
Abstract
Epithelial-mesenchymal transition (EMT) refers to plastic changes in epithelial tissue architecture. Breast cancer stromal cells provide secreted molecules, such as transforming growth factor β (TGFβ), that promote EMT on tumor cells to facilitate breast cancer cell invasion, stemness and metastasis. TGFβ signaling is considered to be abnormal in the context of cancer development; however, TGFβ acting on breast cancer EMT resembles physiological signaling during embryonic development, when EMT generates or patterns new tissues. Interestingly, while EMT promotes metastatic fate, successful metastatic colonization seems to require the inverse process of mesenchymal-epithelial transition (MET). EMT and MET are interconnected in a time-dependent and tissue context-dependent manner and are coordinated by TGFβ, other extracellular proteins, intracellular signaling cascades, non-coding RNAs and chromatin-based molecular alterations. Research on breast cancer EMT/MET aims at delivering biomolecules that can be used diagnostically in cancer pathology and possibly provide ideas for how to improve breast cancer therapy.
Abbreviations
| bHLH | = | basic helix-loop-helix |
| BMP | = | bone morphogenetic protein |
| CSC | = | cancer stem cell |
| DNMT | = | DNA methyltransferase |
| EMT | = | epithelial-mesenchymal transition |
| FGF | = | fibroblast growth factor |
| HDAC | = | histone deacetylase |
| lncRNA | = | long non-coding RNA |
| mTORC | = | mammalian target of rapamycin complex |
| MET | = | mesenchymal-epithelial transition |
| miRNA | = | micro-RNA |
| MAPK | = | mitogen activated protein kinase |
| PDGF | = | platelet derived growth factor |
| PRC | = | polycomb repressive complex |
| TF | = | transcription factor |
| TGFβ | = | transforming growth factor β |
Introduction
Cancer is a complex disease with a long evolutionary trajectory stemming from the introduction of genetic mutations in normal cells to the biochemical alterations of chromatin, signaling pathways and cell biological processes. The final manifestation of this disease involves malignant alterations in tumor cell differentiation, migration and miscommunication with the immune and other systems that survey the homeostasis of the organism. The epithelial-mesenchymal transition (EMT) is a biological process that takes place during organismal development, with prominent examples being the formation of the primitive streak, the neural crest delamination, heart valve differentiation and lung organogenesis.Citation1 EMT has also been described in the pathology of fibrotic disorders and cancer, as a mechanism that links the effects of chronic inflammation to the wounding of tissues, resulting in epithelial cell dedifferentiation, deposition of excessive extracellular matrix and acquisition of motile properties by cells that otherwise are destined to be relatively immotile, as a consequence of gradual loss of proper tissue architecture.Citation2,3 Epithelial cells alter their apico-basal polarity, remodel their intermediate filaments and microfilaments and intercellular adhesions, secrete new matrix proteins and glycosaminoglycans, and acquire a mesenchymal state that correlates to enhanced migratory capacity.Citation1 In addition, in the context of cancer, EMT reprograms epithelial cells and generates cancer stem cells (CSCs) that exhibit resistance to chemical or radiological therapy or senescence induced by oncogenic stimuli, and provide necessary attributes that promote tumor cell metastasis.Citation4-6 An important feature of cells undergoing EMT during embryonic development or cancer progression is the reversibility of the process (mesenchymal-epithelial transition, MET), which generates new epithelial cells that build new embryonic tissues or metastatic colonies of secondary tumor growth. Because of the prominence of cell biological and molecular studies on the process of EMT in breast cancer, the present article focuses on this type of malignancy; however, many of the principles that govern EMT appear to be universal and independent from tissue type.
Growth and Transcriptional Factors Governing the EMT Program
One of the reasons behind the universal principles that guide EMT processes is the dependency of these processes on a cohort of secreted growth factors, cytokines and intracellular transcription factors that execute the signals provided by the growth factors. The list of the extracellular guiding cues that initiate and maintain the EMT process over time is large, and includes environmental factors such as smoking and ultraviolet radiation, tissue-specific pathological conditions such as hypoxia and associated cytokines including the transforming growth factor β (TGFβ), fibroblast growth factor (FGF), Notch, insulin-like growth factor, Wnt, interleukin-like EMT inducer and more.Citation1 This example nicely illustrates the principle of inter-dependent cytokine requirement for EMT to get established in certain cases of breast cancer that exhibit estrogen receptor deregulation: the reduced expression of estrogen receptors in breast cancer and the development of hypoxia lead to induced secretion and extracellular activation of TGFβ, which starts a cascade of signaling events that include the pathways of Notch, interleukin-like EMT inducer and insulin-like growth factor, which cause secretion and activation of the vascular endothelial growth factor, catalyzing neo-angiogenesis that fosters sustained EMT and subsequent tumor cell extravasation and metastasis.Citation7
The EMT program can be triggered by a specific group of transcription factors in response to the above signaling pathways (e.g., TGFβ, FGF, Notch, etc.) during development or cancer progression. These EMT transcription factors (EMT-TFs) coordinately regulate the repression of epithelial genes and upregulation of mesenchymal genes, thus promoting migratory and invasive cellular behavior. The well-studied classical EMT-TFs are the Snail, ZEB and Twist family members, which converge to repress E-cadherin – a key feature of EMT.Citation8,9 Snail1 (Snail) and Snail2 (Slug) are zinc finger proteinsCitation10-12; ZEB1 (δEF1) and ZEB2 (SIP1) are zinc finger and homeobox domain proteins;Citation13,14 and Twist1 and Twist2 are basic helix-loop-helix proteins (bHLH).Citation15,16 Additional bHLH family members, E47 and E2-2, are also known to induce EMT.Citation17
The embryonic chromatin protein high mobility group A2 (HMGA2) integrates EMT signals downstream of TGFβ by induction of Snail1, Snail2, Twist1 and repression of inhibitor of differentiation 2 (ID2).Citation18-20 Another transcriptional regulator induced by TGFβ is Sox4, a transcription factor of the SRY-related HMG-box family and is also involved in embryonic development.Citation21,22 Sox4 regulates the expression of the classical EMT-TFs in mammary epithelial cells during EMT. The EMT-TFs also regulate the expression of each other in a feed-forward loop,Citation18,23,24 establishing the EMT nuclear reprogramming that is reminiscent of the embryonic stem cell fate regulatory circuitry controlled by master transcription factors, Oct4, Nanog and Sox2.Citation25
