
Abstract
Here, we reported a Chinese case of Creutzfeldt-Jakob disease (CJD) with a rare mutation in the prion protein gene (PRNP) leading to an exchange of amino acid from valine (V) to isoleucine (I) at codon 180 (V180I). The 72 year-old Chinese women started with gradual memory loss. On admission, she did not present special abnormality during clinical examinations except bradykinesia in her lower extremities. Myoclonic jerks and increased muscle tone were noticed 3 months after the onset. No periodic activity was recorded at electroencephalography (EEG) and 14-3-3 protein was negative in the cerebrospinal fluid (CSF) sample. Brain diffusion weighted images (DWI) demonstrated high signal intensities in bilateral frontal, parietal, temporal and occipital cortices, especially on the left hemisphere, and high signal intensities were also seen in the left caudate nucleus and the putamen. The patient had no family history of similar symptoms. Her general condition was gradually deteriorative, but the patient was still alive when we performed the follow-up 12 months after onset.
Introduction
Human prion diseases are a group of rare fatal neurodegenerative diseases. They are classified as sporadic, inherited or acquired, including Creutzfeldt–Jakob disease (CJD), Kuru, Gerstmann–Sträussler–Scheinker syndrome (GSS) and fatal familial insomnia (FFI).Citation1 CJD has an incidence of 1 or 2 cases per million per year with rapidly progressive dementia, cerebellar ataxia, myoclonus, and behavioral changes. About 85% of all CJD cases are sporadic, 10–15% are inherited, and less than 1% are infectious.Citation2 Prion diseases are characterized by an accumulation of an abnormally folded isoform (PrPSc) of the host encoded cellular prion protein (PrPC). PrPSc type and genotype at polymorphic PRNP codon 129 are associated with different sporadic CJD phenotypes.Citation3 More than 55 mutations in the PRNP coding sequence have been described associating with human genetic prion diseases, whose clinical manifestations and neuropathological abnormalities may vary largely depending on the various genotypes.Citation4 Till now, more than 50 cases of various genetic prion diseases have been identified by Chinese National CJD Surveillance Network since 2006, among them FFI cases with D178N mutation in PRNP geneCitation5 and T188K genetic CJD (gCJD) casesCitation6 are the most frequent ones. Here, we reported the first Chinese gCJD case with a substitution of an isoleucine for a valine at codon 180 (V180I) by PRNP sequencing. The patient was methionine homozygous at codon 129 and glutamic acid homozygous at codon 219, respectively.
Case Report
A 72-year-old woman was reported to the China National Surveillance Network for CJD (CNSNC) as a CJD suspected case based on the diagnostic criteria recommended by World Health Organization (WHO). The clinical data were collected by hospital neurologists and epidemiologic information was collected by the staff of the local Centers for Disease Control (CDC). Samples of blood and cerebrospinal fluid (CSF) were collected for PRNP sequencing and 14-3-3 protein assay at CNSNC.
The patient showed signs of amnesia about one month before her admittance to the hospital. Approximately 10 d before her admission, she was reported to repeatedly ask the same questions. Also, she was unable to remember whom she talked with on the phone just a couple of minutes before and her memory worsened rapidly. On admission, she had little spontaneous speech and could understand only very simple instructions. Her general physical examination was unremarkable and Bradykinesia was noted in her lower extremities. The neurological examinations of cranial nerves, motor system, sensory system, autonomic nerves, and muscle tone did not reveal any special abnormality. No pathological reflex and meningeal irritation sign was noticed. Blood routine tests, including white blood cells, red blood cells, blood platelets and hemoglobin, were all in normal range. Blood glucose, blood lipids and various enzymes were also in normal range. Her blood anti-thyroglobulin antibody was 74.6 U/ml, which was higher than the normal limit (<30 U/ml).
