerization. It relies on ZMW chip containing numerous holes in the sub wavelength scale that have diameter in tens of nanometer, created by penetrating a slim metal film held on by transparent substrate.Citation31
SMRT approach is based on synthesis by sequencing, utilizing fluorescent labeled nucleotides. In this technique, DNA is necessitated to a minute volume in ZMW and the connate or agnate nucleotides which are fluorescently labeled, present near where the DNA polymerase is to be measured. Wave guides’ dimensions are so minute that light can only permeate the place near the edge, where the sequencing polymerase is bound.Citation32 Nucleotides closer to the polymerase in small volumes can be illuminated and detected by fluorescence. Integrated Nucleotide in the extending strand of DNA spends large amount of time closer to the polymerase. All competent nucleotides are involved in the reaction; each tagged or marked disparate colored fluorescent dyes to discriminate from one another. Bettering base calls apply particular integration time of every nucleotide. Reading of sequences up to thousand of bases, longer than possible with second generation systems are collected in real time for each characteristic molecule.Citation33 But, the current result is less than 100,000 reads per run, so the overall yield of sequence is less than second generation systems. In addition, the raw error rate, 15–20% is particularly increased than any other modern sequencing technologies, for example data for some applications experience problems i.e., variant detection ().
Figure 1. Diagrammatic representation of the complete procedure of SMRT DNA sequencing process.
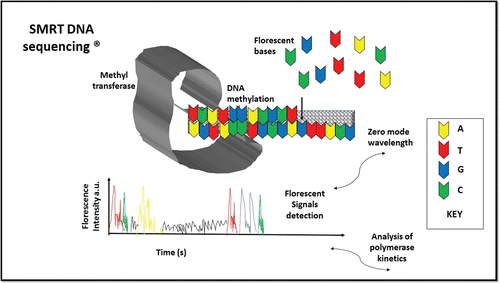
Reads that are much longer such as ‘strobed reads’, can be manufactured by turning off the laser periodically for time periods during sequencing, which averts termination of the nucleotides prematurely and polymerases by laser induced photo damage. If longer reads are not required, the increased raw error rate can be perturbed through ligation of a hairpin oligonucleotide to both the terminals of the DNA, forming a circular template (named SMRTbell for single molecule real time sequencing) and then the same molecule is sequenced repeatedly.Citation34 This process succeeds when relatively short molecules are used; consequently longer reads cannot be utilized by this process, so the high raw error rates are retained. Even the long reads increased raw error rate can be efficaciously applied for joining sequencing contigs. An extra advantage of this system is the tendency to feasibly detect bases which are modified. It is achievable to recognize 5-metylcytosine, although sequencing context role and other factors in affecting the accuracy remains to be clarified. Basically with this system, direct RNA sequencing can be feasible but this has not been recorded for naturally occurring RNA molecules because nucleotides repeatedly attached to the reverse transcriptase before the integration of nucleotides, thereby giving untrue signals with multiple insertions thus causing hindrances in meaningful sequence determination.Citation18 Moreover, this system long read count restrict it to the recognition of common mRNA other than the profiling of quantitative expression or transcription complete coverage both of which demands a much higher read count that is achievable in the coming future. In a nutshell, short turnaround time and the long reads make this technique the most helpful and advantageous in assembling the genomes, asserting the structural analysis of variation analysis, haplotyping, metagenomics and splicing isoforms identification.Citation35
Single-molecule RNAP motion-based real-time sequencing
DNA of every cell contains information that is required to create and sustain life. Nanoscale mechanism that acts as gatekeeper to this information database is RNA Polymerase (RNAP). On the template of DNA, RNAP molecule moves along while creating mRNA (mRNA) through selected portions, hence initiating gene expression. It is similar to that of motor proteins like kinesin and myosin. The RNAP activity is vastly complex. Transcription consists of initiation, elongation and termination.Citation36
To start transcription, a promoter sequence is recognized and attached by RNAP. RNAP specificity for different promoters is influenced by a number of initiation factors. For the creation of transcription bubble in which DNA is locally gelled some of these factors play a key role in the formation of Open Promoter Complex (OPC), showing the template strand DNA bases. During the “abortive initiation,” series of short RNA transcripts are produced in which RNAP is entered, followed by the starting promoter site the return and release of RNAP. Eventually, RNAP forms transcription elongation complex after detaching from the promoter region, now having the full potential to transcribe the entire gene ().
Figure 2. Schematic representation of single molecule RNAP motion based Real time sequencing process.
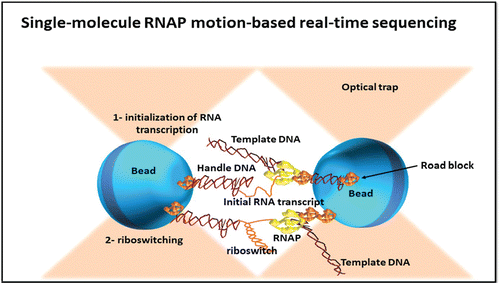
After this elongation is initiated by addition of nucleotides at the 3′ end of the extending RNA followed by termination which causes the release of newly synthesized RNA.
The Single-molecule RNAP motion-based real-time sequencingCitation37 utilizes RNAP to a single polystyrene bead while another bead is attached at the distal end of a DNA fragment. Optical trap is in place with every bead and beads are levitated through a pair of these traps. Polymerase position which are tagged by beads can be sensitively found out through calculation of light scattering from the bead, either using centroid tracking in video images or by laser bead scattering of light which is more precise than former. The RNAP and DNA fragment interaction with one another leads to the change in DNA length between the 2 beadsCitation38 leading to movement of 2 beads with angstrom region precision thus resulting in resolution of single base on a single molecule of DNA. By movement records or alignment of 4 displacement, each with one of the 4 nucleotides lower concentration which is similar to that of primers used in sequencing through Sanger technique and for alignment applying the known sequence flanks to unknown sequenced fragments, hence the sequence information can be deduced for it.Citation39 Enhanced resolution in the DNA length tether is straightened and is allowed for more direct displacement measurements. Other positional resolution of the measurement is in energetically bias steps of the transcription cycle on which force can be applied to move it along the tether. This technique indicates that the very sensitive optical trap method and nucleic acid enzyme movement permits sequence information extraction from single DNA molecule directly.Citation40
Problems associated with reverse transcriptase can be avoided by this process, thus providing unmatched quantitative accuracy for measurements of expression of RNA. Precise expression calculations are made possible with very high read counts per sample with either RNA or cDNA, a mechanism that is impossible with other single molecular techniques of the same molecule can remarkably allow the detection of a very rare variant and improve the error rate also in a mixed sample. For example, a sample of many normal cells containing rare variants and mixture of few tumor cells among themselves might not be detected through DNA, which is amplified. With the repeatedly sequenced same molecule, heterogeneous samples mutations such as tumors can be easily detected. Because of the minimal preparations of sample needed and the potential to use high read count and exceptionally low starting quantities, this technique is ideal for quantitative applications such as copy number variation, ChIP and RNA expression and conditions in which sample quantity is restricted. Nowadays it is much cheaper than second generation systems.Citation41
Nanopore sequencing
Nanopore is one of the techniques applied for Third Generation DNA sequencing (). Nanopore Sequencing is rapid real time technology. Nanopore Systems depend on electronic cognition of DNA sequences. It has the proficiency of high speed, low cost and low sample preparation work.
Figure 3. Nano pore sequencing process; description of sequencing process.