The complexity of signaling crosstalk during EMT can be presented by studying the TGFβ and Wnt pathways. To present deeper analysis of the crosstalk in this section, we diverge slightly and cite EMT cases from organs other than the breast. Components of the Wnt pathway, namely, the adaptor protein axin, the protein kinase glycogen synthase kinase 3β (GSK3β) and β-catenin interact and modulate the activity of Smad proteins of the TGFβ pathway in the cytoplasm or in the nucleus. Cytoplasmic axin associates with Smad3 and facilitates Smad3 phosphorylation and overall efficiency of downstream transcriptional responses to TGFβ.Citation26 On the other hand, cytoplasmic axin brings GSK3β to Smad3, which phosphorylates Smad3 and induces its proteasomal degradation thus keeping the steady state pool of Smad3 under control.Citation27 This mechanism leads to general control of Smad3 signaling by axin/GSK3β. In the mammary EpH4 cell model of EMT, oncogenic activation (e.g., by an inducible c-Fos-estrogen receptor transgene) caused activation of the nuclear β-catenin/T-cell-specific transcription (TCF) 4 complex together with the autocrine activation of TGFβ and downstream Smad nuclear complexes that mediate mesenchymal gene expression.Citation28 During endothelial-mesenchymal transition (EndoMT) that forms the valves in the heart in response to TGFβ2, EMT induced by TGFβ3 in the palate, or EMT developed during lung fibrosis, β-catenin/TCF complexes are critical, together with Smads for the induction of mesenchymal genes in the transforming endothelial cells or for the repression of E-cadherin in the regressing epithelial cells of the palate.Citation29-31 These examples of EndoMT will be discussed further later. In the case of lung EMT after inflammation, a specific protein complex between Smad2 and phosphorylated β-catenin on tyrosine 654 that is mediated by activation of integrin-α3β1 is a crucial determinant that controls E-cadherin repression.Citation29 In a similar manner, Smad3 can facilitate nuclear accumulation of β-catenin during proliferation and maintenance of adult mesenchymal stem cells.Citation32 The TGFβ-Wnt signaling network does not only contribute to the generation of transcriptional complexes between Smads and β-catenin, but also contributes to the regulation of Snail stability and transcriptional activity since Snail is a direct target of phosphorylation by the GSK3β kinase.Citation33 The result of such molecular interactions between TGFβ and Wnt signaling components is evident in numerous cases of epithelial tumorigenesis. In mouse models of breast and intestinal cancer that are driven by Wnt ligand overactivation, a high degree of synergism between Wnt and TGFβ signaling was shown to contribute to the progression of tumorigenesis.Citation34 In breast cancer models, the non-canonical Wnt5a, among all Wnt family ligands, appears to play critical roles in cooperation with or acting upstream of TGFβ signaling. TGFβ signaling controls Wnt5a expression in the mammary gland and Wnt5a signaling depends on TGFβ receptor function.Citation35 According to this mechanism, TGFβ and non-canonical Wnt5a act as tumor suppressors and when these 2 factors are lost, breast cancers gain stable canonical Wnt/β-catenin nuclear activity that promotes tumor growth.Citation36 However, it should be mentioned that Wnt5a is required for tumor cell invasion and CSC properties in basal-like breast cancer.Citation37 In the prostate, upon androgen reduction, TGFβ gets elevated and the resulting hyperproliferation of prostate epithelial cells often leads to tumorigenesis, a mechanism that depends on expression and signaling by multiple Wnt family ligands.Citation38 In vitro EMT caused by the cooperation between Ras and TGFβ signaling leads to the switch from cadherin-dependent adhesion to integrin receptor-dependent adhesion and this process coordinates with the secretion of Wnt5a and the activity of the non-canonical Wnt pathway of planar cell polarity. Finally, the link between EMT and cancer cell stemness in breast cancer cell models has also been shown to be mediated by the coordinate activity of TGFβ, secreted non-canonical Wnt members and inhibitors of bone morphogenetic protein (BMP) signaling that act in an autocrine manner to promote cancer stemness.Citation39 These and numerous other experimental examples enlighten our understanding of the molecular networks that control the EMT process in the breast and many other organs.Citation9
General Aspects of TGFβ-Induced EMT
The links between TGFβ signaling and EMT were developed since the early days of the molecular dissection of this signal transduction pathway and have been firmly established in the field through a large set of solid publications.Citation40,41 Early studies of TGFβ signaling in mouse mammary epithelial cells, a) Namru murine mammary gland, NMuMG cells, which are non-tumorigenic, showed time-dependent and reversible EMT in response to TGFβCitation42-45; b) in EpRas cells, which are mouse mammary EpH4 cells transformed by the H-Ras oncogene, showed a more stable EMT that is dependent on autocrine TGFβ and also developed tumors in mouse xenografts.Citation46 TGFβ could elicit all the hallmarks of EMT such as loss of cell-cell contacts, including adherens and tight junction disassembly, cytoskeletal adaptations including focal adhesion-bound actin stress fibers, intense perinuclear vimentin filaments accompanied by gradual loss of cytokeratin filaments, spindle-like morphology and enhanced motility.Citation43 The complex cell biology of the EMT process can be relatively well understood and classified when viewed as a program of cell differentiation emanating under the guidance of a single growth factor like TGFβ. According to this notion, the EMT program can be divided to specific subprograms () that include the above-mentioned junctional complex and cytoskeletal programs, but also 2 additional subprograms, the secreted factor and extracellular matrix programs.Citation41 In brief, TGFβ induces the synthesis and secretion of several other growth factors, cytokines and chemokines and many of these collectively contribute to the establishment of EMT, tumor cell invasiveness and metastasis. Some of these growth factors optimally operate in a remodeled extracellular matrix that is composed by newly synthesized and deposited proteins or glycosaminoglycans, that mediate new cell-cell interactions, paracrine cell activation and enhanced cell migration.Citation47
Figure 1. Mechanisms in TGFβ-induced EMT and BMP-induced MET. A mammary epithelial cell is shown on top with the 2 sister signaling pathways of TGFβ and BMP. These 2 growth factors are color coded to match the process of EMT and MET respectively. Signaling by the dimeric extracellular proteins is mediated by plasma membrane receptors (type II (RII) and type I (RI), which phosphorylate Smad proteins or activate other non-Smad signaling proteins (mainly protein kinases shown in gray boxes). Phosphorylation events on the type I receptor or on the Smads are shown with a black circled white P. Smads form complexes with Smad4 and together with non-Smad kinases transmit signals into the nucleus where different target genes are regulated positively (black arrows) or negatively (red arrows). Target genes of the pathways are symbolized as circles on the chromatin thread and are divided for simplicity and based on the text in 4 groups: a) miRNA and lncRNA genes (pink); b) co-transcription factor genes (orange), e.g., HMGA2, C/EBP or IDs in the case of BMP signaling; c) EMT-TF genes (purple); d) effector genes including secreted polypeptides, junctional, extracellular matrix and cytoskeletal components (yellow). MiRNAs are shown to negatively regulate various other target mRNAs. LncRNAs are shown to participate with chromatin regulators (black box on the left). These chromatin regulators participate in the chromatin-based epigenetic control of every step in the signaling and gene regulatory cascade (not shown). Make note of the sequential regulatory cascades whereby for example, the effector (yellow) gene is regulated by incoming Smad and non-Smad signaling, co-TFs and EMT-TFs, together with the conserted action of the lncRNA/chromatin factors (not shown). Such signaling mechanisms lead to EMT or MET leading to changes of cell architecture and differentiation as depicted at the bottom of the figure. Platelets and other cell types assist to the process of EMT, whereas the contribution of accessory cell types that promote MET is not well investigated (question mark). The generation of cancer stem cells is indicated with dark pink color and the metastatic breast carcinoma epithelial cells are colored as dark pink to indicate their development via MET from the CSCs. The latter is a speculative hypothesis and not yet demonstrated experimentally. Note the lack of normal basement membrane in the metastatic epithelial colony, which is also hypothetical and aims at emphasizing that the metastatic carcinoma cells and the epithelial cells in the primary cancer may not generate identically organized tissue architecture.