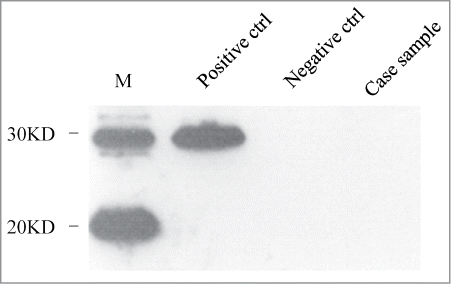
Diffusion weighted imaging (DWI) demonstrated high signal intensities in bilateral frontal, parietal, temporal and occipital cortices, especially on the left hemisphere (, left panel). High signal intensities were also seen in the left caudate nucleus and the putamen (, right panel). Electroencephalogram (EEG) on the second day of admission revealed intermittent 8 Hz slow wave on bilateral hemisphere without periodic synchronous discharge (PSD). Lumbar puncture was performed a couple of days after hospitalization and routine biochemical examination of cerebrospinal fluid (CSF) was normal. CSF 14-3-3 protein was detected by Western blot as described briefly. A total of 20 μl CSF sample was prepared for 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) by addition of 5 times loading buffer. Following electrophoresis, the fractionated proteins were transferred to nitrocellulose (NC) membranes (Whatman, USA) by the semi-dry method in transfering buffer. After blocking, the membrane was incubated with 1:1000 diluted 14-3-3 polyclonal antibody (Santa Cruz Biological, Santa Cruz, USA) at room temperature for 2 hours. Membranes were subsequently incubated with goat anti-rabbit HRP-conjugated secondary antibody and reactive signals were visualized using an enhanced chemiluminescence kit (Amersham-Pharmacia Biotech, USA). The 14-3-3 protein in the patient's CSF sample was negative in 3 independent tests ().
Figure 1. Diffusion weighted brain MRIs show high signal intensity in diffuse cerebral cortex with dominant involvement of left hemisphere (arrows). High signal intensities were also seen in the left caudate nucleus and the putamen (arrowheads).
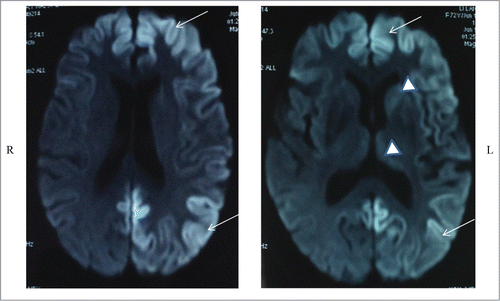
Figure 2. Western blot analysis of 14-3-3 protein in CSF samples. Positive ctrl: 10% goat brain homogenate. Negative ctrl: a human CSF sample previously confirmed to be 14-3-3 negative. Case sample: the CSF sample of the present V180I gCJD case. M: commercial prestained protein molecular markers whose relative molecular weights are shown on the left.
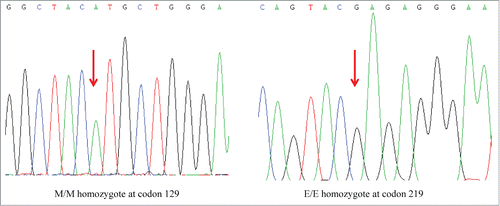
The patient's PRNP gene was amplified by an established polymerase chain reaction (PCR) protocol with the specific primers described beforeCitation7 (forward primer: 5′-GGC AAA CCT TGG ATG CTG G-3′ and reverse primer: 5′-CCC ACT ATC AGG AAG ATG AGG-3′). The genomic DNA was extracted from her peripheral blood leukocytes using a commercial kit (QIAGEN, Germany). Direct sequencing of the PCR product revealed a missense mutation at codon 180 of the PRNP gene (G to A), leading to a substitution from valine (Val) to isoleucine (Ile) (). The polymorphism of codon 129 was methionin homozygote (, left panel) and that of codon 219 was glutamate homozygote (right panel), respectively. No additional nucleotide exchanges were found in other regions of the PRNP sequence.
Figure 3. Graphic presentation of the sequencing analysis of PRNP. The left graph shows the DNA sequence of the patient at codon 180 shows a G to A heterozygous transition at codon 180 in one PRNP allele, leading to an exchange from Val (V) to Ile (I). The right graph shows a control of the DNA sequence of a person containing Val/Val homozygote at codon 180 in PRNP alleles.
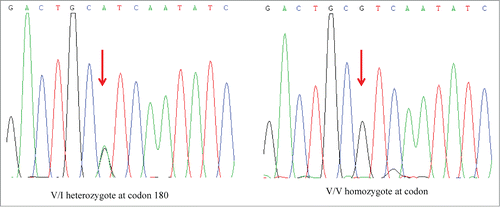
Figure 4. Graphic presentations of homozygosity for methionine at codon 129 (left) and homozygosity for glutamate at codon 219 (right) in the sequencing analysis of PRNP gene of the present V180I gCJD patient.