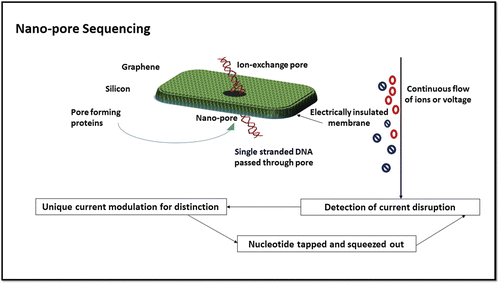
Nanopore can be created by proteins responsible for pore formation or as a hole in man-made materials such as silicon or graphene. Nanopore used for this procedure is a miniature biopore with nanoscale (∼1 to 2 nm) in diameter. Nanopore can be used as a detector when it is placed in membrane, which is electrically insulated. It can be seen in lipid bilayer ingrained with protein channel, which helps in ion exchange.Citation42 Because of nanopore's biological importance any particle motion can disrupt the voltage across the channel. The gist of the idea is that using nanopore in sequencing requires single stranded DNA thread passed through α haemolysin (αHL) pore. αHL is obtained from Staphylococcus aureus, a 33 kD protein that assembles itself to form 7 transmembane channel. It can endure extraordinary voltage up to 100 mV with current 100 pA. This particular characteristic helps in block formation in nanopore.Citation43
During this sequencing, a flow of ions or current is continuously applied. Electrophysiological technique is used for the standard detection of current disruption. Sequence readout is based on the difference in size between all deoxyribosenucleoside monophosphates (dNMPs). Hence, for a particular dNMPs, unique current modulation is represented for distinction. Ionic current is recapitulated after the nucleotide is tapped altogether and is squeezed out.Citation18
Sequencing through nanopore can deliver a number of advantages in the market of next generation sequencing. Basically, it can reach great speeds of 1 bp/ns and can possibly read long lengths of bases >5 kbp. Thus it will be much simpler for researchers to see large-scale patterns such as translocations, in which sections of DNA are removed from one part of the genome to another, and copy number variations, in which DNA sequences are repeated continuously. Translocations are believed to underlie various types of cancer and other illnesses, while copy number variations are connected to a variety of neural and developmental disorders. Furthermore, fluorescent agents are not required in the bases detection it is fluorescent tag free. Thirdly, enzyme involvement is remarkably precluded, other than the use of exonucleases in ssDNA and the cleavage of nucleotides.Citation44 This insinuates that nanopore sequencing throughout the reaction is less perceptive to temperature and outcome maintained can be reliable and definitive. It is also not time consuming and cyclic addition of reagents. In addition, modified bases can be identified by nanopore sequencing, which can directly sequence RNA. The latest goal is to improve base identification by using αHL nanopores, designed by traditional mutagenesis, targeted chemical modification and artificial protein mutagenesis, fit for real-time sequencing to generate DNA reading heads. Moreover, instead of applying polymerization in DNA sequences through nanopore single DNA sequenced by means of DNA strand depolymerization. Thus time for sample prep i.e. amplification and cloning steps can be considerably abridged.Citation45
Nanopore sequencing has so much potential that it is already commercialized for example: Nanopore Technology (Oxford, UK) has marketable products in this direction and it is predicted that DNA sequencing will keep growing at a rapid rate for several years to come.Citation46
Next Generation Sequencing Technology as a Tool for Crop Improvement
Crop improvement with the aid of next generation sequencing technologies have revolutionized plant biology can be used as an effective tool for addressing critical issues regarding crop improvement. With this technology, vast quantity of data can be generated from draft sequences at much shorter time in contrast to the traditional Sanger-based sequencing method.Citation47 With this technology genes or genomic regions with trait of interest can be identified by various techniques such as conventional linkage mapping marker-assisted back crossing, genome wide selection techniques and marker assisted recurrent selection.Citation48
Sanger sequencing of bacterial artificial chromosome (BAC)-based physical maps have been the most dominant approach since 2010, through which several genomes of rice, poplar and maize have been determined ().Citation49 Rice genome was mapped through BAC based mapping, whereas for maize BACs were not completely sequenced. For grapevine, sorghum and soybean whole genome shotgun (WGS) reads and BAC end sequences (BES) were assembled by using powerful techniques.Citation49 These methods that were used to investigate genome prior to next generation sequencing were time consuming and expensive.
Table 1. Comparison of different platform used currently in Next generation of sequencing technologies
The sorghum genome was the first to be sequenced using WGS sequence assemblies.Citation50 There are 2 Joint Genome Institute (JGI) in the USA and the Beijing Genome Institute (BGI, Shenzhen) in China that are currently sequencing the genomes of important food and fuel crops with expert scientists, and in collaboration with science groups all over the world to improve the functional understanding of these genomes.Citation49 A large number of crop genomes have been sequenced since 2012 using highly effective NGS technologies in which Illumina and Roche 454 have achieved prime importance.Citation49 Sugar cane a hybrid between Saccharum officinarum and Saccharum spontaneum is having haploid contents of 8 and 10 chromosomes from each respectively. Its genome size is approximately 15 GbCitation51 determined through WGS strategies.Citation49
Wheat improvement through next generation sequencing
Triticum aestivum is bread wheat that is an essential crop all over the world and is an important source of food. In order to access the heritable traits of a particular plant it is necessary to understand its genome for improvement in order to cope with increasing food demand with changing global climate but if the genome of a plant is complex it gets very difficult for a plant biologist to correlate the genome variations with agronomic traits.Citation52 Although the genomes of various crops including maize, cotton, pineapple, rice and sorghum have been sequencedCitation50 but wheat genome sequencing has been an intricate task. This is due to the fact that wheat has a very complex and large genome as compared to other cereal crops. Wheat genome size has been estimated to be 17Gbp through evaluating the potential BAC-end sequencing.Citation53 The complex genome of Triticum aestivum is due to the allohexaploidy of wheat because it contains 3 distinct diploid genomes that combine to act as diploid together. These diploid donor species diverged approximately 2 to 4 million years ago and are characterized as AA, BB and DD.Citation47 There must have been 2 distinct hybridization events contributed to the development of Triticum aestivum. It is believed that the combination of Triticum urartu (AA) with an unknown relative of Aegilops speltoides (BB) produced the tetraploid Triticum turgidum tetraploid hybridized with Aegilops tauschii (DD) led to the evolution of hexaploid Triticum aestivum.Citation54 The hardness (Ha) locus has been found to control the hardness of Triticum aestivum grain as well as its relatives triticum and aegilops species. This variation of trait is an example that appeared due to gene loss as a result of polyploidization.Citation54 National Science Foundation in mutual collaboration with the US. Department of Agriculture sponsored IWGSC. In this workshop hexaploid wheat sequencing was done using physical mapping and BAC approach.Citation55 This method involves generating BAC library followed by identification of a minimum tilling path then sequencing and generating a physical map, at the end the final assembly of the genome. However this approach had certain limitations that it requires time and resources to a great extent that would scale up significantly for larger genomes.Citation47 Shot gun sequencing of individual chromosomes followed by assembly and identification of contiguous regions on genes that can be oriented and ordered based on synteny analysis with related species has been used for determining the barley genome.Citation56 By applying NGS technology, array hybridization analysis, followed by synteny analysis with related species about 21,766 barley genes has been arranged in a linear array. It has been proposed that there is a mosaic structural similarity between barley (H) genome and bread wheat being hexaploid can be applied to wheat genome.Citation56 The complexity of wheat genome has been reduced by chromosome arm 7DS and identifying genes using this approach.Citation57 Further sequencing of 7BS and 4AL arm suggested that the total gene content of wheat is approximately 77000 genes.Citation57 An attempt to develop chromosome-arm 7DL-specific microsatellite markers in wheat have been possible due to chromosome sequences obtained with Illumina Hi Seq2000 sequencing platform that will help in genetic mapping and positional cloning in wheat.Citation58
Transcriptome is the entire complement of RNA molecules that are present in cells or tissues. With the help of wheat transcriptomics we can determine important genes controlling important traits of wheat crop.Citation59 The first Next generation technology used for transcriptomic studies mainly due to long reads was Roche 454 technology. The second Illumina technology that has gained interest as an important tool for gene discovery and expression study due to deep sequencing coverage and faster in comparison to 454 sequencing.Citation60 But using Illumina, Roche 454, Helicos and SOLiD ABI techniques it has been possible to achieve whole-genome sequencing, targeted resequencing.Citation61 This shows that the next generation technologies have such an exceptional potential in functional genomics.Citation61 It has been found that a chromosomal region QTL (Quantitative Trait Locus) of wheat is associated with rich variation of mRNA transcripts observed during genetic mapping and eQTL mapping a single gene can have multiple or single QTL we can locate genes and their positions on chromosomes. In this way, we can have a better understanding of genes controlling phenotypic traits and their variations.Citation62 Another important aspect of next generation sequencing technology is discovering molecular markers for crop improvement studies such as inferring genome dynamics.Citation48 Marker-assisted selection techniques have become increasingly popular as it can prove to be useful for improving numerous vital economic traits. Marker assisted selection, MABC and Forward Breeding strategies are successfully used in various breeding methods.Citation63 With the help of new technological approaches based on Next Generation Sequencing, technology production and detection of single nucleotide polymorphisms (SNPs) can enable develop novel selection methods.Citation64 NGS with GWAS cannot only be used for identifying genes and genetic variation but a potential tool for identifying molecular markers such as SNPs, gene insertions and deletions and other copy number variation, which are linked to growth and development and stress response.Citation65 SNPs are now used as a principle marker in plant biotechnology for genetic analysis. They signify a single nucleotide difference between 2 individuals at a distinct location. Wheat SNPs are categorized into 2 groups one is homologous and other is varietal where homologous SNPs are differences between the related genomes that are AA, BB, and DD while differences between wheat varieties on a single wheat sub genome are termed as Varietal SNPs. Varietal SNPs are important in sense they are associated with heritable differences between populations and individuals. Varietal SNPs can be identified using homologous SNPs.Citation47 It has been found that a homolog of T. urartu OsGASR7 can be used as a potential candidate for improving wheat yield. Moreover, with the discovery of 2,989,540 SNPs (single nucleotide polymorphisms) future development and characterization of genetic markers have become possible.
Crops that are well adapted to abiotic stresses have become an increasingly important factor in food production due to drastic changes in climatic conditions. In an approach which involved germplasm collection from tolerant crops and their wild relatives has been carried out by identifying and isolating QTL(s), genes(s) and allele(s) that confer important traits such as heat, drought and other abiotic stresses by applying genetic engineering approaches.Citation66
Variation in wheat genome can be detected through Exome sequence analysis of exomic region of tetraploid wheat great extent of differentiation in the exomic region of wild and domesticated wheat species and this differentiation was consistent with the evolutionary process of wheat.Citation67 It has been seen that many SNPs have lost their function due to high level of variation capacity in wheat genome specifically in the coding regions. This has indicated that the ability of tetraploid wheat to adapt to changing environment has not only because of diverse genome but also due to variations produced at increased pace.