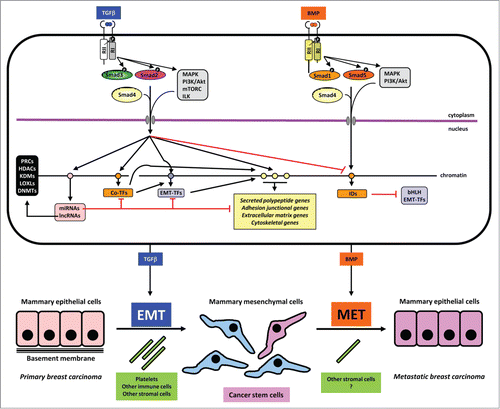
In this section we will diverge again from discussing strictly breast-related EMT. While the majority of experimental studies of EMT focus on the prototype isoform TGFβ1, the first demonstration of a link between TGFβ and EMT came from studies of EndoMT that generates the cushions in the heart valves, a process operating under the guidance of the TGFβ2 isoform.Citation48 Thus, TGFβ2 initiates the dissociation of adherent endothelial cells, and induces expression of TGFβ3, which promotes myoepithelial cell invasion into the developing tissue in chicken embryos. Species-specific differences are known to exist with respect to the necessity and the developmental timing during which these different TGFβ isoforms become activated and functional.Citation48 Accordingly, mouse TGFβ2 appears to be self-sufficient and does not require TGFβ3. Careful analysis of all 3 TGFβ isoforms using the respective knockout mice, showed that normal heart valve EndoMT can occur in the complete absence of TGFβ1 or TGFβ3, whereas knockout of TGFβ2 led to a defective EMT in assays of explanted embryonic heart tissue, which corroborates the in vivo phenotype of the TGFβ2 knockout mice that exhibit enlarged heart cushions.Citation49 Downstream of TGFβ2 lies Snail1 at least in mouse endothelial cells, which contributes to the upregulation of many mesenchymal genes, including α-smooth muscle actin (α-SMA), SM22α and calponin.Citation50 In mice, the TGFβ2 signal also cooperates with the Wnt, β-catenin/TCF4 transcriptional complex to mediate the EndoMT and in vitro experiments using endothelial cells showed that α-SMA expression is regulated by crosstalk between TGFβ2 and β-catenin/TCF4,Citation30 as mentioned above. While TGFβ2 seems to initiate the EndoMT in the heart valves, invasiveness depends on BMP2 signaling derived from myocardial cells and Snail1 activity that is stabilized after BMP2 exposure, both events lying under the control of Notch1 signaling, which operates in the adjacent endocardial tissue.Citation51 These 2 cytokines, TGFβ2 and BMP2, seem to contribute to EndoMT and invasiveness by binding to the type III co-receptor β-glycan, which coordinates signaling by both TGFβ-specific type I receptors, activin receptor-like 5 (ALK5), and BMP-specific type I receptors, ALK2 and ALK3.Citation52 Upstream of the action of TGFβ2 lies the MAP kinase kinase MEKK3, whose signaling in the developing heart of mice regulates the transcriptional induction of TGFβ2.Citation53 Thus, heart valve morphogenesis is a good example of TGFβ2-dependent EMT.
Another tissue that shows isoform-specific dependence for its EMT is the palatal epithelium that undergoes EMT in order for the 2 sides of the epithelium to fuse and generate the intermediary seam. TGFβ3 is the key player in this process, which causes the EMT by repressing E-cadherin via a nuclear Smad-TCF4 transcriptional complex on the E-cadherin promoter region as explained above.Citation31 Not only does TGFβ3 promote the formation of the Smad-β-catenin-TCF4 complexes, but also induces the levels of TCF4 at the transcriptional level.Citation54 In addition to Smads, non-Smad signaling contributes to the stabilization and activation of Snail1 and ZEB2 during palatal EMT, and additionally, the small GTPase Rho and its downstream Rho kinase contribute to mesenchymal phenotypic changes in the palate, including actin cytoskeleton remodeling.Citation55,56
Some additional examples of EMT can be listed based on their sensitivity to different TGFβ isoforms. Similar to the heart, pulmonary valve morphogenesis requires EndoMT, which is driven by TGFβ2 and is antagonized by the pro-angiogenic factor VEGF-A.57 Retinal epithelial cells undergo EMT at least in vitro and in vivo during fibrosis that leads to retinopathies, and this EMT is driven by TGFβ2, which then induces expression of the Notch ligand Jagged1, that synergistically with TGFβ2-Smad and non-Smad signaling promotes the expression of EMT-TFs that mediate the mesenchymal transition.Citation58 In the context of cancer-related EMT, all 3 TGFβ isoforms can induce hepatocarcinoma EMT that is mediated by downstream induction and signaling of the platelet derived growth factor (PDGF) A.Citation59 In EMT and metastatic spreading of breast cancer with oncogenic phosphatidylinositol 3’ kinase (PI3K)/Akt pathway activation, TGFβ2 activity is required; Akt directly phosphorylates Twist1, causing its stabilization and transcriptional activation, and leading to the enhanced expression of the TGFβ2 gene.Citation60 Finally, EMT and MET cycles are controlled by the reciprocal loop between TGFβ and members of the miR-200 family.Citation61,62 In zebrafish, the tyrosine phosphatase PEZ regulates the transcriptional expression of TGFβ3, which then drives several cases of developmental EMT in the heart, pharynx and in somites.Citation61 In several tumor cells that exhibit EMT, ZEB1 directly represses the expression of miR-200 family members, and these microRNAs (miRNAs) negatively regulate expression of TGFβ2, among many other targets.Citation62 Thus, the EMT-TF network facilitates the further expression and activity of specific TGFβ isoforms during cancer progression toward metastasis,Citation62 or during tissue fibrosis such as in the kidney.Citation63
It is worth pointing out 2 aspects of TGFβ cell physiology that relate to the process of EMT and often generate conflicting views on the topic: a) TGFβ is known to arrest the cell cycle of normal mammary epithelial cells at the early G1 phase.Citation45 Thus, at least in vitro, mammary epithelial cells undergo EMT while their cell cycle is arrested. This is in agreement with the general view that EMT correlates with cell cycle arrest and enhanced survival.Citation7 b) However, the pro-EMT action of TGFβ represents a pro-tumorigenic and pro-metastatic aspect of the biology of this growth factor in the context of cancer.Citation64 In the sequence of multi-stage events of cancer progression, the pro-tumorigenic TGFβ actions are usually thought to follow the tumor suppressive effects that TGFβ exhibits as a factor that maintains epithelial tissue homeostasis.Citation64 In other words, the pro-tumorigenic, pro-EMT effects of TGFβ, at the cellular level correlate to one of the hallmarks of the tumor suppressive effects TGFβ has. This complicated concept is explained by the observation that long-term exposure to TGFβ bypasses the initial growth arrest response of mammary epithelial cells in culture, and proceeds with sustained cell proliferation in the presence of a mesenchymal phenotype.Citation42 This adaptive response of breast cells is dependent on multiple signaling inputs in addition to TGFβ.Citation7,42 In other words, while TGFβ arrests the mammary epithelial cell cycle and initiates EMT, concomitant exposure of cells to other cytokines and growth factors in the context of the tumor microenvironment ensure the progression of carcinogenesis and eventually help so that breast cancer cells maintain mesenchymal features while having bypassed the proliferation arrest that is imposed on their epithelial progenitors by TGFβ.
