
Abstract
Previous studies have demonstrated that Shadoo (Sho), a GPI-linked glycoprotein encoded by the Sprn gene with a membrane localization similar to PrPC, is reduced in the brains of rodents with terminal prion disease. To determine the functional significance of Sho in prion disease pathogenesis, Sho-deficient mice were generated by gene targeting. Sho knockout and control wild-type (WT) mice were infected with themouse-adapted scrapie strains 22L or RML. No significant differences in survival, the incubation period of prion disease or other disease features were observed between Sho mutant and WT mice. In this model of prion disease, Sho removal had no effect on disease pathogenesis.
Keywords:
Introduction
Prion diseases are transmissible and lead to fatal neurodegeneration. They occur in humans as Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker disease, fatal familial insomnia and kuru, and in other mammals such as sheep (scrapie) and cattle (bovine spongiform encephalopathy).Citation1 The presumed infectious agent is an improperly folded form of a host-encoded protein, the cellular prion protein (PrPC). Conversion of PrPC into the disease-associated isoform, PrPSc, is thought to be the primary pathogenic event, although the mechanism by which PrPSc causes disease is poorly understood.Citation2 There has been speculation that a host chaperone protein, protein X, is involved in prion disease pathogenesis.Citation3,4 Although PrPC is known to interact with or localize in close proximity to numerous other proteins in the cell membrane,Citation5,6 none of these proteins have been shown to be associated with prion disease pathogenesis.
The mammalian prion protein family consists of 3 members: PrPC, the testis-specific Doppel (Dpl),Citation7 and Shadoo (Sho), a neuronal paralog of PrPC that is encoded by the Sprn gene.Citation6,8 Previous work has shown overlapping expression patterns of Sho and PrPC in the central nervous system; like PrPC, Sho transgenes can counter the neurotoxic effects of Dpl, N-terminal truncated PrPC and reduced levels of Sho in experimental models of prion infection.Citation2 Sho is thus a likely factor influencing prion disease pathogenesis and can indicate the activation of a pathway that includes protease-resistant PrPSc. We previously showed that overexpression of Sho protein in transgenic mice does not affect the pathogenesis of scrapie.Citation9-11 To investigate the relationship between Sho and PrPC and a potential role for Sho in the pathogenesis of prion disease, in this study, a Sho-deficient mouse line was generated, and mutants and wild-type (WT) control mice were inoculated intracerebrally with either the 22L or Rocky Mountain Laboratory (RML) strain of scrapie. The results demonstrate that there is no correlation between Sho expression and the incubation period and progression of scrapie.
Results
Targeting of the Sprn gene
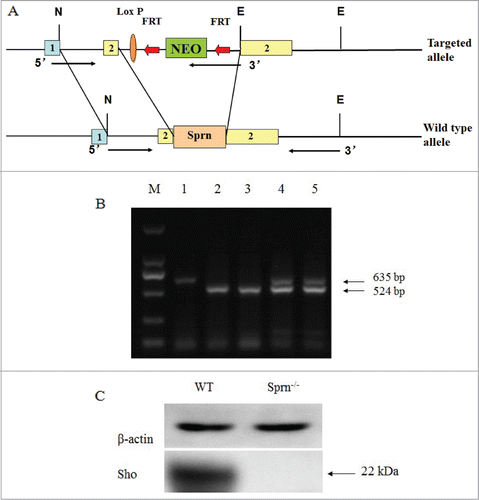
A Sho-deficient mutant mouse line was generated using knockout strategy. A fragment of exon 2 (11,002–11,447 bp; gene ID 45733), representing more than 88% of the Sho coding sequence, was replaced by a neomycin resistance cassette, resulting in the excision of Sprn (). Western blot and PCR genotyping of offspring from the inter-breeding of mice heterozygous for the Sprn deletion () showed that WT, heterozygous KO, and homozygous KO animals were present in the expected 1:2:1 Mendelian ratio.
