
Abstract
Enteropathogenic Escherichia coli (EPEC) binding to human intestinal cells triggers the formation of disease-associated actin rich structures called pedestals. The latter process requires the delivery, via a Type 3 secretion system, of the translocated Intimin receptor (Tir) protein into the host plasma membrane where binding of a host kinase-modified form to the bacterial surface protein Intimin triggers pedestal formation. Tir-Intimin interaction recruits the Nck adaptor to a Tir tyrosine phosphorylated residue where it activates neural Wiskott-Aldrich syndrome protein (N-WASP); initiating the major pathway to actin polymerization mediated by the actin-related protein (Arp) 2/3 complex. Previous studies with Nck-deficient mouse embryonic fibroblasts (MEFs) identified a key role for Nck in pedestal formation, presumed to reflect a lack of N-WASP activation. Here, we show the defect relates to reduced amounts of Tir within Nck-deficient cells. Indeed, Tir delivery and, thus, pedestal formation defects were much greater for MEFs than HeLa (human epithelial) cells. Crucially, the levels of two other effectors (EspB/EspF) within Nck-deficient MEFs were not reduced unlike that of Map (Mitochondrial associated protein) which, like Tir, requires CesT chaperone function for efficient delivery. Interestingly, drugs blocking various host protein degradation pathways failed to increase Tir cellular levels unlike an inhibitor of deacetylase activity (Trichostatin A; TSA). Treatments with TSA resulted in significant recovery of Tir levels, potentiation of actin polymerization and improvement in bacterial attachment to cells. Our findings have important implications for the current model of Tir-mediated actin polymerization and opens new lines of research in this area.
Abbreviations
| Ab | = | Polyclonal antibody |
| A/E | = | Attaching and effacing |
| BFP | = | Bundle-forming pili |
| Cmp | = | Chloramphenicol |
| CrkII | = | CT10 regulator of kinase |
| CrkL | = | Crk-like |
| Ctr. | = | Control |
| EHEC | = | Enterohaemorrhagic Escherichia coli |
| EPEC | = | Enteropathogenic Escherichia coli |
| Esp | = | EPEC-secreted proteins |
| FBS | = | Fetal bovine serum |
| GBD | = | GTPase-binding domain |
| HDAC | = | Histone deacetylases |
| HeLa | = | Human cervical epithelial cancer cell line |
| IRSp53 | = | Insulin receptor tyrosine kinase substrate p53 |
| IRTKS | = | Insulin receptor tyrosine kinase substrate |
| LEE | = | Locus of enterocyte effacement |
| Map | = | Mitochondrial associated protein |
| MEFs | = | Mouse embryonic fibroblasts |
| MoAb | = | Monoclonal antibody |
| MOI | = | Multiplicity of infection |
| Nck | = | Non-catalytic tyrosine kinase |
| NF-kB | = | Nuclear factor kB |
| Nle | = | Non-LEE effectors |
| NPF | = | Nucleation promoting factor |
| N-WASP | = | Neural Wiskott–Aldrich syndrome protein |
| PRD | = | Proline-rich domain |
| SH3 | = | Src homology 3 |
| Tir | = | Translocated Intimin receptor |
| TNF- α | = | Tumor necrosis factor-α |
| TSA | = | Trichostatin A |
| T3SS | = | Type 3 secretion system |
| WB | = | Western Blot |
| WT | = | Wild type |
Introduction
Numerous pathogens have evolved mechanisms to subvert for their own benefit the cellular complexes that control actin polymerization.Citation1 Among such pathogens, enteropathogenic Escherichia coli (EPEC) is one of the leading causes of infantile diarrhea worldwide, especially in developing countries. EPEC is a non-invasive bacterium that colonizes the intestinal epithelium through the formation of characteristic attaching and effacing (A/E) lesions. These lesions are characterized by a localized loss of epithelium microvilli, close adherence of the bacteria to the host cell membrane and the generation of filamentous actin-rich structures beneath these bacteria called pedestals.Citation2 Although they have been described more than two decades ago, the biological purpose of pedestals is not completely understood. Importantly, the disruption of genes critical for the formation of these structures has been shown to diminish colonization and subsequent disease in humansCitation3 and in experimental animals.Citation4
The capacity to generate actin pedestals depends on the translocation of bacterial effector proteins into host cells via a type 3 secretion system (T3SS). During the first steps of infection, EPEC adheres non-intimately to the host epithelium in discrete microcolonies, whose formation is mediated by the type 4 pili termed bundle-forming pili (BFP) owing to their capacity to laterally aggregate into long braided structures.Citation5 Microcolony formation enhances EPEC attachment to host cells and facilitates the injection of effectors via T3SS.Citation6,7 The attached EPEC delivers the translocated Intimin receptor (Tir), which drives the major pathway responsible for regulating actin polymerization. Other translocated effectors include Mitochondrial associated protein (Map) and EPEC-secreted proteins (Esp) H, F, G, and Z that are encoded within a pathogenicity island termed the locus of enterocyte effacement (LEE).Citation8 Upon injection into the cell cytoplasm, Tir is inserted into the plasma membrane in a hairpin-loop conformation, exposing an extracellular loop which interacts with the bacterial surface protein Intimin.Citation9 This binding facilitates extremely tight attachmentCitation10 and results in the clustering of Tir in the plasma membrane that contributes to the downstream signaling events leading to the formation of actin-rich pedestalsCitation11 in a manner that depends on Tir tyrosine phosphorylation.Citation9
Tir is phosphorylated by various host tyrosine kinasesCitation12,13 at tyrosine 474 (Y474)Citation14 within the C-terminal cytoplasmic domain, thereby recruiting the host cell adaptor proteins non-catalytic tyrosine kinase (Nck) 1 and 2 (collectively referred as Nck). Nck in turns recruits the neural Wiskott–Aldrich syndrome protein (N-WASP),Citation15 a member of the WAS family of proteins that promote actin polymerization by binding and activating the actin related protein (Arp) 2/3 complex.Citation16,17 N-WASP presents a closed inactive conformation mainly due to intramolecular autoinhibitory interactions that involve the C-terminal acidic domain and the GTPase-binding domain (GBD).Citation18,19 N-WASP requires the interaction with other proteins through its GBD or proline-rich domain (PRD) and possibly post-translational modifications to be fully active. Thus, Nck binds directly to the numerous proline motifs in the PRD of N-WASP through its Src homology 3 (SH3) domains and activates N-WASP by destabilizing the inhibitory interactions.Citation20 Although it is not clear whether N-WASP is recruited to Tir via direct binding of Nck to N-WASP or indirectly through another cell host protein, it has been shown that N-WASP is absolutely required for pedestal formation by EPEC, as demonstrated by the lack of pedestals on infected N-WASP-deficient mouse embryonic fibroblasts (MEFs).Citation17
EPEC hijacks many signaling pathways to control N-WASP activity. Thus, a second Tir tyrosine residue Y454, phosphorylated at low efficiency, is involved in the recruitment of the insulin receptor tyrosine kinase substrate p53 (IRSp53).Citation21 Tir also interacts with the scaffold protein IQGAP1 contributing to ongoing actin polymerization that results in the formation of pedestals.Citation22,23 Moreover, EspH promotes the recruitment of N-WASP and the Arp2/3 complex independently of Nck or IRSp53/IRTKSCitation24 and EspF binds to the GBD domain of N-WASP contributing to its activation.Citation25
In addition to tyrosine phosphorylation, Tir is phosphorylated on serine residues S434 and S463 linked to increases in apparent molecular mass and efficient pedestal formation.Citation26 It is thought that these shifts in apparent molecular mass indicate changes in the three-dimensional structure of Tir to enable tyrosine phosphorylation and/or promote Tir insertion into the plasma membrane.Citation26,27
The roles in the control of actin polymerization of other host proteins recruited to pedestals are beginning to be elucidated. Thus, cortactin has been implicated in the Tir-Nck-N-WASP signaling pathway.Citation28,29 Another example is the endocytosis-associated proteins, clathrin, epsin1 and Eps15 which are crucial for pedestal formation.Citation30 Remarkably, the first negative regulator of actin polymerization at pedestals has been recently described. CT10 regulator of kinase (CrkII) and Crk-like (CrkL) are adaptor proteins that inhibit actin polymerization by competing with Nck binding to phosphorylated Y474 of Tir as recently reported by Nieto-Pelegrin et al.Citation31
Nck-deficient MEFs have been widely used for studying EPEC-induced signaling pathways.Citation15,32-34 Using this cell line, Nck was initially found to be indispensable for EPEC pedestal formationCitation15 but later studies reported low levels of pedestal formation.Citation32–34 In this study we describe that Nck-deficient cells present unforeseen decreased levels of the EPEC effector Tir. Our study points toward a more complex role for Nck adaptors in EPEC signaling than previously thought.
Results
The levels of Tir are reduced in EPEC-infected cultured cells depleted for Nck
We previously studied how cortactin contributes to the Tir-Nck-N-WASP major pathway by which EPEC initiates actin polymerization using, among other reagents, Nck-deficient MEFs.Citation29 Unexpectedly, we determined by Western Blot (WB) analysis that the levels of Tir found in EPEC-infected Nck-deficient MEFs were significantly reduced and therefore we aimed to analyze this finding ().
Figure 1. Analysis of Tir homeostasis within cells with reduced expression of Nck adaptor. (A) WT, Nck-deficient cells (Nck -/-) and Nck1 reconstituted cells (R) were infected with EPEC for 3 h or left uninfected as a negative control. Cell monolayers were lysed in imidazole buffer and fractionated by centrifugation. The membrane fraction (pellet) was resuspended in Laemmli sample buffer. One third of the membrane fraction and one fiftieth of the cytoplasmic fraction were analyzed by WB with anti-Tir MoAb, anti-EspF Ab and, as a loading control, with anti-actin MoAb. The genotype of the cells used was corroborated by blotting with anti-Nck MoAb. (B) HeLa cells were treated using siRNA with oligonucleotides against Nck or with a control oligonucleotide (Ctr.) and infected with EPEC for 3 h. Cell monolayers were lysed in 1% Triton X-100 lysis buffer and the soluble supernatants (contain cytoplasmic and membrane fractions) were blotted with anti-Tir Ab. The arrow indicates the fully-modified Tir form. Actin and tubulin were used as loading controls and the levels of Nck were analyzed by blotting with anti-Nck MoAb. The graph represents the quantification of the 3 Tir bands normalized to actin. (C) Quantification of the number of pedestals of a total of 100 HeLa cells treated with siRNAs for Nck (black bar) compared to control oligonucleotide treated cells (Ctr., white bar). Graphs represent mean ± SD. Statistical analysis was performed using the Student´s t-test from 3 independent experiments; **, P˂0.01.
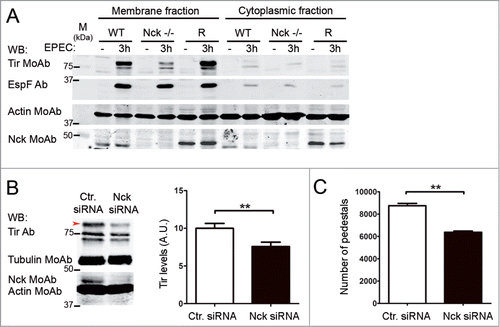
To quantify the reduction we used wild type (WT), Nck-deficient MEFs and those latter MEFs retrovirally reconstituted with eGFP and myc-tagged Nck1 (rescued cells, abbreviated as R). We performed infections with EPEC for 3 hours or left the cells uninfected as a negative control. Then we used a fractionation protocol that enriches in Tir-containing membranes compared to cytoplasmic fraction as detected by WB with a monoclonal antibody (MoAb) against Tir (). Tir appears as a doublet of approximately 78 kDa (unmodified Tir) and 90 kDa (kinase-modified form) as reported.Citation9 Actin was used as a loading control and blotting with anti-Nck MoAb was performed to confirm the genotype of the MEFs used. We found that infected Nck-deficient cells presented significantly reduced Tir levels compared to WT and rescued cells (). We obtained a significant (88 %) reduction of the levels of Tir for infected-Nck-deficient cells compared to WT cells (average of 6 experiments after normalizing for actin; data not shown).
We next wondered whether the reduction would be restricted to the Tir effector or, alternatively, it would also affect other T3SS-secreted EPEC effectors. Therefore, we reprobed the membranes with a polyclonal antibody (Ab) against the EspF effector. In contrast to Tir levels, we found equal amounts of EspF in all cell types analyzed (). To further confirm these results, we analyzed by WB the levels of Tir and EspF in total cell lysates after time-course EPEC infections (data not shown). These results demonstrated that Nck-deficient cells present normal levels of injected EspF protein but significantly reduced levels of Tir.
To illustrate the relevance of our initial observations to human cells, we infected HeLa cells where Nck expression was knock-down with siRNA (Nck siRNA) using commercially available oligonucleotides against human Nck1 and Nck2 and a scrambled sequence nucleotide as a negative control (). shows that 16 hours after transfection with Nck siRNA the levels of Nck were reduced by 85% when cells were infected with EPEC to allow the formation of pedestals. Lysates of EPEC-infected cells were obtained using a Triton X-100-based lysis method to isolate both cytoplasmic and membrane soluble fractions simultaneously (supernatants) while the adherent bacteria fractionate into the Triton X-100 insoluble fraction (pellet).Citation11 WB analysis of the Triton X-100-soluble fraction with an Ab against Tir revealed unmodified and modified species as described.Citation35 The treatment of cells with the Nck siRNA resulted in a statistically significant decrease of Tir levels that was more obvious in the band with slower electrophoretic motility, which represents fully-modified TirCitation35 (, red arrow and graph). Moreover, after quantifying the number of pedestals we determined that pedestals were significantly reduced in cells treated with Nck siRNA (). These results seem to indicate that depletion of human Nck by siRNA in HeLa cells affects the levels of Tir protein and the number of pedestals formed as occurred in MEFs deficient for Nck.
As increasing multiplicity of infection (MOI) accelerates the attachment rate as well as the translocation efficiency of EPEC,Citation6 we infected Nck-deficient cells with increasing MOIs to determine if the levels of Tir delivery would also increase (Fig. S1). However, we found that increasing the MOI resulted only in a slight increase in the levels of Tir within Nck-deficient cells (Fig. S1A). We also found that the levels of EspB effector, a component of the translocation pore,Citation36 were not diminished in Nck-deficient cells, as observed with EspF (Fig. S1A).
It is well established that Nck-deficient cells form significantly less pedestals than WT cells.Citation15,32 In order to analyze the levels of Tir in a cell type also resistant to the formation of pedestals by EPEC, we infected N-WASP-deficient MEFsCitation17 and found that these cells had equal levels of Tir when compared to WT or to N-WASP-deficient cells that were retrovirally reconstituted with N-WASP (R N-WASP) (Fig. S2). This result suggests that the low levels of Tir found in Nck-deficient cells cannot be attributed exclusively to the absent of pedestals.
Decreased EPEC adhesion to Nck-deficient cells
Then, we considered that the difference found in Tir levels could be accompanied with different adhesion levels of the bacteria to the cell types analyzed. As a first approach, we performed an assessment of the number of attached bacteria by counting cell-associated bacteria stained with DAPI, from representative microscopic fields (Fig. S3A). Quantification of attached bacteria on Nck-deficient cells after normalizing to the numbers found in WT cells to 100 %, was found to be 46.9 % compared to 90.8 % for R cells (Fig. S3B). Moreover, our results show a 20-fold decrease in the number of bacteria that form pedestals on Nck-deficient cells compared to WT cells (6 % on Nck-/-, 73 % on R cells, 100 % on WT cells).
Another technique for semi-quantitative determination of bacterial attachment to the cell surface is WB analysis of the Triton X-100-insoluble fraction with a MoAb against DnaK (a bacterial heat-shock protein).Citation37 Moreover, the levels of Tir and that of other effectors detected in the insoluble fraction are an indicator of bacterial attachment to cells.Citation36 We found that the levels of DnaK in the insoluble fraction of Nck-deficient cells were decreased with respect to control cells (asterisk in Fig. S1B). The levels of Tir, EspB and EspF in the insoluble fraction were also reduced in Nck-deficient cells with respect to WT cells. Collectively, these results indicate that Nck-deficient cells show an EPEC adhesion defect compared to WT cells.
Intimin-Tir interaction is more important for EPEC binding to murine MEFs than to human HeLa cells
Considering that EPEC adhesion to Nck-deficient MEFs is decreased (Figs. S1B and S3) and the reduction of Tir levels was more drastic in murine MEFs than in human HeLa cells (), we examined EPEC adhesion to MEFs in greater detail.
We initially focused on Tir-Intimin interaction and performed infection experiments with EPEC strains that lack Intimin or Tir (Δeae and Δtir mutant, respectively) with, as a control, Δtir mutant complemented with WT Tir expressed from a low-copy-number plasmid (Δtir + ptir). In order to compare EPEC adhesion between murine and human cells we infected HeLa cells in parallel with identical MOI before analyzing, by WB, the Triton-X100 soluble (cytoplasmic and membrane fractions) and insoluble fractions (contain the attached EPEC) ().
Figure 2. EPEC adhesion to Nck-deficient cells. WT, Nck-deficient cells (Nck −/−) and Nck1 reconstituted cells (R) were infected for 3 h with EPEC strains Δeae (deleted gene encoding Intimin), Δtir (deleted gene encoding Tir) and, as a control, Δtir + ptir (Δtir complemented with Tir). As a control HeLa cell infections were performed in parallel. Cell monolayers were lysed in 1% Triton X-100 lysis buffer. The soluble supernatants that contain the cytoplasmic and membrane fractions (A) and the insoluble pellets that contain the attached bacteria (B) were subjected to SDS-PAGE and blotted with the indicated antibodies. Actin was used as a loading control. DnaK is an EPEC chaperone used as a control for the bacterial protein content.
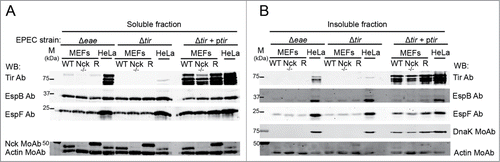
WB analysis of the soluble fraction of Δeae mutant-infected cells shows that the levels of Tir in murine fibroblasts were significantly decreased with respect to human epithelial HeLa cells (). This result denotes differences in translocation efficiencies between both cell types. We next analyzed by WB the levels of DnaK and other effectors of the insoluble fraction. We found that the levels of DnaK, Tir, EspB and EspF of the Δeae mutant strain attached to MEFs were clearly reduced with respect to Hela cells. Similar results were obtained with Δtir mutant (). Collectively, these results show that, compared to HeLa cells, the adhesion of EPEC to murine fibroblasts has a higher dependence on Intimin-Tir interaction.