It is therefore appropriate to briefly discuss the 2 faces of TGFβ biology during cancer progression. The current state of the art proposes that tumor suppressive mechanisms induced by TGFβ in the breast and many other epithelial tissues involve the transcriptional and Smad-mediated upregulation of cell cycle inhibitor genes such as p15 and p21, which block the cell cycle at the early G1 phase.Citation65,66 In addition, a critical prerequisite of cell cycle inhibitor gene upregulation is the transcriptional repression of c-myc, a condition that is often bypassed in breast cancers with oncogenic myc activation or overexpression.Citation65 It is worth emphasizing that epithelial cells where c-myc is overexpressed do not respond anymore to TGFβ in terms of stopping their cell cycle, however, they maintain the EMT response, as c-myc redirects Smad complexes to the Snail1 gene causing the induction of EMT.Citation67 Many tissue-specific knockout mouse models have also been generated that clearly demonstrate the tumor suppressor activity of TGFβ in a variety of organs, including the breast.Citation64 Some of these models have made clear that the more sensitive cell type to the tumor-derived TGFβ are the resident fibroblasts of the tumor stroma or immune cells, such as T lymphocytes that contribute to enhanced tumor growth and cancer progression when TGFβ receptor or Smad signaling is lost in these cells.Citation68
The other face of TGFβ in tumor progression is a pro-tumorigenic that seems to be more universal and usually is linked to every tumor type studied so far. Primary determinants of this activity of TGFβ are: a) the potent immunosuppressive action of TGFβ, which inhibits proliferation and differentiation of B and T lymphocytes, and thus, tumor-derived TGFβ generates a locally immunocompromized microenvironment that is beneficial to the advancement of tumorigenesisCitation68; b) the EMT that can be mediated either in an autocrine or paracrine manner, and associated with EMT, the enhanced abundance of cancer stem cells and metastatic stem cells that disseminate more effectively to distant sites of tumor spreadCitation41; c) the pro-angiogenic effects of TGFβ, which are usually indirect and mediated by the transcriptional induction of VEGFs and Notch family ligands that ensure a more robust tumor vasculature.Citation64 Thus, such pro-tumorigenic and pro-metastatic actions of TGFβ seem to overtake and last for the lengthier period of tumor progression.
Smad and Non-Smad Signaling in EMT
TGFβ makes a remarkable inducer of EMT because it involves both Smad and non-Smad signaling () to regulate genes controlling cell motility and invasion, by remodeling the actin cytoskeleton and extracellular matrix.Citation41,47,69 Smad proteins play a vital role in TGFβ-induced EMT as inhibition of Smad2, Smad3 and Smad4 functions, and overexpression of the negative regulator Smad7, blocked EMT in NMuMG cells.Citation45,70 TIF1γ (TRIM33), a histone binding protein, antagonizes EMT by competing with Smad4 for binding of active Smad2/3 complexes.Citation71 In addition, Smad proteins form complexes with members of the AP1 family to induce genes related to invasiveness, e.g., matrix metalloproteinase genes MMP2 and MMP9, during EMT.Citation72
The robust EMT response to TGFβ in NMuMG cells could be attributed to the genotype of these cells, which harbor one wild-type and one mutant p53 R277C allele. Silencing of the wild-type p53 allele hastened the onset of TGFβ-induced EMT, whereas p53 stabilization by nutlin counteracted the EMT effects.Citation73 The p53 protein blocks EMT by upregulating miR-34 and miR-200, which target Snail1 and ZEBs, respectively.Citation74,75 In agreement with this, molecular analysis of p53-null breast cancers classified them as having the claudin-low tumor gene signature, that is, cancers with dominant EMT and cancer stem cell characteristics.Citation76
In addition, TGFβ directly regulates the majority of the EMT-TFs.Citation41 We found that TGFβ, via Smad signaling, upregulates HMGA2 to drive EMT,Citation19 and this has been complemented independently in vivo as HMGA2 overexpression causes metastasis,Citation77 further reinstating the link between EMT and metastasis. A related family member HMGA1, overexpressed in basal-like breast cancers, is required for EMT and self-renewal to promote metastasis.Citation78 Snail1 is another direct target of TGFβ/Smad signaling and of HMGA2.Citation20,79 Furthermore, Smads and Snail1 interact and are recruited to repress E-cadherin and coxsackie and adenovirus receptor (CAR), a tight junction protein.Citation80 Likewise, ZEB1 and ZEB2 are upregulated by TGFβ and also able to interact with Smad proteins.Citation81,82
EMT can be induced via the non-canonical TGFβ signaling pathway that signals in parallel to Smad activation downstream of the 2 TGFβ receptors, the type II (TβRII) and type I (TβRI)/ALK5 receptor serine/threonine kinases. TGFβ activates the mitogen activated protein kinase (MAPK) and PI3K pathways () to regulate cytoskeleton organization, migration and invasion, which are characteristics of the EMT response.Citation83 TGFβ receptors signal through the ShcA adaptor protein to activate the ERK MAPK and p38 MAPK pathways to promote breast cancer cell proliferation and invasion.Citation84,85 TβRI recruits the TRAF6 ubiquitin ligase and becomes polyubiquitinated, leading to the cleavage of the TβRI intracellular domain, which enters the nucleus to activate Snail1 and MMP2 genes.Citation86 TGFβ upregulates Sox4, in a Smad-independent manner, to regulate a group of mesenchymal genes and the histone methyltransferase EZH2 during EMT.Citation21,22 Furthermore, Sox4 creates a feedback loop by activating canonical TGFβ signaling to maintain EMT.Citation22 Overexpression of Sox4 is often found associated with the aggressive, triple-negative breast cancer subtype.Citation22 TGFβ disrupts cell polarity during EMT when it activates TβRII, which phosphorylates the polarity complex protein Par6 to recruit the Smurf1 ubiquitin ligase. Smurf1 ubiquitinates and targets RhoA for degradation, destabilizing the actin filament network and thereby dissolving the tight junctions.Citation87
Downstream of the PI3K lies the Akt signaling pathway that plays an important role in cell survival, motility and metastasis. Inactivating mutations in PTEN, the phosphatase responsible for counteracting PI3K activity, is commonly found in human malignancies. Although several features, like the mechanism of activation, are shared between the 3 isoforms of Akt (Akt1-3), distinct signaling effects downstream of the specific isoforms are beginning to emerge.Citation88 In a study by Arboleda and colleagues Akt1-3 were overexpressed in breast and ovarian cancer cells. Only Akt2 overexpressing cells displayed increased invasive behavior in an in vitro invasion assay.Citation89 When injected into the mammary fat pad of immunocompromized mice, Akt2 transfected MDA-MB-435 breast cancer cells showed a higher number of lung metastases compared to the parental cell line. This effect was abrogated when a kinase-dead Akt2 was expressed. These findings indicate that Akt2 needs to be activated for metastasis to occur. In agreement, overexpression of active Akt2 in 2 different transgenic mammary carcinoma models was found to markedly increase the incidence of pulmonary metastases, in contrast to overexpression of active Akt1.Citation90 The pro-metastatic properties of Akt2 may reside in its capacity to regulate the activity of Twist1 and Snail1, 2 classical EMT-TFs.Citation91,92 Expression of both Twist1 and Akt2 was found to be elevated in highly invasive breast cancer cells,Citation91 and silencing of Akt2 decreased Twist1-induced migration and invasion. Moreover, Twist1 was found to bind E-box elements in the Akt2 promoter and thereby enhance its expression.Citation91 Snail1 and Akt2 have been shown to directly interact in the cell nucleus, an interaction that requires the C-terminal half of Snail1 and the plekstrin homology-domain in Akt2.Citation92 Snail1 and Akt2 bind together to the E-cadherin promoter and cooperate to repress expression of E-cadherin. Silencing of Akt2 prevents Snail-induced downregulation of E-cadherin.Citation92 Finally, Iliopoulos and colleagues showed that the miR-200a family regulates E-cadherin repression and EMT.Citation93 The amount of miR-200a present in a cell was shown to be dependent on the balance between Akt1 and Akt2 expression. A low ratio of Akt1 to Akt2 promotes EMT by reducing the level of miR-200a, again highlighting Akt2 as a driver of EMT.