Figure 1. Generation and characterization of Sprn−/− mice. (A) Sprn knockout strategy. A region in exon 2 was replaced by a neomycin resistance cassette in the targeting vector, resulting in the deletion of 88% of the Sho coding sequence. N, NotI; E, EcoRI; NEO, neomycin; 1, exon1; 2, exon2; 1-2, intron. (B) PCR genotyping of Sprn−/− mutants. Primers for the neomycin resistance cassette amplified a 524 bp band, while a 635 bp band was amplified by primers recognizing the Sho coding sequence. Lane 1, Sprn+/+; lanes 2 and 3, Sprn−/−; lanes 4 and 5, Sprn+/−. (C) Western blot of mouse brain homogenates, with β-actin used as a loading control. The 22 kDa band representing the Sho protein is absent in Sprn−/− animals.
Viability of Sprn−/− mice
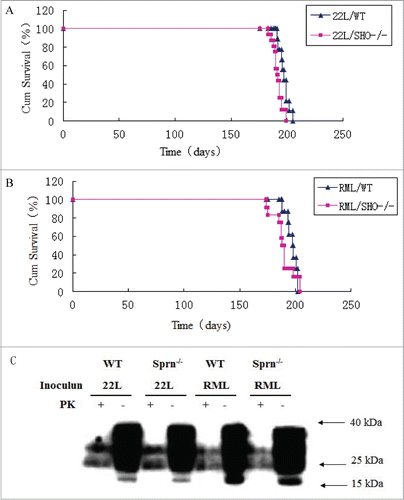
The survival of Sprn−/− mice to adulthood allowed the activity of endogenous Sho in response to a challenge by prions to be examined. Sprn−/−and WT mice were inoculated with 22L () or RML () prions by intracerebral injection. Using a log-rank test, the F-values for one-way ANOVA were 3.28 for 22L and 2.32 for RML, while mean differences were not statistically significant (P-values of 0.083 and 0.145, respectively). Protease-resistant PrPSc species in brain homogenates were not obviously different in mutant and WT animals infected with either prion (). Thus, the absence of Sho does not significantly alter the temporal and clinical manifestation of infections by 22L and RML prion isolates.
Figure 2. Comparable viability and resistance to prion infection of Sprn−/− and WT mice.(A) Survival curves of Sprn−/− and WT mice inoculated with 22L prions. The mean difference was not statistically significant (P = 0.083). (B) Survival curves of Sprn−/− and WT mice inoculated with RML prions. The mean difference was not statistically significant (P = 0.145). (C) PrP and PK-resistant PrPSc levels after infection with 22L or RML prions were similar in Sprn−/− and WT mice, as determined by protein gel blotting.
Comparative brain morphology of Sprn−/− and wild-type mice
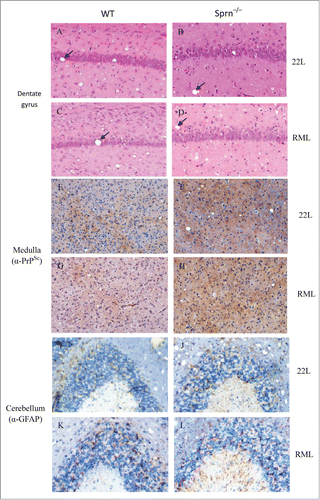
Neuropathological signs of prion disease, including spongiform degeneration and astrogliosis, were similar in the dentate gyrus of prion-infected Sprn−/− and WT mice, irrespective of the prion strain used (). Histopathological changes observed in the brains of prion-infected Sprn−/− and WT mice included vacuolization and neuron and neurite degeneration. In contrast to a recent report,Citation12 PrPSc deposition in the medulla was significantly higher in mutants than in WT (). There were slight differences in the distribution of astrogliosis between mutants and WT, as evidenced by the expression of the astrocyte marker GFAP. In WT, strong GFAP staining was mostly observed in the granular layer, while both molecular and granular layers were enriched in GFAP in Sprn–/– mice ().