Rice improvement through next generation sequencing
Next generation sequencing has been used for the improvement of rice. Complete genome sequencing of rice has been accomplished using a japonica rice cultivar in 2005 by the IRGP traditional Sanger Based sequencing method. High-throughput sequencing method has been used to resequence the rice genome.Citation68 By comparing the 2 genomes of japonica rice cultivar that are Nipponbare and KoshihikariCitation69 about 67,051 Single Nucleotide Polymorphisms have been recognized.Citation68 Next generation sequencing has been used to sequence 10 genomes of Oryza genus by using 454 Life Sequencing platforms. The complete sequence of chromosome arm of Oryza barthii has been accomplished.Citation70 The basic idea was breaking the genome into smaller units by which the assembly complexity and number of sequencing libraries can be greatly reduced. A latest version of 454 sequencing platform has immensely improved the read lengths called the Titanium. In this method collection of BAC clones from MTP, that bare minimum tiling path has been sequenced followed by their assembly to form superior draft sequence of a chromosome arm and then extended to whole genome.Citation70 A high quality reference genome has been updated and validated in 2005 by using optical map records and next generation sequencing strategies with the elimination of 4,886 sequencing errors in previously assembled genome. Few gene insertions and deletions were also recognized using Roche 454 pyrosequencing platform.Citation71 A challenge of sequencing 517 rice landraces with the identification of 3.6 million Single Nucleotide Polymorpisms led a step forward toward shaping the genetic basis of important agronomic traits. High-density haplotype map has been constructed in this way. Furthermore, important agronomic traits have been analyzed in rice sub-species using genome wide association studies. Biparental cross-mapping has proved to be powerful stratagem to scrutinize intricate traits in rice.Citation72 Bacterial grain rot (BGR) is a destructive disease of rice caused by a bacterial pathogen Burkholderia glumae. Different environmental conditions affects the spread of the disease such as temperature, humidity etc. In order to evaluate BGR resistance of diverse rice cultivars quantitative trait locus mapping for BGR resistance on the long arm of chromosome 1 has been done. The results provide genetic analysis of BGR resistance in rice and thus this trait could be targeted for marker-assisted selection for rice improvement.Citation73 Culture of anther studies has been used to attain a homozygous progeny by inducing the doubling of ‘n’ chromosomes in order to enhance agronomical traits selection, amount of DNA polymorphism including SNPs and Indels among related cultivars has been detected by high-throughput sequencer, Jeong et al.Citation68 estimated sequence variety resulting from anther culturing by performing WGRS of 5 Korean including 3 anther culture lines, their progenitor Hwayeong cultivar, and Dongjin japonica cultivar. It was found that the genetic difference amid progenies resulting from anther culture and their parent cultivar was a result owing to somaclonal variations.
Cotton improvement through next generation sequencing
Cotton is the leading cash crop of the world. Trichomes are a specified plant structures that provide shield against biotic as well as abiotic stresses, water and mineral absorption, elimination of waste, enthralling seed dispersal.Citation74 Trichomes of cotton are unicellular termed as “fiber.” Due to its tremendous economic importance, cotton has been termed as a miraculous fiber since no other fiber is comparable to cotton in terms of the advantageous characteristics that cotton possesses. According to the statistics cotton consumption is nearly 27 million metric tons/year around the world while India is second among world's cotton producing countries. Cotton improvement started 8000 y back. The conventional method of cotton improvement was based on selection in which variability in agronomic traits was generated and detected then genotypes with favorable characteristics were selected these characteristics results from recombination event that occur at different loci.Citation75 The genus Gossypium L. contains 45 diploid and 5 allotetraploid species. There are 2 economically significant cultivated species G. barbadense L. and G. hirsutum L. that are allotetraploid these 2 speices were produced by A and D genome.Citation75 G. raimondii and G. herbaceum are 2 species that are ancestors of these A and D genomes.Citation76 It has been found through investigation that the A genome, D genome, and AD genome groups that include G. herbaceum (A1), G. arboretum (A2), G. hirsutum (AD1), and G. barbadense (AD2) received more attention for domestication due to their plentiful seed trichomes and thus laid the basis of the cloth industry.Citation75 Next Generation Sequencing approaches that include trait introgression by marker-assisted selection (MAS), genetic engineering and in vitro mutagenesis have been used effectively to incorporate novel genes to increase genetic diversity. The interspecific trait introgression technique has been successful in analyzing various number that can be modified to comprise many genes, even entire genomes can be targeted by using high-throughput MAS.Citation77 MAS have appeared to be a successful method in cotton improvement program since molecular markers in cotton are 100% heritable. By determining intra-specific analysis and inter-specific linkage analysis, it was found that the cotton genome is approximately 5000 cM. Through genome investigation it is estimated that transcriptome of cotton diploid species consists of approximately 18000 genes. Similarly, transcriptome of allotetraploid cotton species estimated to have approximately 36000 genes including homologues of both genomes.Citation78 Moreover a 3 fiber gene-rich islands linked with fiber synthesis process, for the early to middle elongation stage and for the middle to late elongation stage has been identified on 5, 10 and14 respectively and on chromosome 15 an island for the deposition of secondary cell wall has been determined.Citation79 This shows that a thorough study of cotton genome has been accomplished and further research is still going on at a pace presenting a promising future toward cotton improvement through genetic engineering and advance biotechnological techniques.
Exploring the Biodiversity of Plants Through Exome Sequencing
The treasures of biodiversity are a rich source of almost all desirable traits that we want in crops. It is quiet a challenging task to understand the diversified organisms and the patterns that control variability among them. In order to identify the closely related species morphological analysis is not always the ultimate elucidation therefore more integrated and efficient techniques are required such as the genetic method.Citation6 The Consortium for the Barcode of Life (CBOL) developed to identify species and differences between species. In order to develop a specie specific barcode sequencing of constant regions of DNA are used as a tool to identify different species. Method to identify a single species specific gene such as cytochrome oxidase I (COI) has been appeared to be a potential barcode.Citation80 However it is not very effective for most plant species because cytochrome c oxidase I geneCitation81 evolve very gradually in plants as compared to animals. Therefore, NITSR (nuclear internal transcribed spacer region) and the plastid trnH-psbA have been proposed by Kress et al.Citation81 to study phylogeny with greater degree of interspecies divergence and variability among land plants. In addition to this, a sense rbcl gene and an anti sense trnH-psbA spacer region has also been used for this purpose.Citation82 Moreover, combination of rbcL and matK is a more successful barcode for plants in terms of recoverability of data, superiority of sequences, and extent of species discrimination determined by CBOL Plant Working Group (2009). Up till now DNA fingerprinting and RAPD (Random Amplified Polymorphic DNA) fingerprinting has been used for specie identificationCitation6 due to certain limitations of next generation sequencing technologies such as error rate and generation of shorter reads.Citation47 However through NGS technology rapid detection of Single Nucleotide Polymorphism (SNP) has been developed assessing few subsets of parental inbred position as well traits of concern in bigger populations can be characterized and mapped using genotyping-by-sequencing (GBS) method.Citation83 Next Generation Sequence analysis can revolutionize biodiversity exploration and the discovery of new species with reduced risk of miscategorization of new species. NGS has helped perform Exome sequencing for taxonomic classification of organisms and once all the data is available one can easily check for any candidate species and its homology with other member of the taxonomic classes. As a single gene cannot effectively serve as a “barcode” Exome sequences can be compared across species among the taxonomic classes to identify the most optimal gene(s) for barcoding process. In this way, one can map trait variations caused by single gene variation in the Exome region through comparative genomics.Citation6
To explore the diverse flora and patterns of plant evolution of Australian rainforests a Bicentenary Plant Diversity Program has been funded by the Royal Botanical Gardens and Domain Trust in 2012 with an aim of combining genetic and environmental data to explore various species. According to Dr. Hannah McPherson the Biodiversity Research officer ‘the use of NGS technology have helped us to explore Chloroplast genome for Barcoding, single nucleotide polymorphism discovery and marker development for comparative studies. It has appeared to be a useful tool for evaluating the variation among and within populations and species of rainforest trees with promising results’.