Activation of the PI3K pathway by TGFβ additionally leads to signaling via the mammalian target of rapamycin complexes -1 and -2 (mTORC1 and mTORC2). The activity of mTORC2, not mTORC1, is essential during TGFβ-induced EMT.Citation94,95 However, mTORC1 activation by TGFβ, in addition to increasing protein synthesis and cell size, promotes migratory and invasive behavior of mammary epithelial cells.Citation94 TGFβ also induces integrin-linked kinase (ILK) phosphorylation of Rictor (a core component of mTORC2 that sets it apart from mTORC1) and thereby promotes the interaction of Rictor with ILK to regulate EMT.Citation96 Knockdown experiments showed that Rictor is required during TGFβ-induced EMT but any functional difference between Rictor/mTORC and Rictor/ILK complexes remains unknown.
Epigenetic Rewiring and Noncoding RNAs in EMT
DNA methylation and post-translational modifications of histones, both of which are intrinsically linked, have an impact on gene expression and chromatin function.Citation97 Gene repression is associated with nucleosomal histone 3 di-/tri-methylation on lysine 9 (H3K9me2/3) or tri-methylation on lysine 27 (H3K27me3) and DNA methylation; whereas active genes are associated with histone acetylation and histone H3 tri-methylation on lysine 4 (H3K4me3).Citation98
EMT entails dynamic changes in expression of the epithelial and mesenchymal gene programs. In recent years, there has been a keen interest in understanding the epigenetic regulation during EMT (reviewed in Ref.Citation99,100). Several studies have shown that genome-wide epigenetic changes occur during EMT in response to TGFβ () or during overexpression of EMT-TFs.Citation21,101,102 Histone modification marks, such as H3K9me2 and H3K4me3, were reduced globally by the action of the dual histone demethylase LSD1 that demethylates H3K4 and H3K9 in mouse hepatocytes undergoing TGFβ-induced EMT.Citation102 Similarly, differences in repressive H3K27me3 marks mediated by EZH2, which is part of the polycomb repressive complex 2 (PRC2), were observed globally in mammary epithelial cells that overexpressed Twist1, Snail1 or were stimulated with TGFβ.Citation21,101 Mammary epithelial cells exposed to TGFβ over time gained repressive histone H3K9me2 and lost acetylated H3K9 active marks, an epigenotype also identified in triple-negative breast tumor tissues.Citation103 The gain or loss of specific histone marks are often correlated with the regulation of EMT-related genes.Citation21,101,102 For example, the Snail1 promoter contains high levels of the repressive mark H3K27me3 in epithelial cells. TGFβ upregulates the histone demethylase KDM6B, which removes the methyl groups from H3K27 tails on the Snail1 promoter and thereby allowing Snail1 to be expressed and cause EMT. Furthermore, KDM6B promotes cell invasion and invasive breast carcinomas show high expression levels of KDM6B compared to normal breast tissue.Citation104
Epigenetic modifications may not be determined by specific DNA sequences, but DNA and histone modifying factors often interact and are recruited to the chromatin by, for example, EMT-TFs which recognize E-box DNA elements with the consensus 5’-CANNTG-3’, and therefore exhibit gene or sequence specific functional effects.Citation8 The mechanism of E-cadherin repression by Snail1 is mediated through the recruitment of histone deacetylases (HDACs) and the corepressor SIN3A.Citation105 Several recent studies have shown that Snail1 also interacts with additional histone modifying enzymes (e.g., G9a, Suv39H1, EZH2, LSD1 and LOXL2)Citation103,106-109 and DNA methyltransferases (DNMTs)Citation103 during EMT. An active E-cadherin promoter is marked by H3K4me3 and has to be first demethylated by the demethylase LSD1,Citation108,110 before H3K9 could be di-methylated by G9a,Citation103 or tri-methylated by Suv39H1,Citation106 to mediate repression of E-cadherin. Knockdown of G9a, Suv39H1 or LSD1 in breast cancer cells reduced migration and metastasis in xenograft models.Citation103,106,110
Recently, Snail1 got implicated in the reorganization of heterochromatin by LOXL2, which removes methyl groups from H3K4 by deamination.Citation111 Snail1 pairs with LOXL2 to downregulate non-coding pericentromeric transcripts (major satellites) that are required for heterochromatin formation and this led to a temporal release of HP1α from the heterochromatin at the initial stage of EMT. Forced expression of major satellites inhibits the release of HP1α and thus, blocks TGFβ-induced EMT. In an inducible EMT model, Javaid et. al analyzed the chromatin dynamics upon Snail1 activation in human mammary epithelial cells and found that Snail1 represses epithelial genes at the early phase of EMT and sustained Snail1 expression activates mesenchymal genes at the late stage of EMT.Citation112 The short time frame during which HP1α is displaced from chromatinCitation111 coincides with the early regulation of epithelial genes by Snail1,Citation112 suggesting that heterochromatin rearrangement by Snail1 is not sequence-specific.
Twist1 recruits SET8 to mono-methylate H4K20 at the promoters of both E-cadherin and N-cadherin genes, mediating the cadherin switch during EMT and breast cancer progression.Citation113 However, it is still not clear how the H4K20me1 mark contributes to repression of E-cadherin or activation of N-cadherin gene expression.Citation113 Furthermore, Twist1 could lead to a global change in genes having bivalent marks H3K4me3 and H3K27me3, which could explain the cell plasticity of cells undergoing EMT.Citation101 Twist1 forms a complex with another chromatin regulator, BRD4, to regulate WNT signaling and promote invasion and tumorigenicity in basal-like breast cancer cells.Citation37
PAD4 is a known histone modifier, which citrullinates arginine residues on histones, and has been recently shown to maintain epithelial integrity in mammary epithelial cells by citrullinating and stabilizing GSK3β. Depletion of PAD4 leads to GSK3β destabilization, increased TGFβ signaling and onset of EMT. Low expression levels of PAD4 correlated with invasiveness in breast tumor samples.Citation114 However, the global pattern and functional effects of histone citrullination by PAD4 on chromatin during EMT remain unclear.