Figure 3. Histopathology of scrapie-infected WT and Sprn–/– mice. (A–D) Hematoxylin and eosin staining of dentate gyrus in WT and Sprn–/– mice infected with scrapie and examined at terminal illness. Changes associated with prion disease, including spongiform degeneration (black arrows in panels A–D). (E–H) Immunohistochemical analysis of spongiform degeneration in the medulla in 22L- or RML-infected WT and Sprn–/– mice, as detected using an antibody against PrP. PrPSc deposition was observed in all samples, but was higher in mutants than in WT for RML. (I–L) Immunohistochemical detection of astrogliosis in the cerebellum of 22L- or RML-infected WT and Sprn–/–mice, using an antibody against GFAP. Astrogliosis was confined to the granular layer in WT mice, but was observed in both the molecular and granular layers in mutants.
Discussion
A previous study showed a down-regulation of Sho protein in prion infection which was inversely related to PrPSc accumulation, suggesting that Sho has a neuroprotective function,Citation11 and it was hypothesized that the decrease in Sho expression could contribute to the clinical manifestations of prior disease.Citation11 In this report, we outline the generation and characterization of a novel Sho-deficient mouse line and examined the putative neuroprotective function of Sho by subjecting mutant and WT mice to prion infection. Sprn, the gene encoding Sho, is conserved in mammals.Citation8 Based on its similarities to PrPC, which is implicated in prion disease pathogenesis, we hypothesized that Sho also plays an important role in prion diseases. This was suggested by the fact that 2 patients with early-onset variant Creutzfeldt-Jakob disease were found to harbor inactive Sprn alleles.Citation13 From the present experiments, it was determined that Sho-deficient mice show a disease susceptibility and time course of disease progression after prion infection that are similar to WT mice, irrespective of the strain used for inoculation (). Moreover, western blots detected similar levels of PrPC and PrPSc in terminally ill Sho-deficient mice and their WT counterparts (). These findings argue against a major role for Sho in the pathogenesis of prion diseases,Citation11,14 and are in agreement with previous results obtained in a different Sho knockout line infected with RML.Citation15
PrPSc accumulation and astrogliosis are markers for prion diseases. Hematoxylin and eosin-stained brain samples from terminally ill Sprn–/– mice and WT controls after inoculation with 22L or RML strain prions () showed similar vacuoles in the mutant and WT mice. However, the deposition of PrPSc was higher in the medulla of RML-infected Sprn–/– mice as compared to WT mice (), although the overall PrPSc level in brain homogenates was not obviously different between the 2 genotypes (). This suggests that Sho protein deficiency enhances the accumulation of PrPSc in the medulla, but that this increase may be masked when the whole brain is considered. In rodent models of prion disease, the levels of Sho are reduced in infected tissues;Citation2,10,11 however, it was found that Sho protein deficiency has no effect on the expression of PrPSc. GFAP was expressed in the cerebellum of 22L- or RML-infected mutant and WT mice; however, in WT mice the expression was mainly in granular layer (), while in Sprn–/– mice, strong GFAP immunoreactivity was observed in the granular and molecular layers (). These observations suggest that Sho deficiency affects the manifestation of astrogliosis in a spatially defined manner.
In summary, these results do not provide any evidence for a neuroprotective role of Sho in prion disease pathogenesis, since there was no difference between scrapie-infected Sprn–/–and WT mice in terms of survival rate and disease progression. These negative results could have resulted from a compensatory effect by other proteins, including PrPC, and thus a neuroprotective role for Sho cannot be completely dismissed. However, these findings demonstrate that Sho is not a major modulator of PrPSc accumulation and prion disease pathogenesis.
Materials and Methods
Generation of Sho-deficient mice
The Sho knockout targeting vector was designed to disrupt the expression of Sho via insertion of a neomycin resistance gene into the Sho open reading frame of the Sprn gene. The targeting vector was constructed from a genomic DNA fragment derived from a C57BL/6 genomic bacterial artificial chromosome clone and included the left (3266 bp) and right (3557 bp) arms encompassing an intron and the 3' region of the Sho open reading frame, respectively, which was cloned into the pBR322-MK-MCS vector. The vector was linearized and electroporated into129SV/EV mouse embryonic stem cells (SCR012). Positive clones were selected with G418 and ganciclovir, and verified by PCR analysis. Correctly targeted clones were microinjected into C57BL/6J mouse blastocysts to produce chimeras that were then crossed into a C57 BL/6 J genetic background. The offspring were screened by protein gel blotting and PCR analysis. Sho knockout mice were generated by the Shanghai Nanfang Research Center for Model Organisms (Shanghai, China).