She had a history of diabetes for more than 1 year. She denied having history of hypertension, cardiovascular disease, surgical operation, blood transfusion, alcohol or substance abuse. She never traveled abroad. She had 2 sons and 3 daughters and retrospective review did not found any similar symptoms or other neurological diseases on her family members. Three months after the onset, she lost the ability to speak and did not recognize her family members. At the same time, she experienced myoclonic jerks and increased muscle tone. Six months after onset, she developed severe akinetic mutism and stayed at home. The patient was still alive when we conducted follow-up 12 months after onset of the disease, but lost contact afterwards. The family members did not agree to carry on the blood DNA detection.
Discussion
As a rare inherited prion disease, V180I CJD is a rare type of fCJD worldwide. There is one case reported in South Korea,Citation8 2 cases in Europe countriesCitation9 and 2 in the United states (Geschwind et al. unpublished results). However, this mutation is recognized as the most common cause of fCJD in Japan.Citation10,11,12 Although 2 individuals with V180I from Korea Association Resource (KARE) do not display symptoms of neurodegenerative disorder and V180I heterozygosity is considered not pathogenic factors but rarely observed polymorphisms,Citation13 the World Health Organization has listed CJD180 as familial CJD.Citation14 The case described in this study is the first V180I gCJD in China since the surveillance for human prion diseases began in 2006.
Comparing with those of sCJD, the clinical and laboratory characteristics of CJD patients with the V180I gCJD have been described by Jin et al,Citation15 including (1) older onset age, (2) slower disease progression, (3) unique clinical symptoms such as frequent cognitive dysfunction, but no visual or cerebellar symptoms, (4) reduced rate of brain-specific proteins, such as neuron-specific enolase (NSE) and 14-3-3 protein, in cerebrospinal fluid (CSF) samples, and (5) a lack of PSD in EEG throughout the course of disease. Our patient here seems to fulfill the features of V180I gCJD, with relatively older onset age (72-year old) and gradually progressive dementia. Amnesia is the primary symptoms of the patients and myoclonic jerk appears at the last stage of the disease. Like other reported V180I cases,Citation15 no visual or cerebellar symptoms have been identified in this Chinese patient case. CSF 14-3-3 protein is negative in the sample collected at relatively early stage. Although her EEG shows abnormal, no sCJD typical PSD has been noticed. Additionally, our patient here does not recall any family history of dementia and has relatively long clinical duration (more than 12 months till the last follow-up). Those data reveal again the special features of the patient carrying V180I mutation in PRNP and highlight the significance for screening PRNP sequence in the patients with similar clinical manifestations.
MRI abnormalities are frequently observed in the patients of V180I gCJD, in which diffuse cortical high-intensity DWI signals are characteristic feature.Citation15 Although high signal intensities in various brain regions can be seem in V180I gCJD with conventional MRI such as FLAIR and T2WI, Kono and the colleagues believe that DWI is more sensitive and is useful for both early diagnosis and monitoring disease progress.Citation11 High-intensity DWI signals is assumed to be attributable to spongiform degeneration. However, limited numbers of neuropathological data of V180I gCJD show that spongiform changes are more moderate compared with sCJD patients.Citation16 In contrast, almost all reported V180I gCJD cases appear high intensity DWI signals. The association of cerebral cortical ribbons in MRI images with neuropathological changes in CJD cases needs more detailed clarification.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Chinese National Natural Science Foundation Grants (81301429), China Mega-Project for Infectious Disease (2011ZX10004-101, 2012ZX10004215), and SKLID Development Grant (2012SKLID102, 2011SKLID211).
