
Abstract
We report a Japanese patient with Creutzfeldt-Jakob disease (CJD) with a V203I homozygous mutation of the prion protein gene (PRNP). A 73-year-old woman developed rapidly progressive gait disturbance and cognitive dysfunction. Four months after the onset, she entered a state of an akinetic mutism. Gene analysis revealed a homozygous V203I mutation in the PRNP. Familial CJD with a V203I mutation is rare, and all previously reported cases had a heterozygous mutation showing manifestations similar to those of typical sporadic CJD. Although genetic prion diseases with homozygous PRNP mutations often present with an earlier onset and more rapid clinical course than those with heterozygous mutations, no difference was found in clinical phenotype between our homozygous case and reported heterozygous cases.
Abbreviations
| CJD | = | Creutzfeldt-Jakob disease |
| sCJD | = | sporadic CJD |
| fCJD | = | familial CJD |
| PRNP | = | prion protein gene |
| PrP | = | prion protein |
| PrPSc | = | scrapie prion protein |
| CSF | = | cerebrospinal fluid |
| EEG | = | electroencephalography |
| MRI | = | magnetic resonance imaging |
Introduction
Creutzfeldt-Jakob disease (CJD) is a disease of fatal neurodegenerative conditions pathologically characterized by accumulation of the abnormal prion protein (PrPSc) in the central nervous system. Approximately 85–90 % of CJD cases are sporadic (sCJD) lacking any mutations in the prion protein gene (PRNP), while about 10–15% of the disorders are inherited.Citation1 Familial CJD (fCJD) is associated with at least 20 distinct genetic mutations that are all transmitted as autosomal dominant traits, including point, deletion and insertion mutations.Citation2 Most of the mutations that cause fCJD, are heterozygous. Pathomechanisms of homozygous mutations in the PRNP remain unclear due to the extreme rarity of the disorder. We describe a Japanese fCJD patient with a V203I homozygous point mutation in the PRNP.
Case Presentation
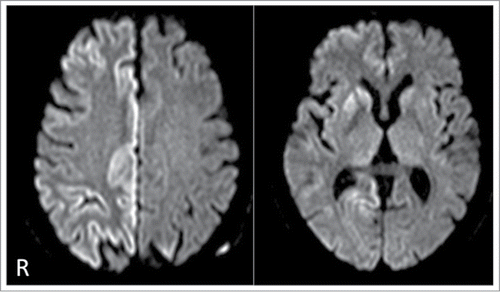
A 73-year-old Japanese woman developed gait disturbance and rapidly progressive cognitive dysfunction. She had no family history of the prion diseases or other neurological disorders; however, her parents were first cousins. Three months after the onset, she became bedridden state. On admission to our hospital, neurological examinations revealed severe cognitive impairment and left-sided hemiparesis. Hyperreflexia with positive plantar reflex was evident in the left extremities. No myoclonus or extrapyramidal signs were obvious. No visual disturbance or cerebellar signs were apparent. Routine hematological examinations and blood chemistry were unremarkable. Cerebrospinal fluid (CSF) study revealed an elevation of tau protein (22,528 pg/ml; normal range, <202 pg/ml) and 14–3–3 protein (5,428 μg/ml; normal range, <500 μg/ml); however, there was neither pleocytosis nor an elevation of protein levels. There was no evidence of infectious diseases in the culture from the CSF. Electroencephalography (EEG) revealed diffuse slowing of the waves without apparent periodic synchronous activities. Diffusion weighted brain magnetic resonance imaging (MRI) showed hyperintensity in the right basal ganglia and the right frontal, parietal, and occipital lobes (). Single photon emission computed tomography images using 99mTc-Ethylcysteinate dimer showed mild hypoperfusion in the right frontal lobe and right parietal lobes which were almost consistent with the areas of hyperintensity on the MRI. Four months after the onset, she entered a state of akinetic mutism state accompanied by frequent myoclonus; moreover, periodic synchronous discharges appeared on the EEG.
Figure 1. Diffusion-weighted magnetic resonance images showing increased signal intensity in the right basal ganglia and the right frontal, parietal, and occipital cortices.