Long-term silencing of genes is achieved through DNA methylation, in which DNMTs methylate the C5 position of cytosine in CpG dinucleotides.Citation97 De novo DNA methylation of several promoters, most notably of the E-cadherin gene, occurs in human mammary epithelial and breast cancer cells undergoing EMT.Citation103,115,116 Thus, TGFβ signaling leads to increased DNA methylation at the E-cadherin promoter.Citation103 Sustained DNA methylation of epithelial genes is maintained by DNMT1 in mammary cells with EMT features, and this involves active TGFβ/Smad signaling as knocking down Smad2, but not Smad4, could prohibit DNMT1 from binding to its target promoters and therefore caused re-establishment of the epithelial phenotype.Citation117
Noncoding RNAs (ncRNAs) are transcribed in many cell types and these transcripts do not encode for any proteins but yet are important in biological processes. The most studied members of ncRNA are the miRNAs that are first synthesized as primary miRNA, cleaved by Drosha into precursor-miRNA and then processed into mature miRNA (averaging 22 nucleotides) by Dicer. MiRNAs inhibit gene expression by targeting mRNA for degradation or by hindering ribosomal translation.Citation118 The miR-200 family (miR-200a/b/c, miR-141 and miR-429) and miR-205 are the most prolific miRNAs in controlling the EMT program, as they negatively regulate ZEB1 and ZEB2 expression.Citation119,120 In a feedback mechanism, ZEB1 binds to the miR-200 promoters to reinforce the mesenchymal phenotype.Citation121 TGFβ signaling decreases the levels of miR-200 family and miR-205 during EMT, through EMT-TFs,Citation121 or through de novo methylation of their promoters,Citation122 and allows the stabilization of ZEB1 and ZEB2 levels. Overexpression of miR-200 in mesenchymal cells led to MET.Citation119,120 Furthermore, BMPs induce miR200 family and miR-205 and promote MET during reprogramming.Citation123 The tumor suppressor p53 maintains MET and decreases invasion of cancer cells by inducing miR-34 to target and degrade Snail1. Similar to the miR-200/ZEB feedback loop, Snail1 can bind the E-boxes in the miR-34 promoter and repress its expression.Citation124 TGFβ upregulates expression of miR-155, whose target is RhoA, leading to the dissolution of tight junctions in mammary epithelial cells.Citation125 High expression of miR-155 correlates with invasive breast cancers. A second target of miR-155 is C/EBPβ, a differentiation transcription factor that activates promoters of epithelial genes like E-cadherin and CAR. C/EBPβ is downregulated by miR-155 during TGFβ-induced EMT and its expression is often lost in triple-negative breast cancers, which are often negative for E-cadherin expression.Citation126
There has been a growing interest in another group of ncRNAs, known as long noncoding RNA (lncRNA), in relation to cancer progression and metastasis. LncRNAs have lengths of up to 100 kb and are implicated in epigenetic regulation of genes.Citation127 An example is the Hotair lncRNA, which binds and guides PRC2 to its gene targets, where PRC2 mediates H3K27 methylation and thereby silencing of genes. Recently, Hotair has been shown to be transcriptionally induced by TGFβ and is required for the EMT process and the gain of cancer stem cell properties. Inhibition of Hotair in breast cancer cells prevented downregulation of E-cadherin.Citation128 Hotair is highly expressed in metastatic breast cancers and correlates with poor prognosis.Citation129 The H19 lncRNA is another target of TGFβ signaling and is also regulated in a feed-forward mechanism by TGFβ-induced Snail2 during EMT. H19 and Snail2, together with miR-675, repress E-cadherin expression and promote metastasis in vivo.Citation130 Upregulation of Malat1 lncRNA by TGFβ has been described in bladder cancer. Malat1 associates with Suz12, another component of PRC2, and regulates EMT genes, and knockdown of either Malat1 or Suz12 inhibits tumor metastasis.Citation131
Thus, the EMT program involves chromatin remodeling, generating a plastic epigenetic landscape characterized by bivalent genes, i.e., genes with both active H3K4me3 and repressive H3K27me3 marks.Citation101 Such “poised” chromatin is then available to undergo the inverse changes during the MET process that is important for somatic reprogramming to induced pluripotent stem cells.Citation132 This could explain the case why EMT has to precede MET during somatic cell reprogramming.Citation133
EMT and Breast Cancer Stem Cells
Data accumulating during recent years support a link between EMT and generation of cancer stem cells. The concept of breast CSCs was first established by the Clarke laboratory and defines a population of cancer cells that has the ability to form new tumors (tumor initiating cells).Citation134 Breast CSCs constitute a minority of the tumor cells and show high expression of the hyaluronan receptor CD44 and low levels of the surface sialoglycoprotein CD24. A few years later it was demonstrated that breast CSCs could be enriched in populations that underwent EMT () due to the high expression of EMT-TFs, like Snail1 and Twist1.Citation5,6 The connection between EMT and stem cell properties may reside in the process of reprogramming that epithelial cells undergo during EMT,Citation99,102 as will be exemplified in this section.
By transfecting differentiated somatic cells with specific transcription factors that promote stemness, Oct4, Klf4, Myc and Sox2, pluripotent stem cells can be induced (iPSCs).Citation135 It was recently shown that optimal reprogramming to stem cells was obtained by step-wise introduction of the factors listed above and involved a sequential EMT-MET process.Citation133 Data from other research groups also confirm the reprogramming to iPSCs as a multistep process, where pluripotency during the early EMT phase is promoted by TGFβ. In the later MET phase, BMP signaling plays a key role by upregulating miR-200 family members that enforce the epithelial phenotype.Citation123 These studies support the notion that reprogramming during EMT is not only relevant to cancer but has direct links to the more physiological process of induced stemness and the generation of pluripotent stem cells. Cycles of EMT and MET seem to play an important role during reprogramming of normal stem cells and cancer cells, suggesting that these processes may provide an adaptive force when cells change their differentiation potential. This is compatible with the findings that TGFβ, BMP and Wnt signaling pathways mediate these transitions in differentiation and stemness in the context of both normal and breast cancer cells.Citation39
Several examples of signaling pathways and transcription factors regulating both EMT and stemness exist. The mesenchymal state of mammary cells is established by the EMT-TF FOXC2, which also contributes to CSC potential at least in part by inducing transcription of the PDGF β-receptor.Citation136 Signaling via PDGFRβ is also required for survival of cancer cells undergoing EMT.Citation137 In agreement, sunitinib, a clinically approved PDGF receptor inhibitor, reduced both the CSC population and the metastatic potential of breast cancer cells with high FOXC2 expression. A second example is the Hippo pathway and its downstream effector protein TAZ, which can inactivate the epithelial polarity protein Scribble, thus causing EMT.Citation138 In addition, TAZ has been shown to promote stemness in breast cancer cells when overexpessed and activated by Hippo pathway kinases. A hallmark feature of cancer cells is their escape from oncogene-induced senescence and apoptosis. Interestingly, EMT-TFs like Twist1 and ZEB1/2 interfere with key pathways regulating cell survival, such as the p53, pRb and PP2A phosphatase, resulting in the development of breast cancers that have characteristic enrichment in stem-like features.Citation139 Resistance to cell death is indeed an important feature not only during the process of EMT, but also for the generation of CSCs.Citation140 Based on a variety of findings, it could be argued that EMT and changes in differentiation induce stem like properties in cancer cells, as opposed to the hypothesis of CSCs being derived from malignant transformation of stem-like progenitor cell types in the mammary gland.
Normal and malignant stem cells are dependent on a tissue microenvironment often called the stem cell niche. Metabolic conditions in the niche are commonly found to regulate cancer stemness and one prominent example in the breast cancer niche is hypoxia. Hypoxia affects multiple molecular pathways, one being the Notch ligand Jagged2 that causes EMT and promotes breast CSC growth; consistent with this observation, Jagged2 expression and active Notch signaling have been found to correlate with poor prognosis and metastasis of human breast cancers.Citation141 The significance of hypoxia for the development of stem cell properties is illustrated by an experiment where breast cancer cells were exposed to cycles of hypoxia and normoxia. This change in microenvironment resulted in enrichment of breast cancer cells with enhanced tumor-initiating potential in mouse transplantation experiments.Citation142 The hypoxia-inducible protein carbonic anhydrase IX (CAIX) contributes to changes in local pH induced by hypoxia.Citation143 CAIX can promote breast CSC proliferation by activation of mTORC1 signaling leading to EMT and high expression of Jagged1 and Notch1. Treatment of mice with a CAIX inhibitor, suppressed CSCs and combination therapy with paclitaxel and the CAIX inhibitor potently blocked metastasis in these mice.Citation143
The concept that EMT links to cancer stemness could potentially offer novel possibilities for improved prognostics and therapeutic intervention for human breast cancer. A recent study addressing the phenotype of circulating tumor cells in the blood of breast cancer patients revealed a diverse cohort of cells exhibiting both epithelial and mesenchymal features.Citation144 Interestingly, a correlation to poor prognosis was suggested for circulating tumor cells harboring mesenchymal features, including TGFβ signaling intermediates and EMT-TFs.