Tir is stable within Nck-deficient MEFs but depends on de novo EPEC protein synthesis for kinase-mediated modification
We examined whether Tir within Nck deficient MEFs is stable or subjected to degradation by treating, post infection, the cells with bactericidal levels of gentamicin as described,Citation38 to kill extra-cellular EPEC and prevent further effector delivery. Thus, we treated 3h-infected MEFs with gentamicin for 6 hours and analyzed the levels of Tir in total cell lysates. Tir was detected even 6 hours after infection of WT and Nck-deficient MEFs. We corroborated that Tir levels in Nck-deficient cells were decreased with respect to WT cells in all studied conditions, but a fraction of Tir is stable even 6 hours after infection in both cell types ().
Figure 3. Stability of Tir present within Nck-deficient MEFs. (A) WT and Nck-deficient cells (Nck−/−) were infected with EPEC for 3 h or left uninfected as a negative control. Then the cells were treated with gentamicin for 6 hour time-course. Cell monolayers from a single well of a 6-well-plate were collected at indicated times by adding Laemmli sample buffer and lysates were blotted with anti-Tir MoAb. (B) WT and Nck−/− cells were infected with EPEC for 15 minutes at an MOI of 300. Cells were then treated with chloramphenicol (+ Cmp.) or vehicle as a control (− Cmp.) for 1, 2 or 3 h. Monolayers were lysed in 1% Triton X-100 lysis buffer. The soluble supernatants that contain the cytoplasmic and membrane fractions were blotted with anti-Tir Ab and anti-EspF Ab. Actin was used as a loading control. For better visualization of Tir the intensity of the image was decreased in the − Cmp WB. Arrows indicate the fully-modified Tir form.
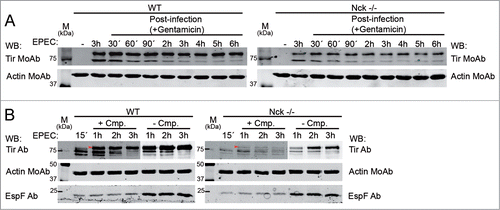
It has been described that 15 to 20 minutes of infection at a high MOI is sufficient for triggering the phosphorylation of injected Tir even when further bacterial protein synthesis is inhibited.Citation39 Accordingly, we examined Tir levels in WT and Nck-deficient cells infected with EPEC (MOI of 300) for 15 minutes before adding the bacterial protein synthesis inhibitor, chloramphenicol, for 1, 2 or 3 hours. WB analysis of the Triton X-100 soluble fractions of WT MEF-infected cells revealed a partially modified Tir form that was converted to the fully-modified Tir form in the presence or absence of chloramphenicol treatment (, left panel). The results are in agreement with previous workCitation14 where only the unmodified (∼78 kDa) and a partially modified form of Tir (∼85 kDa) are apparent at early time points while the fully modified form (90 kDa; latter is substrate for tyrosine phosphorylationCitation39) is evident at later time points. Interestingly, chloramphenicol treatment reveals the gradual conversion of unmodified Tir to the fully modified form.
By contrast, while Nck-deficient cells, as expected, contained less Tir, chloramphenicol treatment inhibited its conversion to the fully-modified Tir form (). In summary, these results suggest that in the absence of Nck, de novo bacterial protein synthesis is necessary at early time points of infection for the conversion of the partially modified to the fully modified Tir form.
Phosphorylation status of Tir and levels of Tir phosphorylation-deficient mutants in Nck-deficient cells
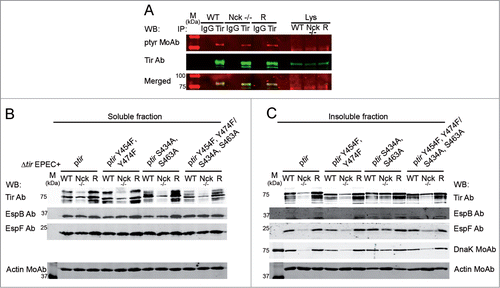
Phosphorylation of Tir at Y474 is crucial for formation of actin pedestals on cultured cellsCitation14 although it is dispensable in infections of explant culture intestinal tissue.Citation40 Therefore we analyzed the tyrosine phosphorylation status of the small but detectable fraction of Tir within Nck-deficient MEFs. We performed immunoprecipitation experiments using an anti-Tir MoAb probing the isolate fraction, on WBs, with a generic phosphotyrosine MoAb and with Tir Ab. This work revealed that some of the Tir present in Nck-deficient cells was tyrosine phosphorylated ().
Figure 4. Phosphorylation status and levels of Tir phosphorylation-deficient mutants within Nck-deficient MEFs. (A) WT, Nck-deficient cells (Nck −/−) and Nck1 reconstituted cells (R) were infected with EPEC for 3 h. Cell monolayers were lysed in modified Ripa buffer and the cell lysates (Lys) were used to perform immunoprecipitation (IP) experiments using an anti-Tir MoAb coupled to magnetic beads. Sequential WB was performed with a generic anti-phosphotyrosine MoAb (in red) and with anti-Tir Ab (in green). The merge of both images is shown. The isotype control IP (IgG) and the cell lysates are shown. (B and C) WT, Nck −/− cells and Nck1 reconstituted cells (R) were infected with EPEC strains for 3 h. EPEC Δtir strain complemented with WT Tir (Δtir + ptir), tyrosine phosphorylation deficient Tir mutant (Δtir + ptir Y454F,Y474F), serine phosphorylation deficient Tir mutant (Δtir + ptir S434A,S463A) and tyrosine and serine phosphorylation deficient Tir mutant (Δtir + ptir Y454F,Y474F/S434,S463A). Cell monolayers were lysed in 1% Triton X-100 lysis buffer. The soluble supernatants that contain the cytoplasmic and membrane fractions (B) and the insoluble pellets that contain the attached bacteria (C) were subjected to SDS-PAGE and blotted with the indicated antibodies. Actin was used as a loading control.
Phosphorylation of Tir at Y474 generates a docking site for Nck. However, Tir also nucleates actin inefficiently through a second phosphorylated-tyrosine (Y454). In addition, two serine residues in Tir (S434 and S463) are phosphorylated.Citation26,41 Serine phosphorylation may induce changes in the three-dimensional structure of Tir that may promote its insertion into the host plasma membrane.Citation14,27 Thus, we explored the role of these phosphorylation substrate residues in altering Tir level within Nck-deficient cells by using strains expressing Tir lacking these phosphorylation substrate residues.
Thus, MEFS were infected with Δtir mutant strains carrying plasmids expressing Tir (ptir), as a control, or non-phosphorylatable Tir forms (ptir Y454F,Y474F, ptir S434A,S463A, ptir Y454F,Y474F/S434A,S463A). WB analysis of the Triton X-100-soluble fractions revealed lower levels of each Tir variant within Nck-deficient cells compared to WT cells (). As before, the delivery levels of EspB and EspF effectors were similar between WT and Nck-deficient cells (). The analysis of the chaperone DnaK on the Triton X-100-insoluble fractions also indicates that the adhesion of the different mutant strains to Nck-deficient cells is diminished when compared to WT cells or Nck1 reconstituted cells ().
These results show that Nck-deficient cells infected with Tir phosphorylation-deficient variants have low levels of Tir that correlate with a decrease in the adhesion of these mutants to Nck-deficient cells, as occurred with infection experiments using WT EPEC ( and Fig. S3). Thus, substitution of tyrosine and serine residues linked to Tir phosphorylation does not impact on the low level of this effector found in Nck-deficient cells.
Reduced levels of the Map effector within Nck-deficient cells
Because of the tight regulation of the process of secretion and translocation, some proteins that are stored in the bacterial cytoplasm require the presence of a dedicated family of T3SS chaperones. The interaction of the effectors with chaperones regulates stability and translocation efficiencies.Citation6 The chaperone CesT binds and enhances the translocation of Tir, Map and other LEE-effectors (EspH, EspJ, EspZ) and non-LEE (Nle) effectors (A, B1, B2, C, G, H1 and H2).Citation7,42 With the aim of analyzing the levels of Map, another CesT-dependent effector, we infected Nck-deficient cells with EPEC which carried a plasmid expressing Map fused in frame, to enable WB detection, with haemagglutinin (HA) and T7 epitope tags (WT+T7-map-HA).
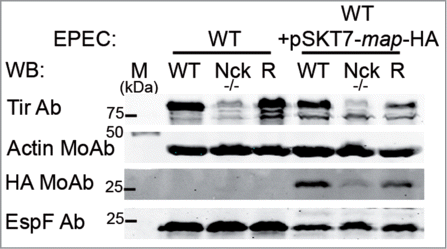
WB analysis of Triton X-100-soluble fraction with a MoAb against HA shows a reduction in the levels of Map within Nck-deficient cells when compared with WT cells (). Moreover, we found that the levels of EspF, whose delivery is dependent on a different chaperone (CesF), were not diminished in Nck-deficient cells infected with EPEC-expressing T7-map-HA (). This result indicates that the levels of at least two CesT-dependent effectors, Tir and Map, are diminished in Nck-deficient cells.
Figure 5. The levels of Map effector within infected-Nck-deficient cells are decreased compared to WT cells. WT, Nck-deficient cells (Nck -/-) and Nck1 reconstituted cells (R) were infected for 3 h with WT EPEC or with WT EPEC that expresses HA tagged-Map (WT+pSKT7-map-HA). Cell monolayers were lysed in 1% Triton X-100 lysis buffer. The soluble supernatants that contain the cytoplasmic and membrane fractions were analyze for WB with anti-Tir Ab, anti-HA MoAb and with anti-EspF Ab. Actin was used as a loading control.
Analysis of the possible degradation of bacterial Tir
The reduced level of Tir, but not EspF or EspB, in Nck-deficient cells was suggestive of a defect in the Tir delivery process or degradation of the majority of the delivered protein. To interrogate the former, we bypassed delivery by ectopically-expressing Tir (as a His-tagged fusion protein) within WT and Nck-deficient cells. Parallel transfections with a plasmid expressing eGFP-tagged cortactin served as a control for transfection efficiency. WB analyses revealed similar amounts of Tir in both cell types (Fig. S4A) suggesting that the reduced level of Tir delivery by EPEC is linked to reduced translocation levels. However, consistent with previous reportsCitation35,38 ectopically-expressed Tir protein underwent little if any kinase-mediated modification (Fig. S4A); events linked to its ability to become correctly insert into the plasma membrane and trigger Intimin-dependent pedestal formation. Thus, the inability of Tir to undergo full modification and/or insert into the plasma membrane may protect it from degradation. Thus, studies refocused on the possibility of Tir degradation.
Degradation by the action of proteasome or cytoplasmic proteases are two mechanisms by which bacterial effector proteins could be degraded in the host cell.Citation43,44 Hence, we tested the effect of chemical inhibitors in order to block a possible degradation of Tir. To this end, we treated the cells with MG-132 proteasome inhibitor for 1 hour prior to and during infection, at a concentration known to prevent EPEC-induced protein degradation in infected cells.Citation38,45 Untreated cells were used as a control. EPEC infection were stopped by adding gentamicin and cellular extracts were collected at indicated times after infection. WB analysis of the cellular extracts revealed that the levels of Tir did not differ between MG-132-treated or untreated Nck-deficient cells (Fig. S4B).
Signaling by the different inflammasome platforms are able to regulate the inflammatory host responses to enteric pathogens. Mice lacking caspase-1, a cysteine protease component of the inflammasome, develop hyper-susceptibility to Citrobacter rodentium infection. Citrobacter rodentium infection of mice is comparable to EPEC infection of humans.Citation46 Hence, we wondered whether the activation of caspases could be implicated in the degradation of Tir. We treated WT and Nck-deficient cells with a general cell-permeable caspase inhibitor (Z-VAD-FMK, abbreviated as Z-VAD) for 2 hours prior to and during infection. Untreated cells were used as a control. As shown in Fig. S4C the treatment with Z-VAD did not increase the levels of Tir in either WT cells or in Nck-deficient cells.
Furthermore, we considered the possible role of other host proteases in the degradation of Tir by examining the impact of treating the cells (4 hours prior to and during infection) with a broad spectrum protease cocktail of inhibitors (P8340). Untreated cells were used as a control. As shown in Fig. S4D the treatment did not increase the levels of Tir either within WT cells or within Nck-deficient cells. We also treated the cells with P8340 for 20 hours pre-infection with similar results (data not shown). Collectively, these results seem to indicate that in EPEC-infected Nck-deficient cells, Tir is not degraded by proteasome, caspases or, in general, by proteases.
Trichostatin A treatment increases the EPEC attachment, Tir delivery and the efficiency of pedestal formation
Trichostatin A (TSA) is a metabolite from Streptomyces hygroscopicusCitation47 that inhibits deacetylase activity and that has been linked to another degradative pathway, autophagy.Citation48 Studies examined its impact on Tir levels within WT and Nck-deficient cells. Thus, cells were treated with TSA for 16 h prior to and during infection. Untreated cells were used as a control. We confirmed treatment-induced increases in the acetylation status of tubulin, by WB analysis (data not shown). WB analysis of the Triton X-100-soluble fractions with a Tir Ab revealed that the levels of Tir were increased within TSA-treated Nck-deficient cells compared to untreated Nck-deficient cells. Moreover, the treatments with TSA also increased Tir levels, as well as the levels of EspB and EspF, within WT and Nck1 reconstituted cells (, soluble fraction). These results indicate that treatments with TSA have a general effect on the levels of T3SS-dependent effectors found in infected cells.
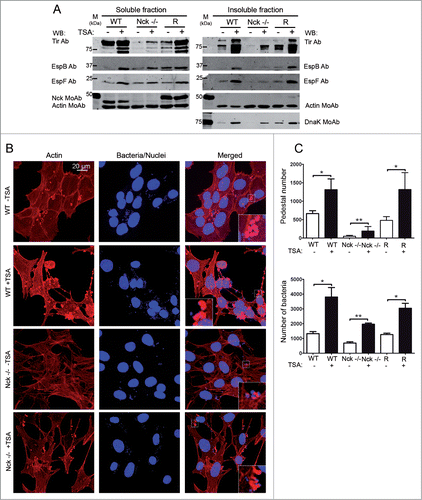
Figure 6. Trichostatin A treatments increase bacterial attachment and the levels of Tir in EPEC-infected MEFs. (A) WT, Nck-deficient cells (Nck−/−) and Nck1 reconstituted cells (R) were treated with Trichostatin A (TSA, 5 μM) for 16 hours prior to and during infection with EPEC for 3h. Cell monolayers were lysed in 1% Triton X-100 lysis buffer. The soluble supernatants that contain the cytoplasmic and membrane fractions and the insoluble pellets that contain the attached bacteria were subjected to SDS-PAGE and blotted with the indicated antibodies. Actin was used as a loading control, blotting with anti-Nck MoAb was performed to confirm the genotype of the MEFs used and with anti-DnaK MoAb as an indicator of bacterial attachment. (B) Confocal immunofluorescence images of WT and Nck −/− cells treated with TSA for 16 h and infected with EPEC for 3 h. Actin staining with TRITC-phalloidin is shown in red. DAPI was used to stain EPEC. The merged images shown in the last column were generated with Leica software. Pictures were taken at 600X magnification and scale bar represents 20 μm. Insets are 4X digital zoom images. (C) Quantification of the number of attached bacteria and pedestals of 100 cells from the immunofluorescence images. Graph represents mean ± SD. Statistical analysis using Student´s t-test for the number of attached bacteria and Mann Whitney test for pedestal number from at least 3 independent experiments is shown. *, P < 0.05; **, P < 0.01.
To determine whether TSA treatments affect the attachment of EPEC to MEFs we analyzed by WB the levels of DnaK in the Triton X-100-insoluble fractions. As shown in , we found a general increment in the levels of EPEC adhesion after TSA treatments. Thus, we can conclude that TSA-treatments increase the levels of Tir and the adhesion of EPEC to MEFs, although we observed that TSA induces a general rearrangement of the actin cytoskeleton and changes the cell morphology.
In order to correlate Tir levels and pedestal formation we quantified the number of pedestals formed in TSA-treated and untreated cells. We stained actin with TRITC-phalloidin to visualized pedestals and bacteria with DAPI () and quantified the number of pedestals (). As shown in the upper graph, the number of pedestals formed in TSA-treated-Nck-deficient cells increased fourfold with respect to untreated Nck-deficient cells. This result implicates that a recovery in Tir levels induced by TSA correlates with higher efficiency of pedestal formation. In addition, the quantification of the number of bacteria attached to cells shows that TSA treatments resulted in a significant increase in EPEC attachment to cells corroborating the DnaK WB results ().
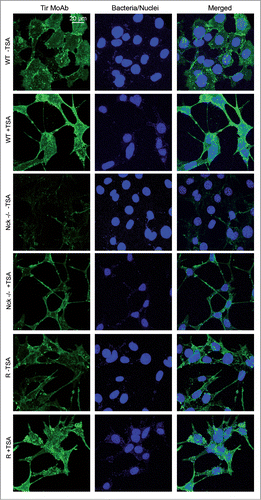
The increase in the levels of Tir after TSA treatments was corroborated by immunofluorescence staining experiments using a MoAb against Tir in TSA-treated or untreated MEFs. As shown in the staining of Tir in Nck-deficient cells is remarkably fainter than in WT cells. This observation corroborates previous results obtained by WB analysis of Tir levels in Nck-deficient MEFs (; Figs. S1, S3, and S6). Importantly, TSA-treated Nck-deficient cells exhibited brighter staining of Tir. In summary, the results obtained using WB and immunofluorescence assays indicate that treatments with TSA increase the levels of Tir, the number of pedestals and the adhesion of EPEC to MEFs.
Figure 7. Immunofluorescence staining of Tir within MEFs treated with Trichostatin. (A) Confocal immunofluorescence images of WT, Nck-deficient cells (Nck −/−) and Nck1 reconstituted cells (R) treated with Trichostatin A (TSA, 5 μM) for 16 h (or left untreated as a control) and infected with EPEC for 3 h. Immunofluorescence staining was done using anti-Tir MoAb (in green) and DAPI to stain EPEC (in blue). The merged images shown in the last column were generated with Leica software. Pictures were taken at 600× magnification and scale bar represents 20 μm.
Discussion
EPEC, like many other pathogens, manipulates host signaling pathways in a redundant manner to promote a successful infection strategy. Thus, numerous EPEC effectors manipulate the host cell mechanisms that control actin polymerization.
Nck adaptor proteins trigger actin polymerization beneath adherent EPEC by activating the actin-nucleation promoting factor (NPF) N-WASP to induce Arp2/3 complex-mediated actin polymerization.Citation15 Indeed, it was elegantly shown that clustering at the plasma membrane of fusion proteins containing all three SH3-domains of Nck induced N-WASP-dependent localized polymerization of actin, including “spots” which could be considered reminiscent of pedestals.Citation49
Knocking-down expression of the Nck1 and Nck2 adaptors significantly reduces EPEC induced pedestal formation in mammalian cell models,Citation15,32-34 presumably because Nck binding to a motif containing the phosphorylated tyrosine 474 of Tir is required to recruit and activate N-WASP. In addition, phosphorylated tyrosine 454 of Tir promotes the nucleation of actin through a secondary Nck-independent pathway.Citation34 However infection studies with human intestinal tissue (provides EPEC's in vivo target; enterocytes) involving a Tir Y454F, Y474F variant lead to actin nucleation and pedestalsCitation40 despite the absence of these Nck-dependent and Nck-independent actin polymerization pathways. Similarly, substitution of the corresponding tyrosine residue of Tir from Citrobacter rodentium (TirCRY471 and Y451) did not affect the recruitment of N-WASP or A/E lesion formation.Citation50 The low levels of Tir effector within Nck-deficient cells described in this work might help explain some of the apparent contradictions between in vitro and ex vivo models, indicating that Nck might be one and not, as thought, “the central” host protein subverted in vivo by EPEC to active N-WASP.
Interestingly, we found that the levels of the Map effector are also reduced within Nck-deficient cells (). At the initial stages of EPEC infection, Map induces the formation of transient filopodia by activating Cdc42.Citation51 Additionally, Map recognizes a local actin rearrangement at the bacterial attachment sites, through an Ebp50-Ezrin-actin complex, establishing a positive feed-back loop that locates activated Cdc42 at the cell membrane leading to localized actin polymerization.Citation52 Therefore, in EPEC-infected Nck-deficient cells, which have reduced levels of Map, this positive feed-back loop might also be broken, representing another underlying cause explaining the reduced number of pedestals in the absence of Nck.
Interestingly, N-WASP-deficient cells, which are also resistant to pedestal formation by EPECCitation16 have normal levels of Tir (Fig. S2). Consequently we can conclude that the low levels of Tir found in Nck-deficient cells cannot be exclusively attributed to the absence of pedestals. We propose that Nck is required for the translocation of TirEPEC and MapEPEC, in a similar way that efficient translocation of TirEHEC and EspFuEHEC requires N-WASP and actin assembly.Citation53
In our studies, EPEC-infected Nck-deficient cells presented a reduction of Tir and pedestals of 88 % and 94 % respectively (; Fig. S3). Our pedestal data is in line with that of Gruenheid et al.Citation15 but their conclusion for a critical Nck role possibly stems from the absence of pedestal quantification as, in our study, only ∼1 in 6 adherent bacteria were associated with pedestals (Fig. S3B and C). However, our findings contrast with the 4-fold decrease in pedestals formation reported by Campellone and co-workers linked to similar (25%) decrease in the level of translocated HA-tagged Tir.Citation34,54 This discrepancy presumably reflects different infection protocols leading to more Tir delivery and, thus, pedestals.
The observed phenotype was not specific to one cell line used since reducing Nck levels (by ∼85% with siRNA) in HeLa cells led to similar results. However, Tir delivery levels and, thus, pedestals were only reduced by ∼20% linked to more efficient adherence of EPEC (). The relative small reduction in pedestals for Nck-knockdown HeLa cells supports a non-essential role for this adaptor. Furthermore, we have corroborated our finding by infecting Nck-deficient cells with Citrobacter rodentium (data not shown), pointing toward a common defect for A/E pathogens.
The diminished adhesion of EPEC to Nck-deficient MEFs (Fig. S3) might be a consequence of the low levels of Tir found in these cells; in agreement with the reported decrease adhesion of EHEC to N-WASP-deficient cells that present decrease levels of TirEHEC.Citation53 However, the major EPEC adherence factor for HeLa cells is the bundle forming pilus followed by Tir-Intimin interaction. Thus, it is likely that the difference in binding reflects the absence of a receptor for this human-specific pathogen on murine cells. In support of this, we found that the adhesion of EPEC to murine cells, compared to human HeLa cells, depends more on the presence of Intimin and Tir, although it should be noted that they are different cell types (fibroblasts and epithelial cells) (). Importantly, no differences were detected in the delivery of two other effectors (EspB and EspF) thereby illustrating that observed cell line data discrepancies did not related to the functioning of EPEC's effector-delivery system.
The analysis of the distinct forms of Tir at early time points of infection deserves further consideration. Thus, we infected cells at high MOIs for a short period of time, reported sufficient to trigger the phosphorylation of Tir.Citation39 Then the cells were treated with chloramphenicol to inhibit bacterial protein synthesis.Citation55 We could detect not only a reduction in the level of Tir within Nck-deficient cells but there was a defect in its modification to the fully-modified Tir form within these cells but not WT infected cells (). The result seems to indicate that, besides de novo bacterial protein synthesis, Nck is required to convert Tir to its fully modified (functional) form.
In order to test the hypothesis of Tir degradation within Nck-deficient cells, we used several chemical inhibitors but these failed to restore Tir levels (Fig. S4). However, treatments with TSA, a known deacetylase and autophagy inhibitor,Citation48,56 lead to a significant increase in the levels of Tir in WT, Nck-deficient and reconstituted MEFs (). Moreover, TSA treatments significantly increased the bacterial attachment and the number of pedestals formed (). Interestingly, we observed a correlation between the levels of Tir and pedestal formation in TSA-treated Nck-deficient cells.
It is difficult to explain how TSA increases the levels of EPEC effectors within infected cells because it affects many cellular processes. In agreement with previous reports,Citation57 we have observed that TSA induces a general rearrangement of the actin cytoskeleton and changes the cell morphology. TSA, apart from inhibiting the activity of histone deacetylases, enhances the expression of the actin-severing protein gelsolinCitation57 which is localized to pedestals.Citation58 It is tempting to speculate that TSA induced rearrangements of the cytoskeleton, which are at least in part mediated by the action of gelsolin, could favor the translocation of EPEC effectors.
In conclusion, we report that EPEC-infected Nck-deficient cells possess reduced cellular levels of Tir and Map, which are two important EPEC effectors that control actin polymerization at pedestals. Our findings might help explain some of the discrepancies found between different infection models used to study the manipulation of the actin cytoskeleton by EPEC, and indicate that Nck adaptors possibly only have an accessory role in activating N-WASP during pedestal formation by EPEC.
Materials and Methods
Cells, bacteria and antibodies
The human cervical epithelial cancer cell line (HeLa) was obtained from the American Type Culture Collection (ATCC). WT MEFs, N-WASP-deficient MEFs, N-WASP-deficient MEFs that were retrovirally reconstituted with N-WASP (Rescued N-WASP),Citation59 Nck-deficient MEFs and Nck-deficient MEFs that were retrovirally reconstituted with eGFP-Myc-tagged Nck1 vector (Rescued Nck)60 were maintained in Iscove's Modified Dulbecco's medium (IMDM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (penicillin/streptomycin).
Enteropathogenic Escherichia coli (ePEC) E2348/69 and anti-Tir 2A5 N-terminal MoAb.Citation61 EPEC strains Δtir,Citation9 Δeae (Intimin) CVD20662and polyclonal Abs anti-Tir,Citation9 anti-EspBCitation63 and anti-EspF.Citation64 EPEC strains Δeae and Δtir are nalidixic acid-resistant (25 μg/ml). Anti-N-WASP Ab was previously described.Citation65 The following commercial antibodies were used: anti-Myc tag 4A6 mouse MoAb and anti-cortactin 4F11 mouse MoAb (Millipore); Anti-phosphotyrosine (p-tyr-100) mouse MoAb (Cell Signaling); Anti-β-actin C4 mouse MoAb (MP Biomedicals). Anti-HA (clone 3F10) rat MoAb was from Roche and anti-α-tubulin rat MoAb was from AbD Serotec. Anti-E.coli DnaK (clone 8E2/2) mouse MoAb was from Enzo Life Sciences. For WB experiments using the Odyssey infrared scanning system (Lycor, Fisher Scientific), the Abs were purchased at a concentration of 1 mg/ml and used at 1:5,000 dilution. The following Abs were used for green signal: IRDye 800CW-labeled goat anti-rabbit and anti-mouse secondary Abs and for red signal: IRDye 680CW-labeled goat anti-rabbit Ab (Fisher Scientific).
Constructs
Constructs encoding Tir and CesT have been described which carry Y454F,Y474FCitation34 or S434A,S463ACitation26 substitutions in Tir. A ∼800bp ClaI/BglII fragment (3’tir to the 5’ cesT) from each construct was transferred into the same sites of ptir (ΔBamHI) encodes Tir and CesTCitation66 to generate ptir Y454F/Y474F and ptir S434A,S463A respectively. The Tir serine residues 434 and 463 were sequentially substituted to alanine using ptir Y454F,Y474F as a template for site directed mutagenesis (Stratagene) with oligonucleotides tir S434A PS FspI PS [CTGAATAGACGTGATGCGCAG] and tir S463A PS AflII PS [GCTCGGA-ATGCCTTAAGT] as per manufactures recommendations. The synonymous introduction of FspI and AflII sites promoted screening of mutations with sequence analyses confirming the specific presence of the changes. Generation of the T7-map-HA expressing plasmid involved swapped BamHI/SalI fragments between pSK-T7mapCitation66 and pSK-mapHA constructs (latter involved oligonucleotide amplification of map gene –from ATG to last codon – into pSK construct that carries the sequence for an HA epitope followed by a stop codon to produce a mapHA inframe gene fusion). Strains carrying the plasmids were produced by electroporation and selected with chloramphenicol (25 μg/ml) or carbenicillin (100 μg/ml).
pcDNA3.1-HisB-Tir was produced by subcloning tir from pGEMT-Tir plasmid and verified by sequencing. peGFP-cortactin was previously described in Nieto-Pelegrin et al.Citation29
Preparation of electrocompetent EPEC cells and electroporation
Mid-log-phase cultures (50 ml, optical density at 600 nm = 0.4 to 0.6) was harvested by centrifugation at 1,200 × g for 15 min at 4°C, and the pellet was washed 3 times with 50 ml of cold sterile water. All manipulations were carried out on ice. The pellet was finally resuspended in 100 μl of cold sterile water. A volume of 40 μl of the resulting electrocompetent cells were electroporated immediately with 6 μl of DNA plasmid at 2KV in 1 mm-gap electroporation cuvettes (Cell Project) using a BTX Electro Cell Manipulator 600. Immediately following electroporation, cells were washed into the bottom of the electroporation chamber with 1 ml of SOC medium. The electroporated cells were allowed to recover for 1 h at 37°C, prior to plating onto the antibiotic-containing LB agar plates.
EPEC infection
EPEC was preactivated by incubating a 1:100 dilution of an overnight culture for 2 h in IMDM supplemented with 10% FBS without antibiotics at 37°C and 5% CO2. After preactivation, the optical density of the suspension at 600 nm was adjusted to 0.2. Cell monolayers were infected with a range of MOI of 3 to 15 for the indicated time in IMDM supplemented with 10 % FBS without antibiotics. For the infections with the different EPEC strains, cell monolayers were infected at an MOI of 60. When indicated, the cells were infected at varying MOIs, such as 30, 150 and 300.
Membrane enrichment and protein extraction procedures
To obtain a membrane enriched fraction, EPEC-infected cells were fractionated as previously described in Nieto-Pelegrin et al.Citation29 For protein extraction, cell monolayers were washed 3 times in cold Dulbecco's phosphate-buffered saline (D-PBS) with calcium and magnesium (Invitrogen), and lysed in 200 μl of 1% Triton X-100 lysis buffer (PBS containing 1% Triton X-100, 0.4 mM Na3VO4, 1 mM NaF and 0.1 mM PMSF) as described previously in Kenny et al.Citation11 After 5 min incubation on ice, the samples were centrifuged at 21,000 × g for 5 min at 4°C to obtain the insoluble fraction, which contained adherent bacteria, host nuclei and cytoskeleton. The soluble supernatants (contain the cytoplasmic and membrane fractions) was kept and the insoluble pellets were washed in 200 μl of PBS. The insoluble pellets were resuspended in 200 μl of 1X Laemmli sample buffer by mixing with a vortex. Both fractions were heated at 100°C for 5 min and a volume of 30 μl of each fraction was subject to 12% SDS-PAGE and analyzed by WB using primary and secondary Abs. Membranes were scanned with the Odyssey Scan system using the red (700 nm) and green (800 nm) channels.
Cell transfection and siRNA treatment
Cells were transfected with plasmids using Lipofectamine 2000 reagent (Invitrogen) as per the manufacturer's instructions. Briefly, HeLa cells were grown to 60–70 % confluence in 6-well plates with heat-sterilized coverslips for immunofluorescence, or without coverslips for WB. Transfection was carried out in IMDM containing 10% FBS without antibiotics for 20 h prior to EPEC infection. As a control for the transfection treatment, cells were treated with the transfection reagent alone.
Inhibition of human Nck1/Nck2 expression in HeLa cells using siRNA was carried out using Silencer Pre-designed mouse siRNA and a scrambled oligonucleotide as negative control (Ambion, Life Technologies) as per the manufacturer's instructions. Briefly, HeLa cells were grown to 50–60% confluency in 6-well plate and transfected with 40 nM of siRNA in the presence of 6 μl of LipofectamineTM RNAiMAX (Invitrogen) per well. Transfections were allowed to proceed in IMDM containing 10% FBS without antibiotics for 20 h prior to EPEC infection.
WB experiments of siRNA treatments and transfections were performed using 6-well plates. Cells were washed once with cold D-PBS and scraped into 200–300 μl 2X Laemmli buffer. Samples were homogenized by 3 passages through a syringe with a 25-gauge needle and centrifugation at 21,000 × g for 15 min at 4°C and resolved by SDS-PAGE.
Immunofluorescence microscopy
Cells were fixed with 10% formalin solution (formaldehyde 4% w/v, Sigma) in PBS at room temperature and permeabilized with 0.1% Triton X-100 for 5 min. After 3 washes with PBS, cells were blocked with 2% BSA in PBS for 10 min and stained at room temperature with anti-Tir 2A5 MoAb for 1 h, washed 3 times with PBS, and finally incubated 1 h with a secondary Alexa 488- conjugated goat anti-mouse Ab (green, Invitrogen) at 1:5,000 dilution. Filamentous actin (F-actin) was visualized by staining during 15 min with 1 μg/ml tetramethyl rhodamine isothiocyanate (TRITC)-phalloidin (Sigma). Bacteria were visualized with DAPI (300 nM). Confocal microscopy was performed at the Parque Científico de Madrid microscopy facility using a Leica Confocal SP2/DM-IRE2 and images were processed with Leica software (version 2.61). Quantification of pedestals and attached bacteria was done by counting representative fields containing a total of 100 cells. Experiments were performed at least 3 times. Statistical analyses were carried out using the 2-tailed Student's t-test (or Mann Whitney test in when the data cannot be assumed to follow a Gaussian distribution in each group) and displayed graphically using GraphPad Prism software v5. Graphs represent mean ± SD.
Immunoprecipitation of Tir
Cells were grown in complete medium to 70–80% confluency on 150-mm plates, then washed once with D-PBS and scraped into 700 μl modified Ripa lysis bufferCitation29 and disrupted by 3 passages through a syringe with a 25-gauge needle, followed by centrifugation at 12,000 × g for 15 min. Per each immuno-precipitation reaction, 25 μl of Magnetic Pan mouse Dynabeads (Invitrogen) were washed and blocked with PBS containing 0.% BSA for 10 min, then incubated with tumbling for 2 h with 4 μl of Tir 2A5 MoAb. After 3 washes with PBS-0.1 % BSA, the beads were added to 300 μl of cell lysate and incubated with tumbling at 4°C for 3 h. The beads were washed 3 times with the help of a magnet (Invitrogen) and 200 μl Ripa lysis buffer diluted 1:10 in PBS. The beads were resuspended in 40 μl 2X Laemmli buffer and heat at 100°C for 5 min before SDS-PAGE.
Gentamicin assays and MG-132 cell treatment
MEFs were grown in 100-mm plates to 70–80% confluency and pretreated for 1 h with proteasome inhibitor MG-132 (Calbiochem) at a final concentration of 50 μM.Citation38 Cells were then infected with EPEC for 3 h in the presence of the inhibitor. After infection, cells were washed 3 times with IMDM supplemented with 10% FBS to remove unattached bacteria. Then, the cells were incubated at 37°C 5% CO2 in IMDM-10% FBS with gentamicin at bactericidal levels (100 μg/ml) and MG-132 for various times after infection (referred as to post-infection times). After the indicated periods of time, cells were washed once with ice-cold D-PBS and collected by scrapping the cells with 300 μl of imidazole buffer.Citation29 Clarified lysates were obtained following the previously described membrane enrichment protocol.
Treatments with chemical inhibitors
MEFs grown in 6-well plates to 70–80% confluency were pretreated for 2 h with the caspase inhibitor Z-VAD-FMK (Promega) at a final concentration of 50 μM.Citation67 The broad specificity protease inhibitor cocktail P8340 (SIGMA-ALDRICH) was used at a concentration of 1:1000 for 4 h or 20 h prior to infections. Cell monolayers were then infected with EPEC for 3 h in the presence of the inhibitors. After infection, cells were washed 3 times with cold D-PBS, collected by adding 200 μl of 2X Laemmli sample buffer and processed as described above.
MEFs cells grown as before were pretreated with the deacetylase inhibitor Trichostatin A (TSA) (from Streptomyces sp, Sigma-Aldrich) for 16 h prior to infectionsCitation68 (5 μM). After 3h-infection in the presence of the inhibitor, cells were washed 3 times in cold D-PBS and lysed by adding 200 μl of 1% Triton X-100 lysis buffer, according to the protocol described above.
Inhibition of bacterial protein synthesis with chloramphenicol
MEFs cells were grown in 6-well plates to 70–80% confluency. Cells were infected during 15 min with EPEC at an approximate MOI of 300. After 15 minutes of infection, chloramphenicol was added to a final concentration of 100 μg/ml.39 As a vehicle control, 70 % ethanol was added to the untreated cells. Cells were then incubated at 37°C 5% CO2 for 1, 2 or 3 hours. Cell monolayers were then washed 3 times with ice-cold D-PBS and lysed by adding 200 μl of 1% Triton X-100 lysis buffer, according to the protocol described above.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are indebted to the following individuals for providing essential reagents that made this work possible: the late Dr. Tony Pawson (Mount Sinai Hospital, Toronto, Canada) for providing Nck-deficient MEFs; Dr. Scott B. Snapper (Massachusetts General Hospital, Boston, USA) for N-WASP-deficient MEFs; Dr. B. Brett Finlay (University of British Columbia, Vancouver, Canada) for EPEC E2348/69 strain and for anti-Tir 2A5 MoAb; Sabine Quitard for Tir and Map construct generation (University of Newcastle, Newcastle, UK); Dr. Chihiro Sasakawa (Institute of Medical Science. University of Tokyo. Japan) for anti-EspF Ab.
KCAM_A_969993_Supplementary_Figures.zip
Download Zip (6.1 MB)Funding
This work was funded by a grant from the Instituto de Salud Carlos III to NM-Q (FIS PS09/0080). EN-P was supported by a grant to NM-Q from Fundación Médica Mutua Madrileña (01754/2008) and by a Complutense University fellowship.
References
- Navarro-Garcia F, Serapio-Palacios A, Ugalde-Silva P, Tapia-Pastrana G, Chavez-Duenas L. Actin cytoskeleton manipulation by effector proteins secreted by diarrheagenic Escherichia coli pathotypes. Biomed Res Int 2013; 2013:374395; PMID:23509714; http://dx.doi.org/10.1155/2013/374395
- Knutton S, Baldwin T, Williams PH, McNeish AS. Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun 1989; 57:1290–8; PMID:2647635
- Donnenberg MS, Tacket CO, James SP, Losonsky G, Nataro JP, Wasserman SS, Wasserman SS, Kaper JB, Levine MM. Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J Clin Invest 1993; 92:1412-7; PMID:8376594; http://dx.doi.org/10.1172/JCI116717
- Marches O, Nougayrede JP, Boullier S, Mainil J, Charlier G, Raymond I, Pohl P, Boury M, De Rycke J, Milon A, et al. Role of tir and intimin in the virulence of rabbit enteropathogenic Escherichia coli serotype O103:H2. Infect Immun 2000; 68:2171-82; PMID:10722617; http://dx.doi.org/10.1128/IAI.68.4.2171-2182.2000
- Giron JA, Ho AS, Schoolnik GK. An inducible bundle-forming pilus of enteropathogenic Escherichia coli. Science 1991; 254:710-3; PMID:1683004; http://dx.doi.org/10.1126/science.1683004
- Mills E, Baruch K, Charpentier X, Kobi S, Rosenshine I. Real-time analysis of effector translocation by the type III secretion system of enteropathogenic Escherichia coli. Cell Host Microbe 2008; 3:104-13; PMID:18312845; http://dx.doi.org/10.1016/j.chom.2007.11.007
- Mills E, Baruch K, Aviv G, Nitzan M, Rosenshine I. Dynamics of the type III secretion system activity of enteropathogenic Escherichia coli. MBio 2013; 4; PMID:23900171; http://dx.doi.org/10.1128/mBio.00303-13
- Elliott SJ, Wainwright LA, McDaniel TK, Jarvis KG, Deng YK, Lai LC, McNamara BP, Donnenberg MS, Kaper JB. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol Microbiol 1998; 28:1-4; PMID:9593291; http://dx.doi.org/10.1046/j.1365-2958.1998.00783.x
- Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 1997; 91:511-20; PMID:9390560; http://dx.doi.org/10.1016/S0092-8674(00)80437-7
- Ross NT, Miller BL. Characterization of the binding surface of the translocated intimin receptor, an essential protein for EPEC and EHEC cell adhesion. Protein Sci 2007; 16:2677-83; PMID:18029421; http://dx.doi.org/10.1110/ps.073128607
- Kenny B, Finlay BB. Intimin-dependent binding of enteropathogenic Escherichia coli to host cells triggers novel signaling events, including tyrosine phosphorylation of phospholipase C-gamma1. Infect Immun 1997; 65:2528-36; PMID:9199415
- Swimm A, Bommarius B, Li Y, Cheng D, Reeves P, Sherman M, Veach D, Bornmann W, Kalman D. Enteropathogenic Escherichia coli use redundant tyrosine kinases to form actin pedestals. Mol Biol Cell 2004; 15:3520-9; PMID:15155808; http://dx.doi.org/10.1091/mbc.E04-02-0093
- Phillips N, Hayward RD, Koronakis V. Phosphorylation of the enteropathogenic E. coli receptor by the Src-family kinase c-Fyn triggers actin pedestal formation. Nat Cell Biol 2004; 6:618-25; PMID:15220932; http://dx.doi.org/10.1038/ncb1148
- Kenny B. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol Microbiol 1999; 31:1229-41; PMID:10096089; http://dx.doi.org/10.1046/j.1365-2958.1999.01265.x
- Gruenheid S, DeVinney R, Bladt F, Goosney D, Gelkop S, Gish GD, Pawson T, Finlay BB. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat Cell Biol 2001; 3:856-9; PMID:11533668; http://dx.doi.org/10.1038/ncb0901-856
- Kalman D, Weiner OD, Goosney DL, Sedat JW, Finlay BB, Abo A, Bishop JM. Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat Cell Biol 1999; 1:389-91; PMID:10559969; http://dx.doi.org/10.1038/14087
- Lommel S, Benesch S, Rottner K, Franz T, Wehland J, Kuhn R. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep 2001; 2:850-7; PMID:11559594; http://dx.doi.org/10.1093/embo-reports/kve197
- Higgs HN, Pollard TD. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J Cell Biol 2000; 150:1311-20; PMID:10995437; http://dx.doi.org/10.1083/jcb.150.6.1311
- Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature 2000; 404:151-8; PMID:10724160; http://dx.doi.org/10.1038/35004513
- Rivero-Lezcano OM, Marcilla A, Sameshima JH, Robbins KC. Wiskott-Aldrich syndrome protein physically associates with Nck through Src homology 3 domains. Mol Cell Biol 1995; 15:5725-31; PMID:7565724
- Weiss SM, Ladwein M, Schmidt D, Ehinger J, Lommel S, Stading K, Beutling U, Disanza A, Frank R, Jänsch L, et al. IRSp53 links the enterohemorrhagic E. coli effectors Tir and EspFU for actin pedestal formation. Cell Host Micro 2009; 5:244-58; http://dx.doi.org/10.1016/j.chom.2009.02.003
- Brown MD, Bry L, Li Z, Sacks DB. Actin pedestal formation by enteropathogenic Escherichia coli is regulated by IQGAP1, calcium, and calmodulin. J Biol Chem 2008; 283:35212-22; PMID:18809683; http://dx.doi.org/10.1074/jbc.M803477200
- Buss C, Muller D, Ruter C, Heusipp G, Schmidt MA. Identification and characterization of Ibe, a novel type III effector protein of A/E pathogens targeting human IQGAP1. Cell Microbiol 2009; 11:661-77; PMID:19134119; http://dx.doi.org/10.1111/j.1462-5822.2009.01284.x
- Wong AR, Raymond B, Collins JW, Crepin VF, Frankel G. The enteropathogenic E. coli effector EspH promotes actin pedestal formation and elongation via WASP-interacting protein (WIP). Cell Microbiol 2012; 14:1051-70; PMID:22372637; http://dx.doi.org/10.1111/j.1462-5822.2012.01778.x
- Alto NM, Weflen AW, Rardin MJ, Yarar D, Lazar CS, Tonikian R, Koller A, Taylor SS, Boone C, Sidhu SS, et al. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J Cell Biol 2007; 178:1265-78; PMID:17893247; http://dx.doi.org/10.1083/jcb.200705021
- Warawa J, Kenny B. Phosphoserine modification of the enteropathogenic Escherichia coli Tir molecule is required to trigger conformational changes in Tir and efficient pedestal elongation. Mol Microbiol 2001; 42:1269-80; PMID:11886558; http://dx.doi.org/10.1046/j.1365-2958.2001.02693.x
- Race PR, Solovyova AS, Banfield MJ. Conformation of the EPEC Tir protein in solution: investigating the impact of serine phosphorylation at positions 434/463. Biophys J 2007; 93:586-96; PMID:17449672; http://dx.doi.org/10.1529/biophysj.106.101766
- Cantarelli VV, Kodama T, Nijstad N, Abolghait SK, Iida T, Honda T. Cortactin is essential for F-actin assembly in enteropathogenic Escherichia coli (EPEC)- and enterohaemorrhagic E. coli (EHEC)-induced pedestals and the alpha-helical region is involved in the localization of cortactin to bacterial attachment sites. Cell Microbiol 2006; 8:769-80; PMID:16611226; http://dx.doi.org/10.1111/j.1462-5822.2005.00664.x
- Nieto-Pelegrin E, Martinez-Quiles N. Distinct phosphorylation requirements regulate cortactin activation by TirEPEC and its binding to N-WASP. Cell Commun Signal 2009; 7:11; PMID:19419567; http://dx.doi.org/10.1186/1478-811X-7-11
- Lin AE, Benmerah A, Guttman JA. Eps15 and Epsin1 are crucial for enteropathogenic Escherichia coli pedestal formation despite the absence of adaptor protein 2. J Infect Dis 2011; 204:695-703; PMID:21810914; http://dx.doi.org/10.1093/infdis/jir386
- Nieto-Pelegrin E, Meiler E, Martin-Villa JM, Benito-Leon M, Martinez-Quiles N. Crk Adaptors Negatively Regulate Actin Polymerization in Pedestals Formed by Enteropathogenic Escherichia coli (EPEC) by Binding to Tir Effector. PLoS Pathog 2014; 10:e1004022; PMID:24675776; http://dx.doi.org/10.1371/journal.ppat.1004022
- Campellone KG, Giese A, Tipper DJ, Leong JM. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol Microbiol 2002; 43:1227-41; PMID:11918809; http://dx.doi.org/10.1046/j.1365-2958.2002.02817.x
- Campellone KG, Rankin S, Pawson T, Kirschner MW, Tipper DJ, Leong JM. Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J Cell Biol 2004; 164:407-16; PMID:14757753; http://dx.doi.org/10.1083/jcb.200306032
- Campellone KG, Leong JM. Nck-independent actin assembly is mediated by two phosphorylated tyrosines within enteropathogenic Escherichia coli Tir. Mol Microbiol 2005; 56:416-32; PMID:15813734; http://dx.doi.org/10.1111/j.1365-2958.2005.04558.x
- Kenny B, Warawa J. Enteropathogenic Escherichia coli (EPEC) Tir receptor molecule does not undergo full modification when introduced into host cells by EPEC-independent mechanisms. Infect Immun 2001; 69:1444-53; PMID:11179311; http://dx.doi.org/10.1128/IAI.69.3.1444-1453.2001
- Wolff C, Nisan I, Hanski E, Frankel G, Rosenshine I. Protein translocation into host epithelial cells by infecting enteropathogenic Escherichia coli. Mol Microbiol 1998; 28:143-55; PMID:9593303; http://dx.doi.org/10.1046/j.1365-2958.1998.00782.x
- Liberek K, Georgopoulos C, Zylicz M. Role of the Escherichia coli DnaK and DnaJ heat shock proteins in the initiation of bacteriophage lambda DNA replication. Proc Natl Acad Sci U S A 1988; 85:6632-6; PMID:2970643; http://dx.doi.org/10.1073/pnas.85.18.6632
- Ruchaud-Sparagano MH, Maresca M, Kenny B. Enteropathogenic Escherichia coli (EPEC) inactivate innate immune responses prior to compromising epithelial barrier function. Cell Microbiol 2007; 9:1909-21; PMID:17388785; http://dx.doi.org/10.1111/j.1462-5822.2007.00923.x
- Rosenshine I, Ruschkowski S, Finlay BB. Expression of attaching/effacing activity by enteropathogenic Escherichia coli depends on growth phase, temperature, and protein synthesis upon contact with epithelial cells. Infect Immun 1996; 64:966-73; PMID:8641808
- Schuller S, Chong Y, Lewin J, Kenny B, Frankel G, Phillips AD. Tir phosphorylation and Nck/N-WASP recruitment by enteropathogenic and enterohaemorrhagic Escherichia coli during ex vivo colonization of human intestinal mucosa is different to cell culture models. Cell Microbiol 2007; 9:1352-64; PMID:17474908; http://dx.doi.org/10.1111/j.1462-5822.2006.00879.x
- Brandt S, Kenny B, Rohde M, Martinez-Quiles N, Backert S. Dual infection system identifies a crucial role for PKA-mediated serine phosphorylation of the EPEC-Tir-injected effector protein in regulating Rac1 function. Cell Microbiol 2009; 11:1254-71; PMID:19438518; http://dx.doi.org/10.1111/j.1462-5822.2009.01330.x
- Creasey EA, Delahay RM, Bishop AA, Shaw RK, Kenny B, Knutton S, Frankel G. CesT is a bivalent enteropathogenic Escherichia coli chaperone required for translocation of both Tir and Map. Mol Microbiol 2003; 47:209-21; PMID:12492865; http://dx.doi.org/10.1046/j.1365-2958.2003.03290.x
- Kubori T, Galan JE. Temporal regulation of Salmonella virulence effector function by proteasome-dependent protein degradation. Cell 2003; 115:333-42; PMID:14636560; http://dx.doi.org/10.1016/S0092-8674(03)00849-3
- Thomas NA, Deng W, Puente JL, Frey EA, Yip CK, Strynadka NC, Finlay BB. CesT is a multi-effector chaperone and recruitment factor required for the efficient type III secretion of both LEE- and non-LEE-encoded effectors of enteropathogenic Escherichia coli. Mol Microbiol 2005; 57:1762-79; PMID:16135239; http://dx.doi.org/10.1111/j.1365-2958.2005.04802.x
- Savkovic SD, Ramaswamy A, Koutsouris A, Hecht G. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am J Physiol 2001; 281:G890-8;
- Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, Deng W, Lamkanfi M, Kanneganti TD. Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem 2012; 287:16955-64; PMID:22461621; http://dx.doi.org/10.1074/jbc.M112.358705
- Tsuji N, Kobayashi M, Nagashima K, Wakisaka Y, Koizumi K. A new antifungal antibiotic, trichostatin. J Antibiot (Tokyo) 1976; 29:1-6; PMID:931784; http://dx.doi.org/10.7164/antibiotics.29.1
- Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. Embo J 2010; 29:969-80; PMID:20075865; http://dx.doi.org/10.1038/emboj.2009.405
- Rivera GM, Briceno CA, Takeshima F, Snapper SB, Mayer BJ. Inducible clustering of membrane-targeted SH3 domains of the adaptor protein Nck triggers localized actin polymerization. Curr Biol: CB 2004; 14:11-22; http://dx.doi.org/10.1016/j.cub.2003.12.033
- Crepin VF, Girard F, Schuller S, Phillips AD, Mousnier A, Frankel G. Dissecting the role of the Tir:Nck and Tir:IRTKS/IRSp53 signalling pathways in vivo. Mol Microbiol 2010; 75:308-23; PMID:19889090; http://dx.doi.org/10.1111/j.1365-2958.2009.06938.x
- Berger CN, Crepin VF, Jepson MA, Arbeloa A, Frankel G. The mechanisms used by enteropathogenic Escherichia coli to control filopodia dynamics. Cell Microbiol 2009; 11:309-22; PMID:19046338; http://dx.doi.org/10.1111/j.1462-5822.2008.01254.x
- Orchard RC, Kittisopikul M, Altschuler SJ, Wu LF, Suel GM, Alto NM. Identification of F-actin as the dynamic hub in a microbial-induced GTPase polarity circuit. Cell 2012; 148:803-15; PMID:22341450; http://dx.doi.org/10.1016/j.cell.2011.11.063
- Vingadassalom D, Campellone KG, Brady MJ, Skehan B, Battle SE, Robbins D, Kapoor A, Hecht G, Snapper SB, Leong JM. Enterohemorrhagic E. coli requires N-WASP for efficient type III translocation but not for EspFU-mediated actin pedestal formation. PLoS Pathog 2010; 6(8):e1001056; PMID:20808845; http://dx.doi.org/10.1371/journal.ppat.1001056
- Campellone KG, Robbins D, Leong JM. EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev cell 2004; 7:217-28; PMID:15296718; http://dx.doi.org/10.1016/j.devcel.2004.07.004
- Rendi R, Ochoa S. Effect of chloramphenicol on protein synthesis in cell-free preparations of Escherichia coli. J Biol Chem 1962; 237:3711-3; PMID:13981641
- Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, Osada H, Komatsu Y, Nishino N, Khochbin S, et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. Embo J 2002; 21:6820-31; PMID:12486003; http://dx.doi.org/10.1093/emboj/cdf682
- Hoshikawa Y, Kwon HJ, Yoshida M, Horinouchi S, Beppu T. Trichostatin A induces morphological changes and gelsolin expression by inhibiting histone deacetylase in human carcinoma cell lines. Exp Cell Res 1994; 214:189-97; PMID:8082721; http://dx.doi.org/10.1006/excr.1994.1248
- Goosney DL, DeVinney R, Finlay BB. Recruitment of cytoskeletal and signaling proteins to enteropathogenic and enterohemorrhagic Escherichia coli pedestals. Infect Immun 2001; 69:3315-22; PMID:11292754; http://dx.doi.org/10.1128/IAI.69.5.3315-3322.2001
- Snapper SB, Takeshima F, Anton I, Liu CH, Thomas SM, Nguyen D, Dudley D, Fraser H, Purich D, Lopez-Ilasaca M, et al. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat Cell Biol 2001; 3:897-904; PMID:11584271; http://dx.doi.org/10.1038/ncb1001-897
- Bladt F, Aippersbach E, Gelkop S, Strasser GA, Nash P, Tafuri A, Gertler FB, Pawson T. The murine Nck SH2/SH3 adaptors are important for the development of mesoderm-derived embryonic structures and for regulating the cellular actin network. Mol Cell Biol 2003; 23:4586-97; PMID:12808099; http://dx.doi.org/10.1128/MCB.23.13.4586-4597.2003
- DeVinney R, Puente JL, Gauthier A, Goosney D, Finlay BB. Enterohaemorrhagic and enteropathogenic Escherichia coli use a different Tir-based mechanism for pedestal formation. Mol Microbiol 2001; 41:1445-58; PMID:11580847; http://dx.doi.org/10.1046/j.1365-2958.2001.02617.x
- Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect Immun 1991; 59:4310-7; PMID:1937792; .
- Kenny B, Abe A, Stein M, Finlay BB. Enteropathogenic Escherichia coli protein secretion is induced in response to conditions similar to those in the gastrointestinal tract. Infect Immun 1997; 65:2606-12; PMID:9199427
- Dean P, Kenny B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Mol Microbiol 2004; 54:665-75; PMID:15491358; http://dx.doi.org/10.1111/j.1365-2958.2004.04308.x
- Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, Kirschner MW. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 1999; 97:221-31; PMID:10219243; http://dx.doi.org/10.1016/S0092-8674(00)80732-1
- Kenny B, Ellis S, Leard AD, Warawa J, Mellor H, Jepson MA. Co-ordinate regulation of distinct host cell signalling pathways by multifunctional enteropathogenic Escherichia coli effector molecules. Mol Microbiol 2002; 44:1095-107; PMID:12046591; http://dx.doi.org/10.1046/j.1365-2958.2002.02952.x
- Ching JC, Jones NL, Ceponis PJ, Karmali MA, Sherman PM. Escherichia coli shiga-like toxins induce apoptosis and cleavage of poly(ADP-ribose) polymerase via in vitro activation of caspases. Infect Immun 2002; 70:4669-77; PMID:12117981; http://dx.doi.org/10.1128/IAI.70.8.4669-4677.2002
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell 2007; 27:197-213; PMID:17643370; http://dx.doi.org/10.1016/j.molcel.2007.05.033