Studying the Plant-Host Pathogen Interactions Through Exome Sequencing
Plants have amazing strategies through which they can identify pathogens such as conserved and variable pathogen associated elicitors on the other hand pathogens evade host defense response by secreting virulence factors.Citation84 These host pathogen interactions are a result of persistently evolving genomic interphases between plants and their specific pathogens. Even a single amino acid alteration in the regulatory protein can alter virulence and susceptibility in the host–pathogen interactions.Citation85 With the arrival of next generation technology genomes of various pathogenic fungi can now be successfully sequenced. In order to study the host pathogen interactions Calvitti et al.Citation86 established a symbiotic association between Aedes albopictus - Wolbachia by artificially transferring wPip strain from Culex pipiens for effective pest control. Illumina's SBS technology has been used successfully for identifying genes involved in host pathogen interactions.Citation87 In an attempt to identify genes involved in host–pathogen interactions in rice and fungus that are expressed by S. homoeocarpa mycelia and creeping bentgrass the sequence analysis has been done using BLAST. Moreover, NGS technology has also been used to study the evolution of plant pathogenesis in Pseudomonas syringae. Sequencing of 25 strains of P. syringae has been done by NGS technology.Citation88 The fungus Puccinia striiformis tritici (PST) is the cause of wheat rust and considerable loss in wheat production all over the world and has been a great concern for plant biologists. NGS has helped in reduction in cost compared to traditional techniques. To access the genomic sequences Illumina sequencing was used for highly virulent PST race 130.Citation89 A plant host-pathogen interaction has also been analyzed using SuperSAGE. SuperSAGE has been applied to rice leaves infected with M. grisea and the analysis of gene expression profiles using entire genomes of both host and the pathogen. This study revealed that a gene called hydrophobin, which is actively transcribed by M. grisea in infected rice leaves might contribute to its pathogenicity. In this way, superSAGE can successfully determine up regulating and down regulating genes during host pathogen interaction. It is clearly seen that NGS technology has proved to be very efficient and quicker in evaluating the genome sequences for its applications in plant pathology and plant-microbe interaction studies.Citation89 In order to develop strategies for crop improvement we need to have a deep insight of molecular mechanisms controlling plant immunity. There are basically 2 immune responses in plants governed by PAMP-triggered immunity (PTI). However, certain pathogens are able to silence PTI signaling through evolution of specific effector proteins. In response, plants have developed cytoplasmic resistance (R) protein receptors, which establish effector-triggered immunity (ETI). To study the transcriptional changes during plant immunity responses to biotic stresses pyrosequencing of expressed genes has been done of banana (Musa acuminate) during infection with fungal pathogen (Mycosphaerella musicola). Using a 454 GS-FLX system pyrosequencer full-length enriched cDNA libraries were sequenced. This has helped identify several expressed genes associated with stresses and responses to biotic stimuli. Moreover expressed resistance gene analogs and defense gene have been identified for further marker development.Citation90
Exploring the Natural Evolution of Crops Through Exome Sequencing
Molecular markers have been extensively used in order to investigate and evaluate crops.Citation6 There are a number of new genomics technologies that have been used for studying the plant variation that have contributed to evolution such as NGS, high-throughput marker genotyping genomics that have proved to be a powerful tools at DNA, RNA and protein level. The genetic variations that appeared in cultivated plants as well as in wild species can be studied by means of advance analytical techniques such as advanced BC QTL analysis.Citation48 Understanding the natural evolution of crops can help us artificially mimic the molecular event to incorporate desired traits into the crop plants. These evolutionary genomic approaches have helped identify genes and genomic regions that are involved in evolution by undergoing natural selection process.Citation91 These studies can also help us identify crop progenitorsCitation92 and can give us a better insight of crop evolution.Citation93 We can determine the domestication events as well as the demographics of domestication.Citation94
This approach has been applied in studying the maize evolution and finally led to the identification of 15 genes that were putatively selected during evolutionary processCitation95 lot of these genes have been co-localized with earlier mapped QTLs linked with traits related to crops.Citation91 In another study it was found that the phenotypic differences between teosinte and maize resulted from genes evolution during selection tend to cluster near QTLs.Citation96 It has been seen when mapping QTL that it controls variability of seed oil content and composition between cultivated and wild sunflower species.Citation97 Another important approach that involves large scale screening of loci that is involved in selection has been used to study evolutionary differences among domesticated and wild crop species.Citation91 A technique that has been used parallel to family-based QTL mapping is the ‘association mapping’ also termed as linkage disequilibrium (LD) mapping that involves the association of alleles at 2 loci. In this method polymorphism in candidate genes correlated with phenotypic variation in varied populations thus also providing a level of precision.Citation98 In family based mapping there is narrow probability for recombination event to occur throughout population growth thus outcome is elevated LD over large physical distances. However, one drawback of this technique is that it requires previous understanding of candidate gene(s) and the phenotypic data in order to carry out evolutionary analysis. On the other hand association mapping is the result of many generations of previous recombination thus reduced LD and consequently increased mapping precision.Citation99
Flavours of various fruits have been analyzed and different chemicals have been found to contribute to give fruits their distinct flavours. It has been found through DNA microarray technique that biogenesis of flavour strawberry occurs in response of expression of a novel alcohol acyltransferase (SAAT) gene expressed in fruits during ripening.Citation100 Crop evolution has been extensively studied using molecular markers. Genes crucial for domestication of rice crop has been extensively evaluated using High-throughput sequencing technologies.Citation101 Phylogenetic relationships of 20 rice cultivars and land races have been revealed using resequencing microarrays to map genome-wide SNP variations.Citation101 A large number of domestication alleles associated with the domestication traits need to be discovered. Exome sequencing technology can be very effective in identification of the domestication alleles and mutagenic events that have favored in the domestication of these traits.Citation6
Exome Sequencing and Symbiotic Cropping Strategy Management
Crop improvement with an objective high yield and forbearance to wide array of environmental stresses have turn out to be the prime attention of plant biotechnologist. Gene pyramiding has been used with an aim of incorporating the agronomically superior traits; this requires a deep consideration of the genetic basis of these traits so by regulating the yield-related genes yield of various crops can be enhanced.Citation102 In order to get the desired yield it is necessary to identify the optimal growth conditions for a crop. This is most commonly done by applying all the given conditions on the crop and optimized accordingly. If one has the exact knowledge about the genetic makeup and genomic architecture of crop plant, the optimal conditions can easily be worked out.Citation6 According to Singh et al.Citation6 “Exome sequencing can help identify the genes and establish the presence of functional gene sets that are involved in symbiotic or other co-existential systems. Such information can be pertinent to planning the crop management and improvement strategies that utilize pre-existing and newly developed co-existential system.” In an approach chloroplast genome has been analyzed and engineered for herbicides, insects, disease and drought resistance in crops.Citation103
Limitations: Crop Improvement Beyond Exome Sequencing
Exome sequencing though offers an efficient method for identification of genomic modification in exons but some critical issues remains uncovered by this technique which comes under whole genome sequencing, transcriptomics, metabolomics and proteomics.
Plant whole genome sequencing
It offers to be an effective tool for genome mapping hence important for assessing allelic/QTLs transmission. MicroRNAs and other non coding RNAs being essential elements of epigenetic process and gene networking required for plant growth and homeostasis play important role in crop improvement.Citation104–105 Non-protein coding RNAs have been found to be responsible for heat stress and powdery mildew infections and can be involved in biotic and abiotic stress in wheat.Citation106 MiRNA169 regulates NSR (nitrogen starvation response) and N starvation causes their down regulation in both shoots and roots.Citation107 Overexpression of miR 169 in transgenic Arabidopsis plants causes less N accumulation and more sensitization to stressCitation107, and increases drought tolerance in transgenic tomatoesCitation108 thus plays an important role in physiological processes and WES is unable to provide such information as these miRNAs are coded most of the time by introns.Citation6
Similarly, untranslated regions have been reported to play major role in localization, functioning and stability regulation. 3′ UTR of GluB-1, a rice storage protein plays crucial role in mRNA stability by functioning as a terminator resulting in higher levels of protein production.Citation109 Point mutation in 5′ UTR of Glb-1 and Glu-A gene increased transcriptional activity in glabrous rice varieties.Citation110 Similarly, mutations in 5′UTR can enhance the protein expression of rice polyubiqutin geneCitation111 and can enhance translation activity in both monocotyledeonous and dicotyledonous plants.Citation112 Alteration of protein expression can be controlled by 3′ and 5′ UTRs that are not part of exons so exome sequencing fails to address these properties vital for crop improvement.
Pesudogene's mRNA are not translated into proteins despite sharing high homology to known functional genes thereby are not counted in exome sequencing although they have been found modulating expression of functional gene by decreasing the concentration of miRNA which can bind to 3′ UTR of functional gene.Citation113 One such example is of IPIS (induced by phosphate starvation) gene, an npc gene from Arabidopsis thaliana, which has a motif with partial sequence complementary to miR-399 (which is Phosphate starvation induced microRNA). It contributes to accumulation of functional mRNA (PHO2) by sequestering miR-399 that can bind to 3′UTR of functional mRNA thereby reducing shoot Pi content.Citation114 Hence, any variation in pseudogene can modify the expression of functional gene activity that surely cannot be detected/identified using exome sequencing.