Platelet-Derived TGFβ and NF-κB Signaling as Inducers of EMT and Metastasis
As described above, TGFβ is a central mediator of EMT and subsequent metastasis. In the tumor microenvironment, TGFβ is expressed by stromal cells such as macrophages but also by the tumor cells themselves. Another major source of TGFβ are megakaryocytes in the bone marrow and their circulating derivatives, platelets.Citation145 TGFβ, along with a plethora of other growth factors and cytokines, are stored in platelet α-granules and released upon activation of the platelets. Tumors generate a pro-thrombotic environment and promote enhanced coagulation and platelet activation primarily due to deregulated expression of tissue factor. As a consequence, cancer patients commonly have an elevated turnover of platelets in their circulation and increased risk of thrombotic occlusion of vessels.Citation146 In a recent study from the group of R.O. Hynes it was demonstrated that direct contact between tumor cells and platelets induces EMT () and primes the tumor cells for metastases.Citation147 Co-culturing of platelets with either breast or colon carcinoma cells induced a more invasive mesenchymal-like phenotype of the tumor cells, which resulted in enhanced seeding of tumor cells in the lungs of mice when injected through the tail vein. By creating a megakaryocyte/platelet-specific knock-out of TGFβ (Pf4-cre;TGFβ1fl/fl), it was elegantly shown that platelet-derived TGFβ is crucial to promote EMT and increased invasiveness. In addition to providing TGFβ, the direct contact with platelets triggered NF-κB signaling in the tumor cells, which was also required for platelet-induced EMT to take place. Platelets have for long been recognized to promote metastasis and potential mechanisms include protection of the tumor cells from immune recognition and shear stress in the circulation, as well as easier tumor cell adherence and subsequent extravasation through the vessel wall when platelets bind to the tumor cells.Citation148 The study by Labelle et al adds another mechanism to the list and provides the first molecular explanation for platelet-induced metastasis. These findings highlight the potential benefit of keeping platelet activity as low as possible in cancer patients, without increasing the risk of bleedings. Indeed, recent clinical data show that patients that have been on low dose aspirin for cardiovascular disease for several years have a dramatically decreased risk to develop metastatic cancer.Citation149,150 This connection has therapeutic relevance since aspirin efficiently prevents platelet activation.
MET and Effective Metastatic Colonization
Although the process of EMT generates mesenchymal and stem-like cells with enhanced invasive and metastatic capacities, it appears that what matters for the effectiveness of metastatic dissemination and secondary tumor growth is the ability of metastatic stem cells to generate new epithelial cancer cells after undergoing MET (). Early studies focusing on signaling by the BMP pathway and its impact on EMT, established that BMPs can counteract TGFβ-induced EMT and promote MET in mammary epithelial cells.Citation151,152 BMP signaling is mediated by distinct receptor serine/threonine kinases and distinct Smads, Smad1, Smad5 and Smad8 (), however, Smad4 also serves as a cofactor of the other Smads in both BMP and TGFβ pathways.Citation41,47,69 Using this canonical Smad signaling pathway, BMP induces the transcription of genes of the ID family, which can negatively regulate the function of pro-EMT TFs with bHLH domains, thus counteracting EMT and possibly stimulating the MET. Accordingly, TGFβ represses the expression of ID family genes () and this is required for the establishment of EMT.Citation151,152 This principle has been nicely exploited therapeutically in mouse models where breast cancer metastasis was effectively blocked by the delivery of BMP ligands to the host animals.Citation153 Recent work has also revealed a more complete and unavoidably more complex picture of the relationship of ID gene regulation and MET in breast cancer metastasis.Citation154 Upon induction of EMT by TGFβ, mesenchymal breast cancer cells also upregulate ID1, which binds to Twist1, inactivates its transcriptional activity and promotes MET at metastatic colonies.Citation154 This is an interesting model that emphasizes the temporal aspect of regulation of key EMT-TFs during the EMT-MET cycle. In the same line of evidence, transgenic mice expressing Twist1 in a conditional manner in the skin and upon a chemical carcinogenesis protocol, demonstrated that transient Twist1 induction is critical for EMT induction and invasive tumor growth, however, when the squamous carcinomas would metastasize, Twist1 had to be silenced so that MET would prevail in order for metastatic epithelial colonies to be established after dissemination through the vasculature.Citation155 However, one should consider that breast cancer metastasis in the absence of MET may well be possible and this clearly applies in certain mouse models such as the EpRas model described above.Citation46 The evidence for a requirement of a succession of EMT-MET cycles suits the current understanding of the multifunctional roles TGFβ and BMP play in breast cancer progression. This evidence also cautions experimentalists about the relative difficulty of applying this basic knowledge of tumor biology to the effective use of novel therapeutics that may perturb the EMT-MET cycle or the corresponding TGFβ-BMP signaling networks.Citation7,156 Careful consideration of the possibilities these alternate signaling processes may offer to the development of therapeutic approaches is warranted.
Perspectives
Here we have highlighted some key examples and processes that explain how cytokines of the TGFβ family promote the EMT and how they may also have an impact on the inverse process of MET. A major need for current investigation in this field is to understand deeper signaling crosstalk between multiple pathways that are initiated by diverse growth factors and cytokines and their relative contribution to EMT and breast cancer metastasis. We need to explore deeper the links between chemical modifications of chromatin, which currently are referred to as epigenetic mechanisms of gene regulation, to the action of specific EMT-TFs and the process of breast cancer stem cell generation. The specific involvement of non-coding RNAs will continue to provide novelty to this area of cancer biology. Of equal importance are studies that aim at clarifying the spatiotemporal control and the molecular analysis of EMT-MET cycles in breast cancer progression, linking the cyclical or reversible processes to the plasticity of chromatin modifications during the metastatic trajectory. The possibility that MET is also driven by the EMT-generated CSCs is an interesting open hypothesis () that promises continues excitement in this booming area of tumor biology.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all past and present members of our groups for their contributions to the scientific work emanating from our laboratories. This article summarizes work from a large number of published papers; however, due to space limitations, we have been unable to include all relevant publications in our discussion. We apologize to those authors whose relevant work has not been included in this review article.
Funding
AM acknowledges funding by the Ludwig Institute for Cancer Research, the Swedish Cancer Society, the Swedish Research Council and the Marie Curie Initial Training Network “IT-Liver” under the European Union FP7 program. A-KO acknowledges funding by the Swedish Cancer Society.