References
- Sikorska B, Knight R, Ironside JW, Liberski PP. Creutzfeldt-Jakob disease. Adv Exp Med Biol 2012; 724:76-90; PMID:22411235; http://dx.doi.org/10.1007/978-1-4614-0653-2_6
- Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363-13383; PMID:9811807; http://dx.doi.org/10.1073/pnas.95.23.13363
- Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999; 46:224-233; PMID:10443888; http://dx.doi.org/10.1002/1531-8249(199908)46:2%3c224::AID-ANA12%3e3.0.CO;2-W
- Zerr I, Poser S. Clinical diagnosis and differential diagnosis of CJD and vCJD. With special emphasis on laboratory tests. APMIS 2002; 110:88-98; PMID:12064260; http://dx.doi.org/10.1034/j.1600-0463.2002.100111.x
- Shi Q, Chen C, Gao C, Tian C, Zhou W, Zhang B, Han J, Dong XP. Clinical and familial characteristics of ten chinese patients with fatal family insomnia. Biomed Environ Sci 2012; 25:471-475; PMID:23026528
- Chen C, Shi Q, Zhou W, Zhang XC, Dong JH, Hu XQ, Song XN, Liu AF, Tian C, Wang JC, et al. Clinical and familial characteristics of eight Chinese patients with T188K genetic Creutzfeldt-Jakob disease. Infect Genet Evol 2013; 14:120-124; PMID:23261545; http://dx.doi.org/10.1016/j.meegid.2012.11.019
- Shi Q, Chen C, Wang XJ, Zhou W, Wang JC, Zhang BY, Gao C, Gao C, Han J, Dong XP. Rare V203I mutation in the PRNP gene of a Chinese patient with Creutzfeldt-Jakob disease. Prion 2013; 7:259-262; PMID:23764840; http://dx.doi.org/10.4161/pri.24674
- Yang TI, Jung DS, Ahn BY, Jeong BH, Cho HJ, Kim YS, Na DL, Geschwind MD, Kim EJ. Familial Creutzfeldt-Jakob disease with V180I mutation. J Korean Med Sci 2010; 25:1097-1100; PMID:20592908; http://dx.doi.org/10.3346/jkms.2010.25.7.1097
- Chasseigneaux S, Haik S, Laffont-Proust I, De Marco O, Lenne M, Brandel JP, Hauw JJ, Laplanche JL, Peoc'h K. V180I mutation of the prion protein gene associated with atypical PrPSc glycosylation. Neurosci Lett 2006; 408:165-169; PMID:17029785; http://dx.doi.org/10.1016/j.neulet.2006.08.008
- Mutsukura K, Satoh K, Shirabe S, Tomita I, Fukutome T, Morikawa M, Iseki M, Sasaki K, Shiaga Y, Kitamoto T, et al. Familial Creutzfeldt-Jakob disease with a V180I mutation: comparative analysis with pathological findings and diffusion-weighted images. Dement Geriatr Cogn Disord 2009; 28:550-557; PMID:20051687; http://dx.doi.org/10.1159/000254842
- Kono S, Manabe Y, Fujii D, Sakai Y, Narai H, Omori N, Kitamoto T, Abe K. Serial diffusion-weighted MRI and SPECT findings in a Creutzfeldt-Jakob disease patient with V180I mutation. J Neurol Sci 2011; 301:100-103; PMID:21094959; http://dx.doi.org/10.1016/j.jns.2010.10.032
- Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F, et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain 2010; 133:3043-3057; PMID:20855418; http://dx.doi.org/10.1093/brain/awq216
- Moe Lee S, Ran Ju Y, Choi BY, Wook Hyeon J, Sun Park J, Kyeong Kim C, Yeon Kim S. Genotype patterns and characteristics of PRNP in the Korean population. Prion 2012; 6:375-382; PMID:22561193; http://dx.doi.org/10.4161/pri.20195
- Jeong BH, Jin HT, Choi EK, Carp RI, Kim YS. Lack of association between 14-3-3 beta gene (YWHAB) polymorphisms and sporadic Creutzfeldt-Jakob disease (CJD). Mol Biol Rep 2012; 39:10647-10653; PMID:23053962; http://dx.doi.org/10.1007/s11033-012-1954-8
- Jin K, Shiga Y, Shibuya S, Chida K, Sato Y, Konno H, Doh-ura K, Kitamoto T, Itoyama Y. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology 2004; 62:502-505; PMID:14872044; http://dx.doi.org/10.1212/01.WNL.0000106954.54011.80
- Deguchi K, Takamiya M, Deguchi S, Morimoto N, Kurata T, Ikeda Y, Abe K. Spreading brain lesions in a familial Creutzfeldt-Jakob disease with V180I mutation over 4 years. BMC Neurol 2012; 12:144; PMID:23176099; http://dx.doi.org/10.1186/1471-2377-12-144