Plant transcriptomics
This addresses the transcription of mRNA at a given point in a given issue.Citation6 Transcriptome being dependent on environment is extremely variable. For analysis and study of entire trancriptome, RNA-Seq approach is used that uses NGS techniques, providing an insight on expression level of transcripts. Transcriptomes of crops e.g. rice, soyabean and grapes has been sequenced using RNA-Seq approach.Citation115-116 Exome sequencing here is of limited importance where transcriptomics stands crucial for crop improvement. RNA-seq has been applied in characterization of drought and salinity stress responses and interaction between host and pathogens.Citation117–118
Expression microarrays technique help in understanding effect of environment on mRNA expression by predicting response of a given genomic architecture to environment. Pattern mapping of expression of OsCESA gene in the life cycle of rice have been done using microarray analysis.Citation119 Expression levels are measured in relation with environmental factors including hormonal and pathogenic stimulation, nutrient starvation, and cardiac rhythms.Citation120 This may have endless applications but it can not overshadow the status of exome sequencing, but it does compliment WES.
Plant metabolomics
Metabolomics explains the interactions between environmental cues and genetic factors represented by metabolite levels serving as an indicator of state of plant health under stress conditions. Gene involved in stress regulation also are responsible for metabolite levels. Overexpression of YK1 gene, for example, where increases production of NADPH and decreases the amount of fructose-1, 6-bisphosphate and G3P also.Citation121-122 Altered levels of metabolites can be determined easily using nuclear magnetic resonance (NMR) technology or mass spectrometry (MS). Such alterations have been thoroughly studied in Arabidopsis in response to environmental factors.Citation123-124 Such behavior of crop plant in relation its surrounding environment can at least be partially anticipated by WES, but outcomes cannot be measured certainly.Citation125
Plant proteomics
As metabolome, proteome also interprets the consequence of interaction between genetic and environmental factors involved in gene expression regulation. Proteomics analysis is used in the identification of proteins involved in regulation of adaptation in response to NaCl stress to ease ion homeostasis in the roots cucumber seedlingCitation126, in the identifications of genes of other crops including rice involved in salt stressCitation127-128 and it has also been used to study acclimation to osmotic stress.Citation129 Molecular mechanisms have been identified using comparative proteomics approach that are involved in physiological processes e.g., to discover the role of oxidative stress on carotenogensis in orange.Citation130 However, without integration of genomic analysis, proteomics is of limited significance requiring analysis that is more complex in order to predict transmission of traits.
Conclusions and Future Prospects
Increasing the research efforts to exome sequencing scale has resulted in generation of vast amount of sequencing data which was unimaginable few years back. Exome sequencing has emerged as an excellent tool for crop improvement which has helped in crop management by understanding its genetic makeup. NGS technologies in plant exomics have served to accelerate crop improvement especially wheat, rice and cotton. Available tools used in ES have been increased dramatically which have aided us to study different critical aspects important for crop improvement including mechanisms for natural evolution of crops, biodiversity and host pathogen interaction. Rapidly available exome data will help improve our understanding toward the future challenges toward crop improvement. However, productive crop improvement requires integration of bioinformatics tools, merging exome sequencing with multiple-omics platforms for best outcomes which exome sequencing can't generate alone. ES however is unable to sequence variants not present in exon portion of genes necessary in controlling transcriptional regulation. Hence, exomes may not be ideal choice for interpreting genome's structural variation.
There is a need to develop large databases specifically for exome to enhance the interpretation of disease management. Analysis techniques need to be developed which will deal with thousands of variants within a genome. Development of technique that will have the ability to combine and integrate data from different variants into one analysis will be high priority. How frequently these suppositions will hold is currently not known and will define the discovery rates in coming years.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Biesecker BB, Peay HL. Genomic sequencing for psychiatric disorders: promise and challenge. Int J Neuropsychopharmacol 2013; 16:1667-2; PMID:23575420; http://dx.doi.org/10.1017/S146114571300014X
- Long SP, Ort DR. More than taking the heat: crops and global change. Curr Opin Plant Biol 2010; 13:240-47; PMID:20494611; http://dx.doi.org/10.1016/j.pbi.2010.04.008
- Varshney RK, Graner A, Sorrells ME. Genomics-assisted breeding for crop improvement. Trend Plant Sci 2005; 10:621-30; PMID:16290213; http://dx.doi.org/10.1016/j.tplants.2005.10.004
- Neale DB, Kremer A. Forest tree genomics: growing resources and applications. Nat Rev Genet 2011; 12:111-22; PMID:21245829; http://dx.doi.org/10.1038/nrg2931
- Harfouche A, Meilan R, Kirst M, Morgante M, Boerjan W, Sabatti M, Scarascia Mugnozza G. Accelerating the domestication of forest trees in a changing world. Trend Plant Sci 2012; 17:64-72; PMID:22209522; http://dx.doi.org/10.1016/j.tplants.2011.11.005
- Singh D, Singh PK, Chaudhary S, Mehla K, Kumar S. Exome sequencing and advances in crop improvement. Adv Genet 2012; 79:87-121; PMID:22989766; http://dx.doi.org/10.1016/B978-0-12-394395-8.00003-7
- Stitziel NO, Kiezun A, Sunyaev S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genom Biol 2011; 12:227; PMID:21920052; http://dx.doi.org/10.1186/gb-2011-12-9-227
- Maxam AM, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA 1977; 74:560-4; PMID:265521; http://dx.doi.org/10.1073/pnas.74.2.560
- Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 1975; 94:441-8; PMID:1100841; http://dx.doi.org/10.1016/0022-2836(75)90213-2
- Smith LM, Sanders JZ, Kaiser RJ, Hughes P, Dodd C, Connell CR, Heiner C, Kent S, Hood LE. Fluorescence detection in automated DNA sequence analysis. Nature 1986; 321:674-9; PMID:3713851; http://dx.doi.org/10.1038/321674a0
- Murphy KM, Berg KD, Eshleman JR. Sequencing of genomic DNA by combined amplification and cycle sequencing reaction. Clin Chem 2005; 51:35-9; PMID:15514094; http://dx.doi.org/10.1373/clinchem.2004.039164
- SenGupta DJ, Cookson BT. SeqSharp: a general approach for improving cycle sequencing that facilitates a robust one-step combined amplification and sequencing method. J Mol Diagn 2010; 12:272-7; PMID:20203000; http://dx.doi.org/10.2353/jmoldx.2010.090134
- Pareek CS, Smoczynski R, Tretyn A. Sequencing technologies and genome sequencing. J Appl Genet 2011; 52:413-35; PMID:21698376; http://dx.doi.org/10.1007/s13353-011-0057-x
- Linnarsson S. Recent advances in DNA sequencing methods-general principles of sample preparation. Exp Cell Res 2010; 316:1339-43; PMID:20211618; http://dx.doi.org/10.1016/j.yexcr.2010.02.036
- Rizzo JM, Buck MJ. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev Res (Phila) 2012; 5-7:887-900; PMID:22617168
- Natrajan R, Reis-Filho JS. Next-generation sequencing applied to molecular diagnostics. Expert Rev Mol Diagn 2011; 11:425-44; PMID:21545259; http://dx.doi.org/10.1586/erm.11.18
- Mardis ER. A decade's perspective on DNA sequencing technology. Nature 2011; 470:198-203; PMID:21307932; http://dx.doi.org/10.1038/nature09796
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet 2010; 11:31-46; PMID:19997069; http://dx.doi.org/10.1038/nrg2626
- Hart C, Lipson D, Ozsolak F, Raz T, Steinmann K, Thompson J, Milos PM. Single-molecule sequencing: sequence methods to enable accurate quantitation. Method Enzymol 2010; 472:407-30; PMID:20580974; http://dx.doi.org/10.1016/S0076-6879(10)72002-4
- Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genom Biol 2011; 12:228; PMID:21920049; http://dx.doi.org/10.1186/gb-2011-12-9-228
- Ansorge WJ. Next-Generation DNA sequencing techniques. New Biotechnol 2009; 25:195-203; PMID:19429539; http://dx.doi.org/10.1016/j.nbt.2008.12.009
- Mardis ER. Next-Generation DNA sequencing methods. Annu Rev Genom Hum Genet 2008; 9:387-402; PMID:18576944; http://dx.doi.org/10.1146/annurev.genom.9.081307.164359
- Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 2007; 8:R143; PMID:17659080; http://dx.doi.org/10.1186/gb-2007-8-7-r143
- Stranneheim H, Lundeberg J. Stepping stones. DNA sequencing. Biotechnol J 2012; 7:1063-73; PMID:22887891; http://dx.doi.org/10.1002/biot.201200153
- Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next generation sequencing systems. J Biomed Biotechnol 2012; 2012:251364; PMID:22829749
- Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. Clinical Chem 2009; 55:641-58; PMID:19246620; http://dx.doi.org/10.1373/clinchem.2008.112789
- Pettersson E, Lundeberg J, Ahmadian A. Generations of sequencing technologies. Genomic 2009; 93:105-11; PMID:18992322; http://dx.doi.org/10.1016/j.ygeno.2008.10.003
- Llaca V. Sequencing technologies and their use in plant biotechnology and breeding. In: Munshi A (ed) DNA Sequencing-Methods And Applications. Rijeka, Croatia: InTech, 2012; 35-60.
- Bragg LM, Stone G, Butler MK, Hugenholtz P, Tyson GW. Shining a light on dark sequencing: characterising errors in ion torrent PGM data. PLoS Comput Biol 2013; 9:e1003031; PMID:23592973; http://dx.doi.org/10.1371/journal.pcbi.1003031
- Anderson MW, Schrijver I. Next generation DNA sequencing and the future of genomic medicine. Genes 2010; 1:38-69; PMID:24710010; http://dx.doi.org/10.3390/genes1010038
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. Real-time DNA sequencing from single polymerase molecules. Science 2008; 323:133-8; PMID:19023044; http://dx.doi.org/10.1126/science.1162986
- Flusberg BA, Webster D, Lee J, Travers K, Olivares E, Clark TA, Korlach J, Turner SW. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nature 2010; 7:461-5; PMID:20453866; http://dx.doi.org/10.1038/nmeth.1459
- Pushkarev D, Neff NF, Quake SR. Single-molecule sequencing of an individual human genome. Nat Biotechnol 2009; 27:847-50; PMID:19668243; http://dx.doi.org/10.1038/nbt.1561
- Clark TA, Murray IA, Morgan RD, Kislyuk AO, Spittle KE, Boitano M, Fomenkov A, Roberts RJ, Korlach J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucl Acids Res 2011; 40:e29; PMID:22156058; http://dx.doi.org/10.1093/nar/gkr1146
- Roberts RJ, Carneiro MO, Schatz MC. The advantages of SMRT sequencing. Genome Biol 2013; 14:405; PMID:23822731; http://dx.doi.org/10.1186/gb-2013-14-6-405
- Vilfan ID, Tsai YC, Clark TA, Wegener J, Dai Q, Yi C, Pan T, Turner SW, Korlach J. Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription. J Nanobiotechnol 2013; 11:8; PMID:23552456; http://dx.doi.org/10.1186/1477-3155-11-8
- Greenleaf WJ, Block SM. Single-molecule, motion-based DNA sequencing using RNA polymerase. Science 2006; 313:801; PMID:16902131; http://dx.doi.org/10.1126/science.1130105
- Ding F, Manosas M, Spiering M, Benkovic SJ, Bensimon D, Jean-François A, Croquette V. Single-molecule mechanical identification and sequencing. Nat Method 2012; 9:367-72; PMID:22406857; http://dx.doi.org/10.1038/nmeth.1925
- Mardis ER. Next-generation sequencing platforms. Annu Rev Anal Chem 2013; 6:287-303; PMID:23560931; http://dx.doi.org/10.1146/annurev-anchem-062012-092628
- Tinnefeld P. Single-molecule detection: breaking the concentration barrier. Nat Nanotechnol 2013; 8:480-2; PMID:23770809; http://dx.doi.org/10.1038/nnano.2013.122
- Ozsolak F. Third-generation sequencing techniques and applications to drug discovery. Expert Opin Drug Discov 2012; 7:231-43; PMID:22468954; http://dx.doi.org/10.1517/17460441.2012.660145
- Markosyan S, Cuervo JE, Noskov S. Towards nanopore sequencing: in-silico studies on the interactions between alpha-hemolysin and SS DNA molecules. Biophysical J 2013; 104:334; http://dx.doi.org/10.1016/j.bpj.2012.11.1859
- Nivala J, Marks DB, Akeson M. Unfoldase-mediated protein translocation through an α-hemolysin nanopore. Nat Biotechnol 2013; 31:247-50; PMID:23376966; http://dx.doi.org/10.1038/nbt.2503
- Clarke J, Hai-Chen W, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol 2009; 4:265-70; PMID:19350039; http://dx.doi.org/10.1038/nnano.2009.12
- Rusk N. Cheap third-generation sequencing. Nat Method 2009; 6:244; http://dx.doi.org/10.1038/nmeth0409-244a
- Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Ventra MD, Garaj S, Hibbs A, Huang X, et al. The potential and challenges of nanopore sequencing. Nat Biotechnol 2008; 26:1146-53; PMID:18846088; http://dx.doi.org/10.1038/nbt.1495
- Berkman PJ, Skarshewski A, Manoli S, Lorenc MT, Stiller J, Smits L, Lai K, Campbell E, Kubaláková M, Simková H, et al. Sequencing wheat chromosome arm 7BS delimits the 7BS4AL translocation and reveals homoeologous gene conservation. Theor Appl Genet 2012; 124:423-32; PMID:22001910; http://dx.doi.org/10.1007/s00122-011-1717-2
- Varshney RK, Dubey A. Novel genomic tools and modern genetic and breeding approaches for crop improvement. J Plant Biochem Biotechnol 2009; 18:127-38; http://dx.doi.org/10.1007/BF03263311
- Bevan MW, Uauy C. Genomics reveals new landscapes for crop improvement. Genom Biol 2013; 14:206; PMID:23796126; http://dx.doi.org/10.1186/gb-2013-14-6-206
- Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood, J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov, A, et al. The sorghum bicolor genome and the diversification of grasses. Nature 2009; 457:551-6; PMID:19189423; http://dx.doi.org/10.1038/nature07723
- Grivet L, Arruda P. Sugarcane genomics: depicting the complex genome of an important tropical crop. Curr Opin Plant Biol 2002; 5:122-7; PMID:11856607; http://dx.doi.org/10.1016/S1369-5266(02)00234-0
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. The B73 maize genome: complexity, diversity, and dynamics. Science 2009; 326:1112-5; PMID:19965430; http://dx.doi.org/10.1126/science.1178534
- Paux E, Roger D, Badaeva E, Gay G, Bernard M, Sourdille P, Feuillet C. Characterizing the composition and evolution of homoeologous genomes in hexaploid wheat through BAC-end sequencing on chromosome 3B. Plant J 2006; 48:463-74; PMID:17010109; http://dx.doi.org/10.1111/j.1365-313X.2006.02891.x
- Chantret N, Salse J, Sabot F, Rahman S, Bellec A, Laubin B, Dubois I, Dossat C, Sourdille P, Joudrier P, et al. Molecular basis of evolutionary events that shaped the hardness locus in diploid and polyploid wheat species (Triticum and Aegilops). Plant Cell 2005; 17:1033-45; PMID:15749759; http://dx.doi.org/10.1105/tpc.104.029181
- Gill BS, Appels R, Botha-Oberholster AM, Buell CR, Bennetzen JL, Chalhoub B, Chumley F, Dvorák J, Iwanaga M, Keller B, et al. A workshop report on wheat genome sequencing: international genome research on wheat consortium. Genetic 2004; 168:1087-96; PMID:15514080; http://dx.doi.org/10.1534/genetics.104.034769
- Mayer KF, Taudien S, Martis M, Simkova H, Suchánkov P, Gundlach H, Wicker T, Petzold A, Felder M, Steuernagel B, Scholz U, et al. Gene content and virtual gene order of barley chromosome 1H. Plant Physiol 2009; 151:496-505; PMID:19692534; http://dx.doi.org/10.1104/pp.109.142612
- Berkman PJ, Skarshewski A, Lorenc M, Lai K, Duran C, Ling EYS, Stiller J, Smits L, Imelfort M, Manoli S, et al. Sequencing and assembly of low copy and genic regions of isolated Triticum aestivum chromosome arm 7DS. Plant Biotechnol J 2011; 9:768-75; PMID:21356002; http://dx.doi.org/10.1111/j.1467-7652.2010.00587.x
- Nie X, Bianli Li, Wang L, Peixun L, Biradar S S, Li T, Doležel J, Edwards D, Luo M, Weining S. Development of chromosome-arm-specific microsatellite markers in Triticum aestivum (Poaceae) using NGS technology. Amer J Bot 2012; 99:369-71; PMID:22935363; http://dx.doi.org/10.3732/ajb.1200077
- Coram TE, Brown-Guedira G, Chen X. Using transcriptomics to understand the wheat genome. CAB Rev: Perspect Agr, Vet Sci, Nutri Nat Resour 2008; 83:1-9.
- Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trend Biotechnol 2009; 27:522-30; PMID:19679362; http://dx.doi.org/10.1016/j.tibtech.2009.05.006
- Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomic 2008; 92:255-64; PMID:18703132; http://dx.doi.org/10.1016/j.ygeno.2008.07.001
- Druka A, Potokina E, Luo Z, Jiang N, Chen X, Kearsey M, Waugh R. Expression quantitative trait loci analysis in plants. Plant Biotechnol J 2010; 8:10-27; PMID:20055957; http://dx.doi.org/10.1111/j.1467-7652.2009.00460.x
- Gupta PK, Roy JK, Prasad M. Single nucleotide polymorphisms: a new paradigm for molecular marker technology and DNA polymorphism detection with emphasis on their use in plant. Curr Sci 2001; 80:524-35.
- Paux E, Sourdille P, Mackay I, Feuillet C. Sequence-based marker development in wheat: advances and applications to breeding. Biotechnol 2012; 30:1071-88.
- Ma Y1, Qin F, Tran LS. Contribution of genomics to gene discovery in plant abiotic stress responses. Mol Plant 2012; 5:1176-8; PMID:22930735; http://dx.doi.org/10.1093/mp/sss085
- Varshney RK, Bansal KC, Aggarwal PK, Datta SK, Craufurd PQ. Agricultural biotechnology for crop improvement in a variable climate: hope or hype? Trend Plant Sci 2011; 16:363-71; PMID:21497543; http://dx.doi.org/10.1016/j.tplants.2011.03.004
- Saintenac C, Jiang D, Akhunov E. Targeted analysis of nucleotide and copy number variation by exon capture in allotetraploid wheat genome. Genom Biol 2011; 12:R88; PMID:21917144; http://dx.doi.org/10.1186/gb-2011-12-9-r88
- Jeong IS, Yoon UH, Lee GS, Ji HS, Lee HJ, Han CD, Hahn JH, An G, Kim TH. SNP-based analysis of genetic diversity in anther-derived rice by whole genome sequencing. Rice 2013; 6:6; PMID:24280451; http://dx.doi.org/10.1186/1939-8433-6-6
- Yamamoto T, Nagasaki H, Yonemaru J, Ebana K, Nakajima M, Shibaya T, Yano M. Fine definition of the pedigree haplotypes of closely related rice cultivars by means of genome-wide discovery of single-nucleotide polymorphisms. BMC Genom 2010; 11:267; PMID:20423466; http://dx.doi.org/10.1186/1471-2164-11-267
- Rounsley S, Marri PR, Yu Y, He R. De novo next generation sequencing of plant genomes. Rice 2009; 2:35-43; http://dx.doi.org/10.1007/s12284-009-9025-z
- Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013; 6:4; PMID:24280374; http://dx.doi.org/10.1186/1939-8433-6-4
- Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010; 42:961-7; PMID:20972439; http://dx.doi.org/10.1038/ng.695
- Mizobuchi R, Sato H, Fukuoka S, Tanabata T, Tsushima S, Imbe T, Yano M. Mapping a quantitative trait locus for resistance to bacterial grain rot in rice. Rice 2013; 6:13; PMID:24280270; http://dx.doi.org/10.1186/1939-8433-6-13
- Chen ZJ, Scheffler BE, Dennis E, Triplett BA, Zhang T, Guo W, Chen X, Stelly DM, Rabinowicz PD, Town CD, et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol 2007; 145:1303-10; PMID:18056866; http://dx.doi.org/10.1104/pp.107.107672
- Boopathi NM, Thiyagu K, Urbi B, Santhoshkumar M, Gopikrishnan A, Aravind S, Swapnashri G, avikesavan R. Marker-assisted breeding as next-generation strategy for genetic improvement of productivity and quality: can it be realized in cotton?. Int J Plant Genom 2011; 2011: Article ID 670104;
- Reinisch AJ, Dong JM, Brubaker CL, Stelly DM, Wendel JF, Paterson AH. A detailed RFLP map of cotton, Gossypium hirsutum × Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome. Genetic 1994; 138:829-47.
- Saha S, Jenkins JN, Wu J. McCarty JC, Gutiérrez OA, Percy RG, Cantrell RG, Stelly DM. Effects of chromosome specific introgression in upland cotton on fiber and agronomic traits. Genetic 2006; 172:1927-38; PMID:16387867; http://dx.doi.org/10.1534/genetics.105.053371
- Reddy OUK, Pepper AE, Abdurakhmonov I. Saha S, Jenkins JN, Brooks T, Bolek Y, El-Zik KM. New dinucleotide and trinucleotide microsatellite marker resources for cotton genome research. J Cotton Sci 2001; 5:103-13.
- Xu Z, Kohel RJ, Song G, Cho J, Alabady M, Yu J, Koo P, Chu J, Yu S, Wilkins TA, et al. Gene-rich islands for fiber development in the cotton genome. Genomic 2008; 92:173-83; PMID:18619771; http://dx.doi.org/10.1016/j.ygeno.2008.05.010
- Hebert PD, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci 2003; 7: 270 Suppl 1:S96-9; PMID:12952648
- Kress WJ, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH. Use of DNA barcodes to identify flowering plants. Proc Nat Acad Sci USA 2005; 102:8369-74; PMID:15928076; http://dx.doi.org/10.1073/pnas.0503123102
- Kress WJ, Erickson DL. A Two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS ONE 2007; 2:e508; PMID:17551588; http://dx.doi.org/10.1371/journal.pone.0000508
- Deschamps S, Victor L, Gregory DM. Genotyping-by-sequencing in plants. Biol 2012; 1:460-83; PMID:24832503; http://dx.doi.org/10.3390/biology1030460
- Dodds PN, Rathjen JP. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 2010; 11:539-48; PMID:20585331; http://dx.doi.org/10.1038/nrg2812
- Carroll RK, Shelburne SA 3rd, Olsen RJ, Suber B, Sahasrabhojane P, Kumaraswami M, Beres SB, Shea PR, Flores AR, Musser JM. Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J Clin Invest 2011; 121:1956-68; PMID:21490401; http://dx.doi.org/10.1172/JCI45169
- Calvitti M, Moretti R, Lampazzi E, Bellini R, Dobson SL. Characterization of a new Aedes albopictus (Diptera: Culicidae)-Wolbachia pipientis (Rickettsiales: Rickettsiaceae) symbiotic association generated by artificial transfer of the wPip strain from Culex pipiens (Diptera: Culicidae). J Med Entomol 2010; 47:179-87; PMID:20380298; http://dx.doi.org/10.1603/ME09140
- Venu RC, Zhang Y, Weaver B, Carswell P, Mitchell TK, Meyers BC, Boehm MJ, Wang GL. Large scale identification of genes involved in plant fungal interactions using Illumina's sequencing-by-synthesis technology. Method Mol Biol 2011; 722:167-78; PMID:21590420; http://dx.doi.org/10.1007/978-1-61779-040-9_12
- Lindeberg M, Myers CR, Collmer A, Schneider DJ. Roadmap to new virulence determinants in Pseudomonas syringae: insights from comparative genomics and genome organization. Mol Plant Micro Inter 2008; 21:685-700; PMID:18624633; http://dx.doi.org/10.1094/MPMI-21-6-0685
- Cantu D, Govindarajulu M, Kozik A, Wang M, Chen X, Kojima KK, Jurka J, Michelmore RW, Dubcovsky J. Next generation sequencing provides rapid access to the genome of Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust. PLoS One 2011; 6:e24230; PMID:21909385; http://dx.doi.org/10.1371/journal.pone.0024230
- Passos MAN, Emediato FL, Cruz VO, de Camargo Teixeira C, de Alencar Figueiredo LF, Martins NF, Togawa RC, Costa MMC, Silva O Jr, Pappas GJ Jr, et al. Understanding plant immunity: transcriptome profiling in Musa-pathogen interactions using next generation sequencing. Acta Hort 2013; 986:227-40.
- Burke JM, Burger JC, Chapman MA. Crop evolution: from genetics to genomics. Curr Opin Genet Developt 2007; 17:525-32; PMID:17933510; http://dx.doi.org/10.1016/j.gde.2007.09.003
- Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M. An SNP caused loss of seed shattering during rice domestication. Science 2006; 312:1392-6; PMID:16614172; http://dx.doi.org/10.1126/science.1126410
- Li C, Zhou A, Sang T. Rice domestication by reducing shattering. Science 2006; 311:1936-9; PMID:16527928; http://dx.doi.org/10.1126/science.1123604
- Liu A, Burke JM. Patterns of nucleotide diversity in wild and cultivated sunflower. Genetic 2006; 173:321-30; PMID:16322511; http://dx.doi.org/10.1534/genetics.105.051110
- Vigouroux Y, Jaqueth JS, Matsuoka Y, Smith OS, Beavis WD, Smith JSC, Doebley J. Rate and pattern of mutation at microsatellite loci in maize. Mol Biol Evolu 2002; 19:1251-60; PMID:12140237; http://dx.doi.org/10.1093/oxfordjournals.molbev.a004186
- Wright SI, Bi IV, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut BS. The effects of artificial selection on the maize genome. Science 2005; 308:1310-4; PMID:15919994; http://dx.doi.org/10.1126/science.1107891
- Burke JM, Knapp SJ, Rieseberg LH. Genetic consequences of selection during the evolution of cultivated sunflower. Genetics 2005; 171:1933-40; PMID:15965259; http://dx.doi.org/10.1534/genetics.104.039057
- Buckler ES, Thornsberry J. Plant molecular diversity and applications to genomics. Curr Opin Plant Biol 2002; 5:107-11; PMID:11856604; http://dx.doi.org/10.1016/S1369-5266(02)00238-8
- Burger JC, Chapman MA, Burke JM. Molecular insights into the evolution of crop plants. Amer J Bot 2008; 95:113-22; PMID:21632337; http://dx.doi.org/10.3732/ajb.95.2.113
- Aharoni A, Keizer LCP, Bouwmeester HJ, Sun Z, Alvarez-Huerta M, Verhoeven HA, Blaas J, Houwelingen AMMLV, De Vos RCH, et al. Identification of the SAAT gene involved in strawberry flavour biogenesis by use of DNA microarrays. Plant Cell 2000; 12:647-61; PMID:10810141; http://dx.doi.org/10.1105/tpc.12.5.647
- Doebley JF, Brandon SG, Smith BD. The molecular genetics of crop domestication. Cell 2006; 127:1309-21; PMID:17190597; http://dx.doi.org/10.1016/j.cell.2006.12.006
- Tripathi AK, Pareek A, Sopory SK, Singla-Pareek SL. Narrowing down the targets for yield improvement in rice under normal and abiotic stress conditions via expression profiling of yield-related genes. Rice 2012; 5:37; PMID:24280046; http://dx.doi.org/10.1186/1939-8433-5-37
- Daniell H, Khan MS, Allison L. Milestones in chloroplast genetic engineering: an environmentally friendly era in biotechnology. Trend Plant Sci 2002; 7:84-91; PMID:11832280; http://dx.doi.org/10.1016/S1360-1385(01)02193-8
- Ruiz-Ferrer V, Voinnet O. Roles of plant small RNAs in biotic stress responses. Annu Rev Plant Biol 2009; 60:485-510; PMID:19519217; http://dx.doi.org/10.1146/annurev.arplant.043008.092111
- Zhang X, Zou Z, Zhang J, Zhang Y, Han Q, Hu T, Xu X, Liu H, Li H, Ye Z. Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant. FEBS Lett 2011b; 585:435-9; PMID:21187095; http://dx.doi.org/10.1016/j.febslet.2010.12.036
- Xin M, Wang Y, Yao Y, Song N, Hu Z, Qin D, Xie C, Peng H, Ni Z, Sun Q. Identification and characterization of wheat long non-protein coding RNAs responsive to powdery mildew infection and heat stress by using microarray analysis and SBS sequencing. BMC Plant Biol 2011; 11:61; PMID:21473757; http://dx.doi.org/10.1186/1471-2229-11-61
- Zhao M, Ding H, Zhu J K, Zhang F, Li W X. Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol 2011; 190:906-15; PMID:21348874; http://dx.doi.org/10.1111/j.1469-8137.2011.03647.x
- Zhang X, Zou Z, Gong P, Zhang J, Ziaf K, Li H, Xiao F, Ye Z. Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol Lett 2011a; 33:403-9; PMID:20960221; http://dx.doi.org/10.1007/s10529-010-0436-0
- Yang, L, Wakasa Y, Kawakatsu T, Takaiwa F. The 3′-untranslated region of rice glutelin GluB-1 affects accumulation of heterologous protein in transgenic rice. Biotechnol Lett 2009; 31:1625-31; PMID:19547924; http://dx.doi.org/10.1007/s10529-009-0056-8
- Liu WX, Liu HL, Chai ZJ, Xu XP, Song YR, Qu le Q. Evaluation of seed storage-protein gene 5′ untranslated regions in enhancing gene expression in transgenic rice seed. Theor Appl Genet 2010; 121:1267-74; PMID:20563548; http://dx.doi.org/10.1007/s00122-010-1386-6
- Lu J, Sivamani E, Li X, Qu R. Activity of the 5′ regulatory regions of the rice polyubiquitin rubi3 gene in transgenic rice plants as analysed by both GUS and GFP reporter genes. Plant Cell Rep 2008; 27:1587-600; PMID:18636262; http://dx.doi.org/10.1007/s00299-008-0577-y
- Sugio T, Satoh J, Matsuura H, Shinmyo A, Kato K. The 5′-untranslated region of the Oryza sativa alcohol dehydrogenase gene functions as a translational enhancer in monocotyledonous plant cells. J Biosci Bioeng 2008; 105:300-2; PMID:18397784; http://dx.doi.org/10.1263/jbb.105.300
- Ebert MS, Sharp PA. Emerging roles for natural microRNA sponges. Curr Biol 2010; 20:858-61; PMID:20937476; http://dx.doi.org/10.1016/j.cub.2010.08.052
- Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio- Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 2007; 39:1033-7; PMID:17643101; http://dx.doi.org/10.1038/ng2079
- Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, et al. RNA-Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol 2010; 10:160; PMID:20687943; http://dx.doi.org/10.1186/1471-2229-10-160
- Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, Bellin D, Pezzotti M, Delledonne M. Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-Seq. Plant Physiol 2010; 152:1787-95; PMID:20118272; http://dx.doi.org/10.1104/pp.109.149716
- Deyholos MK. Making the most of drought and salinity transcriptomics. Plant Cell Environ 2010; 33:648-54; PMID:20002333; http://dx.doi.org/10.1111/j.1365-3040.2009.02092.x
- Di Guistini S, Wang Y, Liao N Y, Taylor G, Tanguay P, Feau N, Henrissat B, Chan S K, Hesse-Orce, U, Alamouti SM, et al. Genome and transcriptome analyses of the mountain pine beetle-fungal symbiont Grosmannia clavigera, a lodgepole pine pathogen. Proc Natl Acad Sci USA 2011; 108:2504-9; PMID:21262841; http://dx.doi.org/10.1073/pnas.1011289108
- Wang B, Guo K, Li Y, Tu Y, Hu H, Wang B, Cui X, Peng L. Expression profiling and integrative analysis of the CESACSL superfamily in rice. BMC Plant Biol 2010; 10:282; PMID:21167079; http://dx.doi.org/10.1186/1471-2229-10-282
- Qiu X, Xie W, Lian X, Zhang Q; Molecular analyses of the rice glutamate dehydrogenase gene family and their response to nitrogen and phosphorous deprivation. Plant Cell Rep 2009; 28:1115-26; PMID:19430792; http://dx.doi.org/10.1007/s00299-009-0709-z
- Hayashi M, Takahashi H, Tamura K, Huang J, Yu LH, Kawai-Yamada M, Tezuka T, Uchimiya H. Enhanced dihydroflavonol-4-reductase activity and NAD homeostasis leading to cell death tolerance in transgenic rice. Proc Natl Acad Sci USA 2005; 102:7020-5; PMID:15863611; http://dx.doi.org/10.1073/pnas.0502556102
- Takahashi H, Hayashi M, Goto F, Sato S, Soga T, Nishioka T, Tomita M, Kawai-Yamada M, Uchimiya H. Evaluation of metabolic alteration in transgenic rice overexpressing dihydroflavonol-4-reductase. Ann Bot 2006; 98:819-25; PMID:16849376; http://dx.doi.org/10.1093/aob/mcl162
- Fukushima A, Kusano M, Redestig H, Arita M, Saito K. Metabolomic correlation-network modules in Arabidopsis based on a graph-clustering approach. BMC Syst Biol 2011; 5:1; PMID:21194489; http://dx.doi.org/10.1186/1752-0509-5-1
- Kusano M, Tohge T, Fukushima A, Kobayashi M, Hayashi N, Otsuki H, Kondou Y, Goto H, Kawashima M, Matsuda F, et al. Metabolomics reveals comprehensive reprogramming involving two independent metabolic responses of Arabidopsis to UV-B light. Plant J 2011; 67:354-69; PMID:21466600; http://dx.doi.org/10.1111/j.1365-313X.2011.04599.x
- Sanchez DH, Pieckenstain FL, Szymanski J, Erban A, Bromke M, Hannah MA, Kraemer U, Kopka J, Udvardi MK. Comparative functional genomics of salt stress in related model and cultivated plants identifies and overcomes limitations to translational genomics. PLoS One 2011; 6:e17094; PMID:21347266; http://dx.doi.org/10.1371/journal.pone.0017094
- Du CX, Fan HF, Guo SR, Tezuka T, Li J. Proteomic analysis of cucumber seedling roots subjected to salt stress. Phytochemistry 2010; 71:1450-9; PMID:20580043; http://dx.doi.org/10.1016/j.phytochem.2010.05.020
- Guo Y, Song Y. Differential proteomic analysis of apoplastic proteins during initial phase of salt stress in rice. Plant Signal Behav 2009; 4:121-2; PMID:19714920; http://dx.doi.org/10.4161/psb.4.2.7544
- Li XJ, Yang MF, Chen H, Qu LQ, Chen F, Shen SH. Abscisic acid pretreatment enhances salt tolerance of rice seedlings: proteomic evidence. Biochim Biophys Acta 2010; 1804:929-40; PMID:20079886; http://dx.doi.org/10.1016/j.bbapap.2010.01.004
- Carpentier SC, Witters E, Laukens K, Van Onckelen H, Swennen R, Panis B. Banana (Musa spp.) as a model to study the meristem proteome: acclimation to osmotic stress. Proteom 2007; 7:92-105; PMID:17149779; http://dx.doi.org/10.1002/pmic.200600533
- Pan Z, Liu Q, Yun Z, Guan R, Zeng W, Xu Q, Deng X. Comparative proteomics of a lycopene-accumulating mutant reveals the important role of oxidative stress on carotenogenesis in sweet orange (Citrus sinensis [L.] osbeck). Proteom 2009; 9:5455-70; PMID:19834898; http://dx.doi.org/10.1002/pmic.200900092