References
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139(5):871-90; PMID:19945376; http://dx.doi.org/10.1016/j.cell.2009.11.007
- Bissell MJ, Rizki A, Mian IS. Tissue architecture: the ultimate regulator of breast epithelial function. Curr Opin Cell Biol 2003; 15(6):753-62; PMID:14644202; http://dx.doi.org/10.1016/j.ceb.2003.10.016
- Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) 1995; 154(1):8-20; PMID:8714286; http://dx.doi.org/10.1159/000147748
- Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008; 14(1):79-89; PMID:18598946; http://dx.doi.org/10.1016/j.ccr.2008.06.005
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133(4):704-15; PMID:18485877; http://dx.doi.org/10.1016/j.cell.2008.03.027
- Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One 2008; 3(8):e2888; PMID:18682804; http://dx.doi.org/10.1371/journal.pone.0002888
- Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol 2011; 27:347-76; PMID:21740232; http://dx.doi.org/10.1146/annurev-cellbio-092910-154036
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007; 7(6):415-28; PMID:17508028; http://dx.doi.org/10.1038/nrc2131
- De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer 2013; 13(2):97-110; PMID:23344542; http://dx.doi.org/10.1038/nrc3447
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2(2):84-9; PMID:10655587; http://dx.doi.org/10.1038/35000034
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2(2):76-83; PMID:10655586; http://dx.doi.org/10.1038/35000025
- Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 2002; 62(6):1613-8; PMID:11912130
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005; 24(14):2375-85; PMID:15674322; http://dx.doi.org/10.1038/sj.onc.1208429
- Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G. SIP1ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res 2005; 33(20):6566-78; PMID:16314317; http://dx.doi.org/10.1093/nar/gki965
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117(7):927-39; PMID:15210113; http://dx.doi.org/10.1016/j.cell.2004.06.006
- Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang Z, Yang CJ, Yuan L, Ouyang G. Twist2 contributes to breast cancer progression by promoting an epithelial-mesenchymal transition and cancer stem-like cell self-renewal. Oncogene 2011; 30(47):4707-20; PMID:21602879; http://dx.doi.org/10.1038/onc.2011.181
- Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A. A new role for E12E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem 2001; 276(29):27424-31; PMID:11309385; http://dx.doi.org/10.1074/jbc.M100827200
- Tan E-J, Thuault S, Caja L, Carletti T, Heldin C-H, Moustakas A. Regulation of transcription factor Twist expression by the DNA architectural protein high mobility group A2 during epithelial-to-mesenchymal transition. J Biol Chem 2012; 287(10):7134-45; PMID:22241470; http://dx.doi.org/10.1074/jbc.M111.291385
- Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin C-H, Moustakas A. Transforming growth factor-b employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol 2006; 174(2):175-83; PMID:16831886; http://dx.doi.org/10.1083/jcb.200512110
- Thuault S, Tan E-J, Peinado H, Cano A, Heldin C-H, Moustakas A. HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J Biol Chem 2008; 283(48):33437-46; PMID:18832382; http://dx.doi.org/10.1074/jbc.M802016200
- Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, Meyer-Schaller N, Schubeler D, van Nimwegen E, Christofori G. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 2013; 23(6):768-83; PMID:23764001; http://dx.doi.org/10.1016/j.ccr.2013.04.020
- Zhang J, Liang Q, Lei Y, Yao M, Li L, Gao X, Feng J, Zhang Y, Gao H, Liu DX, et al. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res 2012; 72(17):4597-608; PMID:22787120; http://dx.doi.org/10.1158/0008-5472.CAN-12-1045
- Dave N, Guaita-Esteruelas S, Gutarra S, Frias A, Beltran M, Peiro S, de Herreros AG. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem 2011; 286(14):12024-32; PMID:21317430; http://dx.doi.org/10.1074/jbc.M110.168625
- Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 2010; 107(35):15449-54; PMID:20713713; http://dx.doi.org/10.1073/pnas.1004900107
- Young RA. Control of the embryonic stem cell state. Cell 2011; 144(6):940-54; PMID:21414485; http://dx.doi.org/10.1016/j.cell.2011.01.032
- Furuhashi M, Yagi K, Yamamoto H, Furukawa Y, Shimada S, Nakamura Y, Kikuchi A, Miyazono K, Kato M, Axin facilitates Smad3 activation in the transforming growth factor β signaling pathway. Mol Cell Biol 2001; 21(15):5132-41; PMID:11438668; http://dx.doi.org/10.1128/MCB.21.15.5132-5141.2001
- Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang X-F. Axin and GSK3-b control Smad3 protein stability and modulate TGF-b signaling. Genes Dev 2008; 22(1):106-20; PMID:18172167; http://dx.doi.org/10.1101/gad.1590908
- Eger A, Stockinger A, Park J, Langkopf E, Mikula M, Gotzmann J, Mikulits W, Beug H, Foisner R. b-Catenin and TGFb signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene 2004; 23(15):2672-80; PMID:14755243; http://dx.doi.org/10.1038/sj.onc.1207416
- Kim Y, Kugler MC, Wei Y, Kim KK, Li X, Brumwell AN, Chapman HA. Integrin a3b1-dependent b-catenin phosphorylation links epithelial Smad signaling to cell contacts. J Cell Biol 2009; 184(2):309-22; PMID:19171760; http://dx.doi.org/10.1083/jcb.200806067
- Liebner S, Cattelino A, Gallini R, Rudini N, Iurlaro M, Piccolo S, Dejana E. B-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 2004; 166(3):359-67; PMID:15289495; http://dx.doi.org/10.1083/jcb.200403050
- Nawshad A, Medici D, Liu CC, Hay ED. TGFb3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J Cell Sci 2007; 120(Pt 9):1646-53; PMID:17452626; http://dx.doi.org/10.1242/jcs.003129
- Jian H, Shen X, Liu I, Semenov M, He X, Wang X-F. Smad3-dependent nuclear translocation of b-catenin is required for TGF-b1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev 2006; 20(6):666-74; PMID:16543220; http://dx.doi.org/10.1101/gad.1388806
- Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, et al. A Wnt-Axin2-GSK3b cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol 2006; 8(12):1398-406; PMID:17072303; http://dx.doi.org/10.1038/ncb1508
- Labbé E, Lock L, Letamendia A, Gorska AE, Gryfe R, Gallinger S, Moses HL, Attisano L. Transcriptional cooperation between the transforming growth factor-b and Wnt pathways in mammary and intestinal tumorigenesis. Cancer Res 2007; 67(1):75-84; PMID:17210685; http://dx.doi.org/10.1158/0008-5472.CAN-06-2559
- Roarty K, Serra R. Wnt5a is required for proper mammary gland development and TGF-b-mediated inhibition of ductal growth. Development 2007; 134(21):3929-39; PMID:17898001; http://dx.doi.org/10.1242/dev.008250
- Roarty K, Baxley SE, Crowley MR, Frost AR, Serra R. Loss of TGF-b or Wnt5a results in an increase in Wntb-catenin activity and redirects mammary tumour phenotype. Breast Cancer Res 2009; 11(2):R19; PMID:19344510; http://dx.doi.org/10.1186/bcr2244
- Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014; 25(2):210-25; PMID:24525235; http://dx.doi.org/10.1016/j.ccr.2014.01.028
- Placencio VR, Sharif-Afshar AR, Li X, Huang H, Uwamariya C, Neilson EG, Shen MM, Matusik RJ, Hayward SW, Bhowmick NA. Stromal transforming growth factor-b signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Res 2008; 68(12):4709-18; PMID:18559517; http://dx.doi.org/10.1158/0008-5472.CAN-07-6289
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011; 145(6):926-40; PMID:21663795; http://dx.doi.org/10.1016/j.cell.2011.04.029
- Katsuno Y, Lamouille S, Derynck R. TGF-b signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol 2013; 25(1):76-84; PMID:23197193; http://dx.doi.org/10.1097/CCO.0b013e32835b6371
- Moustakas A, Heldin C-H. Induction of epithelial-mesenchymal transition by transforming growth factor b. Semin Cancer Biol 2012; 22(5-6):446-54; PMID:22548724; http://dx.doi.org/10.1016/j.semcancer.2012.04.002
- Gal A, Sjöblom T, