
Abstract
The sequestration of misfolded proteins into aggregates is an integral pathway of the protein quality control network that becomes particularly prominent during proteotoxic stress and in various pathologies. Methods for systematic analysis of cellular aggregate content are still largely limited to fluorescence microscopy and to separation by biochemical techniques. Here, we describe an alternative approach, using flow cytometric analysis, applied to protein aggregates released from their intracellular milieu by mild lysis of yeast cells. Protein aggregates were induced in yeast by heat shock or by chaperone deprivation and labeled using GFP- or mCherry-tagged quality control substrate proteins and chaperones. The fluorescence-labeled aggregate particles were distinguishable from cell debris by flow cytometry. The assay was used to quantify the number of fluorescent aggregates per μg of cell lysate protein and for monitoring changes in the cellular content and properties of aggregates, induced by stress. The results were normalized to the frequencies of fluorescent reporter expression in the cell population, allowing quantitative comparison. The assay also provided a quantitative measure of co-localization of aggregate components, such as chaperones and quality control substrates, within the same aggregate particle. This approach may be extended by fluorescence-activated sorting and isolation of various protein aggregates, including those harboring proteins associated with conformation disorders.
Abbreviations
| FACS | = | fluorescence-activated cell sorting |
| GFP | = | green fluorescent protein |
| Hsp | = | heat shock protein |
Introduction
The formation and properties of intracellular aggregates, formed by misfolded or intrinsically aggregation-prone proteins, have been the subject of great interest, particularly because of the suspected involvement of such aggregates in a variety of neurodegenerative diseases.Citation1,2 Moreover, sequestration of misfolded proteins in discrete particles or aggregates can occur even in the absence of cellular or environmental stress, indicating that it is a general protein quality control pathway that is complementary to ubiquitin-proteasome degradation.Citation3 A generic mechanism of misfolded protein aggregate formation involves the initial recruitment of Hsp70/40 chaperones to the misfolded protein, eventually leading to either refolding or ubiquitylation and ultimate degradation.Citation4 While these processes have been the focus of numerous studies of protein misfolding, a number of questions remain open, such as (i) what factors determine the diversion of a misfolded protein from the degradation to the sequestration pathway under normal, stress-free conditions, (ii) what factors are involved in the maintenance and subsequently in the dissolution of the aggregates, and (iii) what determines the number, size, and properties of different aggregates. A convenient method for quantifying intracellular aggregates could provide a useful tool toward answering these questions.
Current approaches for analyzing and quantifying protein aggregates commonly rely on fluorescence microscopy and biochemical methods employing precipitation or separation by detergent insolubility and high molecular mass, both of which suffer from a variety of limitations. Fluorescent microscopy often requires manually-assisted aggregate counting, as image-analysis algorithms are highly dependent on background corrections and may be inadequate in cases of high background fluorescence or small aggregate size. Similarly, co-localization of fluorescently-labeled proteins is generally visual-based and therefore prone to artifacts as well as random error and bias.Citation5,6 On the other hand, aggregate analysis by biochemical methods such as differential centrifugation, detergent insolubility, or size exclusion by gel filtration are usually not suited for monitoring time dependent changes within multiple samples, particularly when there is variability of reporter expression in the population. Furthermore, analysis of aggregate protein composition by biochemical methods has various drawbacks, as aggregate purification by treatment with the anionic surfactant SDS causes dissociation of a significant portion of the associated proteins, while density gradient isolation of aggregates can result in the adhesion of non-specifically-associated polypeptides.
Here we introduce a flow cytometry-based method for characterizing protein aggregates isolated from the cellular environment. Its principal advantages are relative simplicity, medium throughput capability and applicability to quantitative comparisons of aggregate content between different yeast strains with variable levels of reporter expression. In addition, it enables the assessment of co-localization of multiple reporter proteins on the same isolated aggregate.
Materials and Methods
Yeast strains
S. cerevisiae strains used in this study were grown in Synthetic defined (SD) media containing casamino acid (BD Biosciences). Yeast strains used in this study are listed in supplementary Table S1. Unless indicated otherwise, the genetic background for yeast strains used in this study was that of W303 (α, his3–11,15, leu2–3,112, ura3–52, trp1–1, ade2–1, can 1–100).
Plasmids
Plasmids used in this study are listed in Supplementary Table S2. The plasmids were constructed using standard molecular biology techniques. Primers for constructing plasmids and strains using PCR amplification techniques are available upon request.
Fluorescence imaging
Images of cells and lysates were obtained by confocal microscopy using a Bio-Rad (Hercules, CA) MRC-1024 workstation attached to a Zeiss (Jena, Germany) Axiovert 135 M microscope equipped with a 63 × /1.4 objective.
Cell preparation
Cells were grown at the indicated temperatures to a density of 0.75–1.0 OD 600 units, then centrifuged and resuspended in PBS containing 25% glycerol at a concentration of 0.5 OD 600 units/ml. Aliquots of 100 μl of the cell suspension (0.5 OD 600 units) were frozen at −80°C for future use.
Cell disruption
Prior to disruption, the thawed cells were centrifuged and the cell pellet was resuspended in 320 μl buffer containing 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 5% Glycerol, 100 mM KCl and 0.1% NP-40 (Igepal CA-630, Sigma Chem. Co., St. Louis, MO) that had been filtered through a 0.2 μm microfilter to remove contaminating particles that could interfere with flow cytometry. Protease inhibitors were not routinely included in the buffer, as preliminary experiments showed that they did not affect the yield of aggregates. All steps were done at 4°C, up to flow cytometric analysis. Prior to lysis, a 10 μl sample of the cell suspension was removed, diluted in 200 μl PBS and subsequently analyzed by flow cytometry to measure the fraction of fluorescent cells in the population (this value is essential for normalizing aggregate numbers to the proportion of cells expressing the relevant reporter). The residual 310 μl of cell suspension were then added to 200 μl of glass beads (425–600 μm from Sigma Chem. Co., St. Louis, MO) in 0.5 ml snap-cap tubes. The glass beads had been prewashed in 5 volumes of 3.7% HCl for 1 hr with occasional swirling, then with DDW until pH was neutral and stored as slurry in DDW at 4°C. The bead slurry was aliquoted into 0.5 ml microfuge tubes and prior to the addition of cells to the beads, excess liquid was aspirated from the beads by suction via a 27 G needle. The cells were lysed in a bead beater (Fast Prep-24, MP Biomedicals, Santa Ana, CA) with 2 30-s bursts with a 30-s interval in between. Following disruption, the samples were centrifuged in a free-swinging bucket at 500 × g for 5 min to remove unbroken cells, and 150 μl of the bead supernatant comprising the cleared cell lysate were removed for analysis by flow cytometry and for determination of protein concentration with Bradford reagent (from Sigma Chem. Co., St. Louis, MO).
Flow cytometry
Flow cytometry analysis was performed using a FACS Aria III flow cytometer-sorter equipped with 488 nm and 531 nm lasers (BD Biosciences, San Jose, CA) and all flow cytometry data were analyzed with FACSDiva software (BD Biosciences). Side and forward scatter of aggregates in cell lysates were determined using log scale SSC/FSC plots with respective thresholds of 200 and 1000. Voltage settings for the SSC, FSC, GFP and mCherry channels were kept constant for all experiments described. Cell lysates were analyzed at a flow rate of 10 μl per minute for a period of 30 sec to quantify the number of aggregates per 5 μl of cell lysate.
Results
Flow cytometric identification of subcellular particles in yeast cell lysates
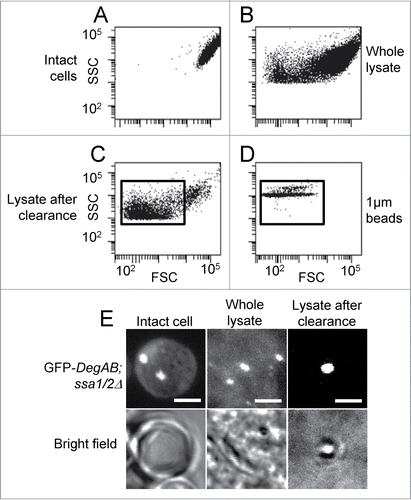
Flow cytometry analysis of cell lysates indicated the presence of a substantial number of subcellular particles with a wide forward scatter distribution, indicative of variable particle sizes (). This fraction comprised cellular debris and organelles, as well as aggregates. These were clearly distinguishable from whole cells by their significantly smaller side- and forward- scatter values (compare ). Low-speed centrifugation of the lysates removed most of the larger particles (compare ) comprising larger cell fragments and organelles, as corroborated by confocal microscopy (, bright field). The gate used to select particles corresponding to protein aggregates was chosen to exclude nuclei and whole cells and encompass particles with ∼1 μm diameter, as determined using 1 μm polystyrene beads (). The aggregate diameters observed by fluorescent microscopy (, upper panel) were in the range of 0.5–1 μm (), which roughly corresponds to the size of the particles included in the gate shown in .
Figure 1. Identification of aggregates in cell lysates by flow cytometry and fluorescent imaging. ssa1/2Δ yeast cells expressing GFP-DegAB were lysed as described in the Materials and Methods section, followed by low-speed centrifugation. Crude and purified lysates were analyzed by flow cytometry and visualized by confocal microscopy. (A–D) Dot plots of side versus forward scatter (SSC vs. FSC) of (A) intact cells, (B) total crude lysates, (C) lysates after centrifugation of 1000 × g for 5 min, and (D) 1 μm polystyrene beads. (E) Visualization by fluorescence confocal microscopy of the samples shown in A, B, and C.
Detection of heat shock-induced protein aggregates in cell lysates by flow cytometry
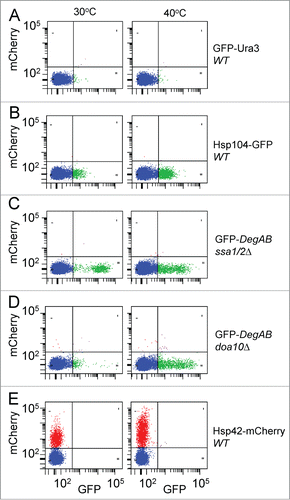
In order to assess in qualitative terms the capacity of this approach to detect aggregates, we compared lysates from normal and heat-shocked cells, expressing fluorescent proteins known to be associated with aggregates. These included the chaperones Hsp104-GFP and Hsp42-mCherry in wild type (WT) strain backgrounds and the misfolded protein GFP-DegAB in the background of deletion of both the Hsp70s Ssa1 and Ssa2 (ssa1/2Δ) or the E3 ubiquitin ligase doa10 (doa10Δ)Citation6-9 (). The soluble cytosolic fusion protein Ura3-GFP served as a negative control and was used to establish the appropriate gates for differentiating between non-fluorescent particles and fluorescent aggregates. A 2-dimensional dot-plot was used to enable discrimination between auto- and authentic fluorescence. Visual inspection of dot plots of mCherry vs. GFP fluorescence revealed the presence of fluorescently-labeled aggregates of Hsp104-GFP and Hsp42-mCherry even at the normal growth temperature of 30°C, though these were not observed with Ura3-GFP. Aggregate distribution similar to Hsp104-GFP and Hsp42-mCherry was also observed in Hsp70 depleted cells (ssa1/2Δ) expressing the misfolded fusion protein GFP-DegAB. The aggregation of GFP-DegAB strongly depends on its polyubiquitylation by the quality control E3 ligase Doa10.Citation9 Accordingly, the number of GFP-DegAB aggregates was markedly reduced in doa10Δ cells, in agreement with our previous observations.Citation9 Finally, heat shock at 40°C resulted in a visible increase in the number of fluorescent aggregates in all the systems except 2: Ura3-GFP, expressed in WT cells and GFP-DegAB expressed in ssa1/2Δ cells. The Ura3-GFP fusion protein demonstrated very little aggregation, at both 30°C and 40°C, presumably due to factors such as high solubility and dilution upon lysis. GFP-DegAB expressed in ssa1/2Δ cells showed high levels of aggregation, at both 30°C and 40°C. Indeed, Hsp70 depletion by itself leads to increased protein aggregation, regardless of heat shock.
Figure 2. Detection of heat shock-induced protein aggregates by flow cytometry. Yeast cells expressing GFP- and mCherry-fusion proteins were incubated during the logarithmic growth phase at either 30°C (left graphs) or 40°C (right graphs) for 3 hours and then lysed and processed for flow cytometry as described in the Materials and Methods section. The aggregate population was detected and gated in SSC vs. FSC plots as shown in . The various fluorescent aggregate populations are shown in dot plots of mCherry vs. GFP fluorescence of 5 μl of cell lysates of: (A) GFP-Ura3, expressed in WT cells, (B) Hsp104-GFP, expressed in WT cells, (C) GFP-DegAB, expressed in ssa1/2Δ cells, (D) GFP-DegAB, expressed in doa10Δ cells, and (E) Hsp42-mCherry, expressed in WT cells.
Characterization of protein aggregate size and fluorescence intensity
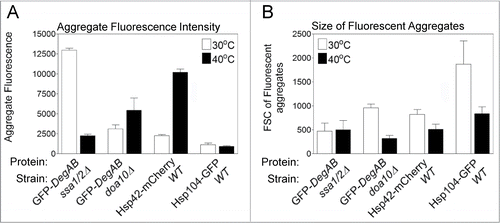
Flow cytometry enables the characterization of several aggregate features that are not easily obtained by fluorescence microscopy, such as mean fluorescence intensity and size of aggregates. As shown in , the mean fluorescence intensity of the aggregates was increased 4.5-fold at 40°C relative to 30°C in the case of Hsp42-mCherry-containing aggregates, whereas it was decreased by 5.7-fold in GFP-DegAB -containing aggregates (in the ssa1/2Δ background) (). In contrast to the sharp change in mean fluorescence intensity of these 2 reporters, their relative size showed insignificant change, as determined by their forward scatter, FSC (). A different pattern was obtained for GFP-DegAB aggregates in the doa10 Δ background and Hsp104 in the WT background, both of which showed little change in fluorescence intensity, but a 2- and 3-fold decrease in size at 40°C. We attribute these shifts in aggregate properties to changes in the number of reporter protein molecules per aggregate as well as in protein composition, upon heat shock.
Figure 3. Characterization of the size and fluorescence intensity of protein aggregates. The various fluorescent aggregate populations defined by the gates shown in the dot plots in were used to derive values of mean fluorescence intensity and size of the aggregates. (A) The mean fluorescence intensity of the aggregates. (B) The mean forward scatter (FSC) values of the fluorescent aggregates.
Biochemical characterization of protein aggregates by flow cytometry
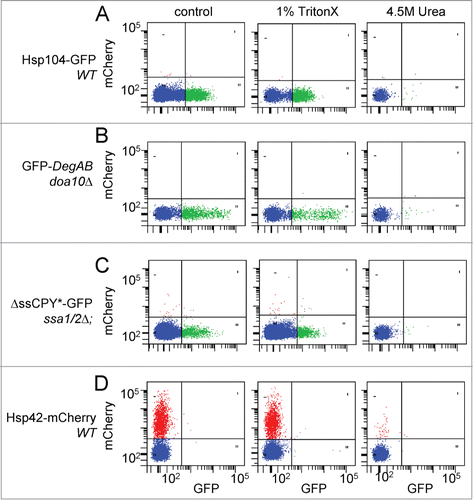
The applicability of the flow cytometry method for biochemical characterization of aggregates is illustrated in . Lysates of cells expressing the misfolded model proteins ΔssCPY*-GFPCitation10 or GFP-DegAB, or the Hsp104-GFP or Hsp42-mCherry chaperones, in the indicated cell backgrounds, were treated with either Triton X-100, SDS or 4.5 M urea and then subjected to flow cytometry analysis. Addition of the non-ionic detergent Triton X-100 (1% v/v) either during or after cell lysis did not significantly affect the number or fluorescence of the detected aggregates, induced at 40°C, indicating that these aggregates are Triton X-100 insoluble. Similarly, the addition of SDS (0.1% w/v) caused a ≤20% decrease in aggregate numbers (not shown). Thus the characteristics of the aggregates obtained by flow cytometry are typical to quality control aggregates, similar to the previously described IPOD (Insoluble Protein Deposit),Citation11 polyglutamine aggregatesCitation12 or the terminally misfolded reporter protein, short-lived GFP (slGFP), obtained following chaperone depletion.Citation13 Conversely, treatment of cell lysates with 4.5 M urea, an effective protein-disaggregating agent, eliminated virtually all of the aggregate-associated fluorescence (). We assume that this was due to aggregate disintegration and not to GFP denaturation, since GFP is stable up to 8 M urea.Citation14 The same applies to mCherry-labeled aggregates.Citation11
Figure 4. Effects of Triton X-100 and Urea on aggregate integrity. Yeast cell lysates were prepared as described in the Materials and Methods section, from cells expressing the following protein markers: (A) Hsp104-GFP, expressed in WT cells; (B) GFP-DegAB, expressed in doa10Δ cells; (C) ΔssCPY*-GFP, expressed in ssa1/2Δ cells; and (D) Hsp42-mCherry, expressed in WT cells. The cells in samples A, B, and D were incubated at 40°C for 3 hrs to increase the overall aggregate number. Cell lysates were treated at 4°C for 30 min as follows: Left panels, no treatment; Middle panels, 1% Triton X-100; Right panel, 4.5M Urea. Shown are dot plot analyses of mCherry vs. GFP fluorescence of 5 μl of cell lysates.
Quantification of relative aggregate content in cells
As shown in , flow cytometric analysis of cell lysates provides a qualitative measure of changes in aggregate concentration in the lysates. In order to perform reliable quantification of cellular aggregate content we took into account 2 additional factors: (1) the proportion of cells expressing the fluorescent reporter, measured by flow cytometry of intact cells, and (2) the efficiency of cell lysis, measured by the release of cell proteins to the lysate. The importance of the first factor is illustrated in , which shows the high variability in fluorescence distribution in the intact cell population, even in the case of Hsp104-GFP which was integrated into the yeast genome (see Hsp104-GFP distribution in WT cells, top right). Next, we assessed if a direct proportionality exists between the number of fluorescent aggregates per μg protein in the lysate, and the percentage of cells expressing the fluorescent-reporter. This was achieved by mixing fluorescent reporter-expressing cells with control, non-fluorescent cells at different ratios and performing aggregate analysis (). The linear relationship observed between these 2 parameters in 3 separate examples, indicated that the number of aggregates/ μg lysate protein is proportional to the fraction of reporter-expressing cells. In summary, the final calculation of aggregate content takes into account 3 parameters: a) the number of fluorescent aggregates per μl of cell lysate; b) Total protein concentration (μg protein per μl cell lysate); c) the fraction of cells expressing the fluorescent reporter. It is obtained by the formula a/b*c and the result is expressed as ‘Fluorescent aggregates/ μg protein’.
Figure 5. Determination of relative aggregate content in cells. (A) Fluorescence distribution of aggregates in WT cells expressing no reporter, Hsp104-GFP, Hsp42-mCherry, or both fluorescent reporters. The proportion of cells expressing the fluorescent reporters was determined by flow cytometry analysis, as described in the text. (B) Direct correlation between the number of fluorescent aggregates/μg lysate protein and the percentage of cells expressing the fluorescent-reporter in the population. Cells expressing the reporters: GFP-DegAB in ssa1/2Δ cells (left panel), ΔssCPY*-GFP in ssa1/2Δ cells (middle panel), or Hsp42-mCherry in WT cells after heat shock of 40°C for 3 hrs (right panel), were mixed with control, unlabeled cells at different ratios to obtain various percentage levels of fluorescent cells. Cells and aggregates were analyzed by flow cytometry. (C) Quantitation of fluorescent aggregate content in cells expressing the indicated fluorescent reporters. The values of fluorescent aggregates/μg lysate protein were compensated for fluorescent reporter expression, using the calculation described in the text. Data are from 3 or more separate cell preparations.
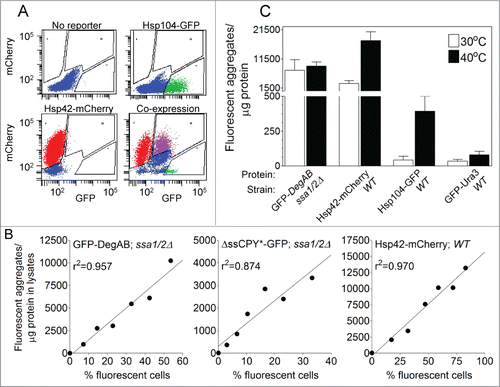
Shown in is an illustration of aggregate quantification. Aggregates were quantified in WT and ssa1/2Δ cells that were either grown at 30°C or heat-shocked at 40°C. Heat shock markedly increased the total aggregate content in WT cells expressing Hsp104-GFP or Hsp42-mCherry (by 8.7- and 5.8-fold, respectively), while the control GFP-Ura3 showed virtually no aggregates. The quality control substrate protein GFP-DegAB expressed in ssa1/2Δ cells showed similar high levels of aggregation, at both 30°C and 40°C. The latter result is not surprising, as depletion of the Hsp70s ssa1 and 2 by itself leads to high levels of GFP-DegAB aggregation, regardless of heat shock.
Evaluating the co-localization of aggregate components
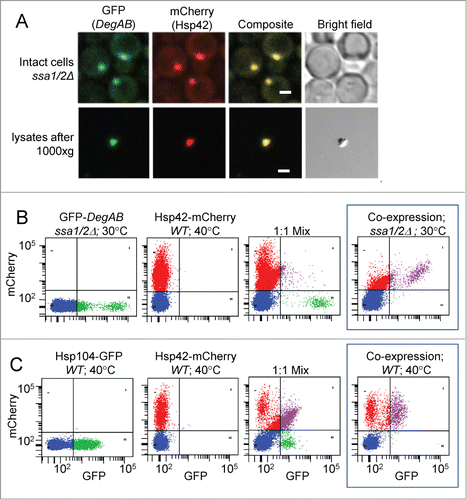
A potentially valuable application of the present assay is the quantification of protein co-localization within aggregates (). Fluorescence microscopy of ssa1/2Δ cells co-expressing GFP-DegAB and Hsp42-mCherry indicate co-localization of the proteins in both intact cells and cell lysates (). However, as shown in , Boxed panel, aggregates from cells co-expressing GFP-DegAB and Hsp42-mCherry in ssa1/2Δ strain are comprised of 2 principal populations: those containing only Hsp42-mCherry and those with both reporters. This enables the comparison between the relative numbers of singly- versus doubly-labeled aggregates. In this system, aggregates with only GFP-DegAB were undetectable, as the reporter completely shifted into the doubly-labeled population. In order to confirm that reporter co-localization in aggregates is not an artifact caused by redistribution of proteins during cell lysis, we mixed together 2 types of cells, each expressing an individual reporter, to produce a 1:1 mixture and analyzed the aggregate content (, left and middle panels). GFP-DegAB and Hsp42-mCherry demonstrated negligible intermixing (, right panel). Similarly, only minor intermixing was observed when cells expressing Hsp42-mCherry were mixed and co-lysed with aggregate-containing cells expressing ΔssCPY*-GFP (data not shown). However, lysis of a mixture of cells expressing Hsp42-mCherry with cells expressing Hsp104-GFP resulted in significant co-localization of the reporters (, right panel), indicative of redistribution during lysis, which is not readily distinguishable from co-localization observed in cells co-expressing both reporters (, boxed panel).
Figure 6. Co-localization of fluorescent reporter proteins in aggregates. (A) Imaging of GFP-DegAB and Hsp42-mCherry, co-expressed in ssa1/2Δ cells by fluorescence confocal microscopy. Upper panels: cellular localization of the 2 reporter proteins; lower panels: co-localization of the 2 reporter proteins in an aggregate, in cleared lysates. Scale bar: 2.5 μm. (B and C) Dot plot analyses of mCherry vs. GFP fluorescence of 5 μl of cell lysates. (B) Aggregates from cells expressing one fluorescent reporter, either GFP-DegAB (Left panel) or Hsp42-mCherry (middle panel). Right panel shows the aggregate distribution of a 1:1 mixture of the 2 cell types. Boxed panel shows the aggregate distribution in cells co-expressing the 2 fluorescent reporters indicated. (C) Aggregates from cells expressing one fluorescent reporter, either Hsp104-GFP (left panel) or Hsp42-mCherry (middle panel). Right panel shows the aggregate distribution of a 1:1 mixture of the 2 cell types. Boxed panel shows the aggregate distribution in cells co-expressing the 2 fluorescent reporters indicated.
Discussion
Here, we describe a method for the characterization and quantification of fluorescently labeled protein aggregates in yeast cells. The method is based on physical disruption of the cells and analysis of the released aggregates by flow cytometry. This method allows aggregates to be easily detected as fluorescent particles, while soluble reporters are highly diluted in the buffer, well below the detection threshold of the system. Thus, the method allows direct analysis of lysed cells without requiring further aggregate purification.
We adopted this approach in an attempt to overcome some of the limitations of existing methods, which are based mainly on fluorescence microscopy or biochemical separation depending on detergent insolubility and high molecular mass. The present method enables objective quantification of aggregates that are easily obtained by cell lysis without requiring further purification steps. It is independent of the complex image-processing algorithms, background corrections and specialized analysis algorithms required by standard microscopy-based methods for aggregate detection and it has the potential to enable the recovery of distinct aggregate populations for further analysis by fluorescence activated sorting. Furthermore, this method allows for the comparison of aggregate content between different strains with variable levels of fluorescent reporter expression. The problem of variable reporter expression is often exacerbated in stressed or chaperone-deficient strains, which are of particular interest in studies of aggregate formation. In addition, when applied to double-labeled cells, this method enables the quantitative, physical co-localization and distribution of the fluorescent reporters among different aggregate populations. It enables detection of small or weakly fluorescent aggregates, which may be missed by microscopy or biochemical methods due to high background fluorescence or soluble, unaggregated forms of the reporter. Furthermore, it allows comparison of the relative fluorescence of different aggregates and can be potentially used to determine the size of fluorescently labeled aggregates on the basis of their forward scattering properties.
Flow cytometry has been largely applied to the study of aggregates formed by polyglutamine-containing Huntingtin proteins in cultured mammalian cells. Mitsui et al.,Citation14 utilized cell disruption followed by purification of the released aggregates by FACS sorting. The sorted aggregates were then further purified by several biochemical methods and analyzed by mass spectrometry.Citation14 A more recent method, termed PulSA (pulse-shape analysis), utilizes the large reduction in the detected pulse width and an increase in pulse height associated with fluorescence within aggregates compared to diffuse cellular fluorescence, to detect Huntingtin polyglutamine aggregates in intact cells.Citation15,16 However, we have not found it applicable to yeast cells, possibly due to the small size of the cells and their aggregates. Likewise, it is not clear whether the detection threshold of the PulSA system is low enough to detect smaller aggregates in mammalian cells.
Several observations support our assumption that the particles detected in cell lysates by flow cytometry represent genuine protein aggregates, rather than cell debris or organelles. Firstly, heat shock at 40°C resulted in an increase in the number of aggregates containing the stress-induced chaperones Hsp42 and Hsp104 ( and ). Secondly, the particles were resistant to mild non-ionic detergents such as NP-40 (0.1%) and TritonX-100 (1%), but were eliminated by high concentrations of Urea (), an effective protein-disaggregating agent. Thirdly, cells expressing high levels of soluble GFP-labeled proteins, such as Ura-GFP in WT background or GFP-DegAB in the background of doa10Δ,Citation9 show negligible levels of fluorescently labeled particles under normal conditions, as the soluble reporters are largely undetected by flow cytometry ( and ).
The efficient release and preservation of aggregates during cell lysis is a critical factor in the present procedure. In order to find the optimal method of aggregate release from cells we compared 3 different methods of cell disruption, using ssa1/2Δ cells expressing aggregated GFP-DegAB: (i) shearing in a high-pressure microfluidizer (ii) partial cell wall digestion with zymolyase, followed by 2 min vortexing with glass beads and addition of 1% Triton X-100, or (iii) vigorous agitation with glass beads in a bead beater. The yields, in terms of GFP-containing aggregates/μg protein, were 25,350 with microfluidization, 5610 with zymolase/glass bead vortexing and 7559 with glass bead beating. However, method (i) is not suitable for analysis of more than a limited number of samples and requires relatively large number of cells, while method (ii) is lengthy and not easily reproducible due to differences in cell wall digestion kinetics between different cell types. In contrast, method (iii) has the advantages of convenience and reproducibility and it can easily become adapted to high throughput analysis. Therefore, we decided to focus on this method and further optimize it for the purposes of the present assay.
Of particular concern in cell lysis by physical disruption is fragmentation of aggregates due to excessive shearing force exerted by glass bead agitation. In preliminary experiments, we sought for disruption conditions that produce significant cell lysis while generating sufficient aggregate numbers for detection by flow cytometry. We found 2 factors in the lysis procedure that profoundly influence the yield of detected aggregates: test tube size and buffer. Using sample volumes of 0.51 ml (including the glass beads), aggregates were detectable only when bead-beating was carried out in 0.5 ml microcentrifuge tubes. No aggregates were detected when 1.5 or 2.0 ml tubes were used, even after shortening the duration of bead beating to 15 sec. We conclude that in order to prevent aggregate destruction the displacement distance of the beads must be minimal. We compared the effect of several lysis buffers on aggregate yield. Low-ionic strength buffers or removal of divalent cations with EDTA resulted in significant depletion of aggregates relative to buffers with high salt and divalent cation concentration (100 mM KCl, 5 mM Mg2+). Therefore, we adopted the latter, which have the additional advantages of approximating the intracellular ionic environment and favoring preservation of nuclei. Finally, aggregate loss during cell lysis was mitigated by restricting the duration of bead-beating to a total of 1 min, compared to several minutes in standard protocols. Under these conditions, > 80% of the cells were rendered permeable to propidium iodide (not shown), indicative of cell wall rupture. In spite of using a mild lysis procedure aimed at maximizing aggregate preservation, we cannot rule out the possibility that some fragmentation of aggregates did occur, which could lead to either under- or overestimation, of aggregate numbers. While this should not affect comparisons among cells harboring aggregates with similar biochemical properties, it should be taken into account in cases where different aggregate types are evaluated.
A potential drawback of any assay involving cell disruption is the possibility of relocation and intermixing of cellular components, which, in the present case, could lead to adhesion or loss of fluorescent reporter proteins to or from aggregates. One means of testing for intermixing of aggregate components is illustrated in , using cells individually labeled by 2 different reporters (i.e. fusion proteins with GFP and mCherry) and comparing them to cells co-expressing the 2 reporters. Mixtures of cells expressing the aggregate-associated chaperone Hsp42-mCherryCitation6 with cells expressing the misfolded model proteins ΔssCPY*-GFP or GFP-DegAB, followed by lysis of the mixture, showed negligible intermixing of fluorescence, as indicated by the low numbers of double-labeled aggregates. In contrast, lysis of mixture of cells expressing Hsp42-mCherry with lysates from cells expressing Hsp104-GFP showed significant co-localization of the 2 proteins in doubly-labeled aggregates, although these levels were significantly lower than in co-expressing cells. This is attributable to spontaneous adhesion of Hsp-104-GFP to Hsp42-mCherry containing aggregates in the mixtures of lysates (, right panel). While such behavior may be expected from Hsp104, which functions as a general chaperone/ disaggregase that shuttles among aggregates,Citation7,17,18 we assume it is less likely to occur with most other proteins, as we observed only negligible spontaneous adhesion between Hsp42-mCherry and Δss-CPY*-GFP or DegAB-GFP. The negligible spontaneous adhesion allowed us to evaluate the co-localization of the sequestration factor Hsp42-mCherry and model misfolded proteins (either Δss-CPY*-GFP or DegAB-GFP), in aggregates released from cells co-expressing the 2 reporters. Thus, this system provides a valuable tool for analysis of aggregate protein composition.
An important potential application of the present assay is the unbiased quantification of the relative cellular aggregate content in terms of the number of fluorescent particles per μg protein of cell lysates. As an illustration, we showed the quantification of aggregates in normal and heat-stressed cells expressing several fluorescently labeled model misfolded proteins and chaperones (). In a similar fashion, the assay can be applied to comparing the effect of cell manipulations on aggregate formation and on the density of reporter molecules within the aggregates. Thus, it may provide a novel alternative approach of aggregate analysis. The current mainstay of aggregate analysis in yeast cells is fluorescent microscopy combined with image analysis, which provides quantitative data in terms of number of particles per given number of cells. However, it may not be sufficiently reliable when applied to cells exhibiting high background fluorescence or small aggregate size or both. In the present method the problems of cellular autofluorescence or background fluorescence due to soluble, unaggregated fluorescent reporters are avoided, since flow cytometry of cell lysates is inherently poorly sensitive to the presence of dissolved fluorophores.
Another feature of the assay is its capacity to provide a measure of the distribution of fluorescent reporter proteins among aggregates in double-labeled cells. As discussed above, protein intermixing may occur during cell disruption, but this problem can be controlled for by analysis of mixtures of singly-labeled cells (). Flow cytometry of lysates from double-labeled cells can readily distinguish and quantify the relative proportions of aggregates containing a single reporter protein vs. those containing both proteins. Furthermore, co-localization of 2 differently labeled proteins in aggregates as observed by the present method indicates a stable physical interaction of the proteins of interest within aggregates. In contrast, co-localization observed by fluorescent microscopy indicates proximity between the 2 proteins, though not necessarily a stable interaction.
A variety of potential applications of this assay can be envisioned. It can be combined with general aggregate-binding dyes such as thioflavin and its analogs to analyze the association of a variety of labeled proteins with aggregates.Citation19 It could be used to monitor the kinetics of aggregate accumulation or elimination in cells under different conditions. Also, the adaptation of the technique to analysis of aggregates in mammalian cells is under development and may prove useful with aggregate types that are not amenable to analysis by PulSA. The requirement for cell lysis limits the assay to medium-throughput applications, however, techniques can be devised for streamlining this step, which should improve its efficiency.
In summary, the method offers a potentially useful alternative to existing modes of aggregate analysis and could eventually facilitate the study of the role of protein aggregates in various misfolding disorders.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
KPRN_A_968445_Table_2_Supplemental.pdf
Download PDF (302.8 KB)KPRN_A_968445_Table_1_Supplemental.pdf
Download PDF (307.5 KB)Acknowledgments
We thank DH Wolf, D Kaganovich, E Craig, M Hochstrasser, and JL Brodsky for strains or plasmids.
Funding
This study was funded by the Niedersachsen-Israel research cooperation program.
References
- Soto C, Estrada LD. PRotein misfolding and neurodegeneration. Arch Neurol 2008; 65:184-9; PMID:18268186; http://dx.doi.org/10.1001/archneurol.2007.56
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med 2004; 10 Suppl:S10-7; http://dx.doi.org/10.1038/nm1066
- Sontag EM, Vonk WI, Frydman J. Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr Opin Cell Biol 2014; 26:139-46; PMID:24463332; http://dx.doi.org/10.1016/j.ceb.2013.12.006
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell 2006; 125:443-51; PMID:16678092; http://dx.doi.org/10.1016/j.cell.2006.04.014
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J 2004; 86:3993-4003; PMID:15189895; http://dx.doi.org/10.1529/biophysj.103.038422
- Specht S, Miller SB, Mogk A, Bukau B. Hsp42 is required for sequestration of protein aggregates into deposition sites in Saccharomyces cerevisiae. J Cell Biol 2011; 195:617-29; PMID:22065637; http://dx.doi.org/10.1083/jcb.201106037
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci USA 2000; 97:1589-94; PMID:10677504; http://dx.doi.org/10.1073/pnas.97.4.1589
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998; 143:1883-98; PMID:9864362; http://dx.doi.org/10.1083/jcb.143.7.1883
- Shiber A, Breuer W, Brandeis M, Ravid T. Ubiquitin conjugation triggers misfolded protein sequestration into quality control foci when Hsp70 chaperone levels are limiting. Mol Biol Cell 2013; 24:2076-87; PMID:23637465; http://dx.doi.org/10.1091/mbc.E13-01-0010
- Park S-H, Bolender N, Eisele F, Kostova Z, Takeuchi J, Coffino P, Wolf DH. The cytoplasmic Hsp70 chaperone machinery subjects misfolded and endoplasmic reticulum import-incompetent proteins to degradation via the ubiquitin-proteasome system. Mol Biol Cell 2007; 18:153-65; PMID:17065559; http://dx.doi.org/10.1091/mbc.E06-04-0338
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008; 454:1088-95; PMID:18756251; http://dx.doi.org/10.1038/nature07195
- Wang Y, Meriin AB, Costello CE, Sherman MY. Characterization of proteins associated with polyglutamine aggregates: a novel approach towards isolation of sggregates from protein conformation disorders. Prion 2007; 1:128-35; PMID:19164926; http://dx.doi.org/10.4161/pri.1.2.4440
- Summers DW, Wolfe KJ, Ren HY, Cyr DM. The type II Hsp40 Sis1 cooperates with Hsp70 and the E3 ligase Ubr1 to promote degradation of terminally misfolded cytosolic protein. PLoS One 2013; 8:e52099; PMID:23341891; http://dx.doi.org/10.1371/journal.pone.0052099
- Mitsui K, Doi H, Nukina N. Proteomics of Polyglutamine aggregates. In: Indu K, Ronald W, eds. Methods in Enzymology. New York: Academic Press, 2006:63-76
- Ramdzan YM, Polling S, Chia CP, Ng IH, Ormsby AR, Croft NP, Purcell AW, Bogoyevitch MA, Ng DC, Gleeson PA, Hatters DM. Tracking protein aggregation and mislocalization in cells with flow cytometry. Nat Methods 2012; 9:467-70; PMID:22426490; http://dx.doi.org/10.1038/nmeth.1930
- Bersuker K, Hipp MS, Calamini B, Morimoto RI, Kopito RR. Heat shock response activation exacerbates inclusion body formation in a cellular model of huntington disease. J Biol Chem 2013; 288:23633-8; PMID:23839939; http://dx.doi.org/10.1074/jbc.C113.481945
- Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol 2012; 179:152-60; PMID:22580344; http://dx.doi.org/10.1016/j.jsb.2012.05.002
- Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 1995; 268:880-4
- Ban T, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of amyloid fibril growth monitored by thioflavin T fluorescence. J Biol Chem 2003; 278:16462-5; PMID:12646572; http://dx.doi.org/10.1074/jbc.C300049200