Scrapie inoculation
Sho knockout mice and WT C57BL/6 control mice (3–4 weeks of age; 6–8 animals per group) were injected intracerebrally with 25 μl of 0.1% mouse-adapted scrapie (22L-infected brain homogenate diluted in phosphate-buffered saline [PBS], or RML prion inoculum prepared from brains of terminally ill BALB/c mice). The inoculated mice were housed in a facility for infected animals and provided with water and standard mouse chow ad libitum. A diagnosis of scrapie was made by the appearance of typical features such as ataxia, hunched posture, rigid tail, and severe weight loss. Terminal disease was independently confirmed by an additional researcher before the mice were euthanized by CO2 asphyxiation. Half of the brain was immediately dissected and frozen, while the other half was immersion-fixed in 10% neutral-buffered formalin. Tissue blocks were embedded in paraffin blocks and cut into 5-μm-thick sections that were stained with hematoxyline and eosin or periodic acid-Schiff reagent.
All procedures were approved by an animal ethics committee operating under the China Code of Practice for the Care and Use of Animals for Scientific Purposes.
Western blot analysis
Mouse brains were homogenized in lysis buffer containing 150 mM NaCl, 0.5% NP-40, 0.5% deoxycholate, and 50 mM Tris-HCl (pH 7.5). For the detection of protease-resistant PrPSc, brain homogenates were digested with proteinase K (PK; Roche, Mannheim, Germany) at a final concentration of 18.6 μg/ml for 60 min at 37°C. Digestion reactions were terminated by the addition of 2 mM phenylmethylsulfonyl fluoride. Untreated and PK-treated brain homogenates were boiled in 6× Laemmli buffer for 5 min, resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride membranes (GE Healthcare, Little Chalfont, UK). Antiserum to Sho was produced by immunizing rabbits with peptide containing amino acid residues 86–100 of mouse Sho (Bioss, Beijing, China). Following a blocking reaction, the membrane was first incubated with the primary anti-Sho antibody (1:2000), followed by HRP-conjugated secondary antibody with Renaissance chemiluminescent reagent (Amersham Life Science, UK) according to manufacturer's recommendations. For western blotting, monoclonal antibodies against PrP (BAR221; Cayman Chemical, Ann Arbor, MI, USA), and β-actin (Boster Biological Technology Co., Ltd., Wuhan, China) were applied.
Quantitation of band intensities in Western blots was performed using Image J software (NIH, Bethesda, USA).
Histology and immunohistochemistry
Brains were fixed in 4% paraformaldehyde at 4°C for 48 h, embedded in paraffin blocks, and cut with a microtome (Leica, Germany) into 5-μm sections that were mounted onto silanized glass slides.Citation2 Sections were deparaffinized in xylene, immersed in 100% ethanol, rehydrated through a graded ethanol series and rinsed in PBS. After deparaffinization, sections were either stained with hematoxylin and eosin for morphological evaluation or processed for immunohistochemistry as follows. Sections were subjected to antigen retrieval by autoclaving at >95°C for 20 min in citric acid buffer (pH 6.0), followed by incubation in concentrated formic acid and guanidine isothiocyanate for 1 min. Endogenous peroxidase activity was quenched by incubation with 1% H2O2 for 10 min. The slides were washed with PBS (pH 7.2) and incubated with primary antibodies BAR221 or anti-GFAP (Beyotime, Haimen, China) in PBS at 37ºC for 20 min. After PBS rinses, immunoreactivity was visualized by reaction with DAB, after which the tissue sections were counterstained with Harris's hematoxylin. Slides were viewed with an LX71 light microscope (Olympus, Tokyo, Japan).Citation12
Statistical analysis
Data was analyzed with SSPS Statistics 17.0 software by performing by one-way ANOVA. Differences in mean survival times between different groups of animals were evaluated with the log-rank test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Natural Science Foundation of China (no. 31072148, 31201714, 31101782 and 31201937) and National Key Technologies R & D Program (no. 2013BAD12B04).