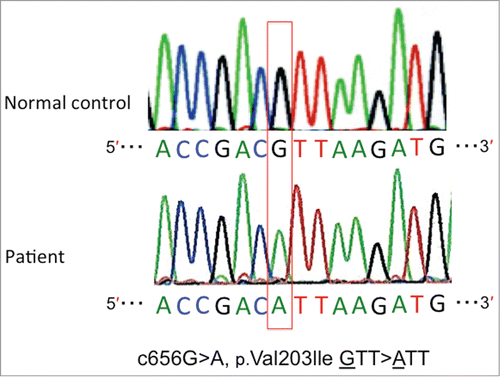
A genomic study of the PRNP approved by the Ethics Committee for Human Genome/Gene Analysis Research at Kanazawa University Graduate School of Medical Sciences was performed after obtaining an informed consent from the family. The gene analysis revealed a homozygous base substitution: c.656G>A (p.V203I) (). The polymorphisms of the PRNP showed methionine homozygosity at codon 129 and glutamine homozygosity at codon 219. The V203I mutation was not observed in any of the 100 healthy Japanese control subjects. We thus made a diagnosis of fCJD with a V203I homozygous mutation. The patient died 24 months after the onset. An autopsy was not performed.
Figure 2. Sequence chromatogram of the homozygous c.656G>A (p.V203I) mutation in the prion protein gene (PRNP).
Discussion
In this report, we have described a Japanese fCJD patient with a V203I homozygous mutation in the PRNP. Although no histopathological examinations were performed, the clinical course to akinetic mutism, findings of the brain MRI and EEG, and elevation of tau and 14–3–3 proteins in the CSF were consistent with those typical of CJD patients.Citation3 The genotype frequency of the V203I mutation in the genetic prion diseases has been reported as 0.9% in the Japanese (2/216 cases)Citation4 and 1.2% in Europeans (5/425 cases).Citation5
There have been 3 case reports of fCJD associated with the V203I heterozygous mutation so far;Citation6-8 however, there have been no reports of fCJD with the V203I homozygous mutation. The clinical characteristics and laboratory features of 3 reported cases and our patient are summarized in . All the reported cases with this mutation lacked a family history of the prion diseases.Citation6-8 The clinical manifestations of the patients with fCJD who have a V203I heterozygous mutation are consistent with those of patients with typical sCJD. This includes rapidly progressive dementia, ataxia, tremor, and myoclonus.Citation6–8 All patients died within 2 months of onset. An elevation of 14–3–3 protein in the CSF was seen in all the reported cases. Two out of the 3 patients showed typical periodic sharp waves on the EEG. Brain MRI also showed gyriform hyper intensity in the cerebral cortex in T2-weighted and diffusion-weighted images.Citation6 As regards of polymorphisms at codon 129 in the PRNP, one patient presented both methionine and valine heterozygosity, while, 2 patients were methionine homozygous only. Histopathological investigation was performed in only one case (the Korean female).Citation6 This patient showed astrogliosis, vacuolization with spongiform changes and large vacuoles, neuronal loss and PrPSc deposition in the cerebrum, basal ganglia and cerebellum;Citation6 Western blot analysis of proteinase K-treated brain samples of the case showed a typical PrP type 1 pattern in addition to a band at 17 kDa.Citation6
Table 1 Clinical and investigational features of patients with the V203I mutation in PRNP
Our patient presented similar neurological and laboratory findings to those of patients with typical sCJD and fCJD with a V203I heterozygous mutation.Citation6-8 The influence of a homozygous mutation in the PRNP on the disease phenotype is uncertain. The substitution of lysine for glutamate at codon 200 (E200K) is the only mutation which has been reported to be homozygous in some cases.Citation9 E200K homozygous patients showed earlier development of the diseases in comparison to those with a heterozygous E200K mutation; however, there were no distinguishing clinical symptoms between the homozygous and heterozygous patients.Citation9 The lack of wild type prion protein in patients with homozygous mutations in the PRNP might be susceptible to the generation of PrPSc and subsequent propagation of the protein. Our patient presented with a long disease history compared to patients with a V203I heterozygous mutation; Citation6-8 however, the longer survival after akinetic mutism in Japanese patients with CJD could be attributable to careful nutritional and medical support.Citation10 Although the amino acid residue of codon 203 is located in the hydrophobic core of PrP,Citation11 details of the pathomechanisms underlying this point mutation remain unknown. Further comprehensive studies are essential to clarify the influence of this mutation on disease phenotype.