References
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Ann Rev Neurosci 2001; 24:519-50; PMID:11283320; http://dx.doi.org/10.1146/annurev.neuro.24.1.519
- Watts JC, Drisaldi B, Ng V, Yang J, Strome B, Horne P, Sy MS, Yoong L, Young R, Mastrangelo P, et al. The CNS glycoprotein Shadoo has PrPC-like protective properties and displays reduced levels in prion infections. EMBO J 2007; 26:4038-50; PMID:17703189; http://dx.doi.org/10.1038/sj.emboj.7601830
- Fasano C, Campana V, Zurzolo C. Prions: protein only or something more? Overview of potential prion cofactors. J Mol Neurosci 2006; 29:195-214; PMID:17085779; http://dx.doi.org/10.1385/JMN:29:3:195
- Kaneko K, Zulianello L, Scott M, Cooper CM, Wallace AC, James TL, Cohen FE, Prusiner SB. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A 1997; 94:10069-74; PMID:9294164; http://dx.doi.org/10.1073/pnas.94.19.10069
- Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA. Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol 2004; 22:724-31; PMID:15146195; http://dx.doi.org/10.1038/nbt969
- Watts JC, Huo H, Bai Y, Ehsani S, Jeon AH, Shi T, Daude N, Lau A, Young R, Xu L, et al. Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 2009; 5:e1000608; PMID:19798432; http://dx.doi.org/10.1371/journal.ppat.1000608
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, et al. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 1999; 292:797-817; PMID:10525406; http://dx.doi.org/10.1006/jmbi.1999.3108
- Premzl M, Sangiorgio L, Strumbo B, Marshall Graves JA, Simonic T, Gready JE. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 2003; 314:89-102; PMID:14527721; http://dx.doi.org/10.1016/S0378-1119(03)00707-8
- Wang H, Wan J, Wang W, Wang D, Li S, Liao P, Hao Z, Wu S, Xu J, Li N, et al. Overexpression of Shadoo protein in transgenic mice does not impact the pathogenesis of scrapie. Neurosci Lett 2011; 496:1-4; PMID:21458534; http://dx.doi.org/10.1016/j.neulet.2011.03.073
- Watts JC, Stöhr J, Bhardwaj S, Wille H, Oehler A, Dearmond SJ, Giles K, Prusiner SB. Protease-resistant prions selectively decrease Shadoo protein. PLoS Pathog 2011; 7:e1002382; PMID:22163178; http://dx.doi.org/10.1371/journal.ppat.1002382
- Westaway D, Genovesi S, Daude N, Brown R, Lau A, Lee I, Mays CE, Coomaraswamy J, Canine B, Pitstick R, et al. Downregulation of Shadoo in prion infections traces a pre-clinical event inversely related to PrPSc accumulation. PLoS Pathog 2011; 7:e1002391; PMID:22114562; http://dx.doi.org/10.1371/journal.ppat.1002391
- Vidal E, Tortosa R, Márquez M, Serafin A, Hidalgo J, Pumarola M. Infection of metallothionein 1+2 knockout mice with Rocky Mountain Laboratory scrapie. Brain Res 2008; 1196:140-50; PMID:18221736; http://dx.doi.org/10.1016/j.brainres.2007.12.034
- Beck JA, Campbell TA, Adamson G, Poulter M, Uphill JB, Molou E, Collinge J, Mead S. Association of a null allele of SPRN with variant Creutzfeldt-Jakob disease. J Med Genet 2008; 45:813-7; PMID:18805828; http://dx.doi.org/10.1136/jmg.2008.061804
- Gurgul A, Polak MP, Larska M, Słota E. PRNP and SPRN genes polymorphism in atypical bovine spongiform encephalopathy cases diagnosed in Polish cattle. J Appl Genet 2012; 53:337-42; PMID:22723200; http://dx.doi.org/10.1007/s13353-012-0102-4
- Daude N, Wohlgemuth S, Brown R, Pitstick R, Gapeshina H, Yang J, Carlson GA, Westaway D. Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrPC-deficiency. Proc Natl Acad Sci U S A 2012; 109:9035-40; PMID:22619325; http://dx.doi.org/10.1073/pnas.1202130109