Conclusion
We report the first case of fCJD with a V203I homozygous mutation in the PRNP. The onset age and neurological and laboratory findings were similar to those reported in patients with V203I heterozygous mutation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Dr. Katsuya Sato at Nagasaki University for providing assistance with the cerebrospinal fluid examination, and Ms. Yukari Yamaguchi at Kanazawa University for providing technical assistance.
Funding
This work was supported by a grant-in-aid from the Research Committee of Prion Disease and Slow Virus Infection, the Ministry of Health, Labor and Welfare of Japan, and from the Research Committee of Surveillance and Infection Control of Prion Disease, the Ministry of Health, Labor and Welfare of Japan.
References
- Bechtel K, Geschwind MD. Ethics in prion disease. Prog Neurobiol 2013; [Epub ahead of print] doi: 10.1016/j.pneurobio.2013.07.001.; PMID:23906487
- Parchi P, Gambetti P, Capellari S. Neurodegeneration 2nd ed. Oxford: Wiley-Blackwell; 2011. Chapter 33, Genetic Creutzfeldt–Jakob disease; 336-45
- Budka H, Mark W, James W, Gambetti P, Parchi P, Tagliavini F. Neurodegeneration 2nd ed. Oxford: Wiley-Blackwell; 2011. Chapter 32, Sporadic Creutzfeldt–Jakob disease; 322-35.
- Nozaki I, Hamaguchi T, Sanjo N, Noguchi-Shinohara M, Sakai K, Nakamura Y, Sato T, Kitamoto T, Mizusawa H, Moriwaka F et al. Prospective 10-year surveillance of human prion disease in Japan. Brain 2010; 133:3043-57; PMID:20855418; http://dx.doi.org/10.1093/brain/awq216
- Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, Collins SJ, Boyd A, Giulivi A, Coulthart M, et al. Genetic prion disease: the EUROCJD experience. Hum Genet 2005; 118:166-74; http://dx.doi.org/10.1007/s00439-005-0020-1
- Jeong BH, Jeon YC, Lee YJ, Cho HJ, Park SJ, Chung DI, Kim J, Kim SH, Kim HT, Choi EK, et al. Creutzfeldt-Jakob disease with the V203I mutation and M129V polymorphism of the prion protein gene (PRNP) and a 17 kDa prion protein fragment. Neuropathol and Appl Neurobiol 2010; 36:558-63; http://dx.doi.org/10.1111/j.1365-2990.2010.01094.x
- Peoc’h K, Manivet P, Beaudry P, Attane F, Besson G, Hannequin D, Delasnerie-Lauprêtre N, Laplanche JL. Identification of Three Novel Mutations (E196K, V203I, E211Q) in the Prion Protein Gene (PRNP) in inherited prion diseases with Creutzfeldt-Jakob disease phenotype. Hum Mutat 2000; 15:482; http://dx.doi.org/10.1002/(SICI)1098-1004(200005)15:5%3c482::AID-HUMU16%3e3.0.CO;2-1
- Shi Q, Chen C, Wang XJ, Zhou W, Wang JC, Zhang BY, Gao C, Gao C, Han J, Dong XP. Rare V203I mutation in the PRNP gene of a Chinese patient with Creutzfeldt-Jakob disease. Prion 2013; 7:259-62; PMID:23764840; http://dx.doi.org/10.4161/pri.24674
- Simon ES, Kahana E, Chapman J, Treves TA, Gabizon R, Rosenmann H, Zilber N, Korczyn AD. Creutzfeldt-Jakob disease profile in patients homozygous for the PRNP E200K mutation. Ann Neurol 2000; 47:257-60; PMID:10665501; http://dx.doi.org/10.1002/1531-8249(200002)47:2%3c257::AID-ANA20%3e3.0.CO;2-U
- Iwasaki Y, Mori K, Ito M. Investigation of the clinical course and treatment of prion disease patients in the akinetic mutism state in Japan. Rinsho Shinkeigaku 2012; 52:314-9; PMID:22688110; http://dx.doi.org/10.5692/clinicalneurol.52.314
- Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wüthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci U S A 1998; 95:11667-72; PMID:9751723; http://dx.doi.org/10.1073/pnas.95.20.11667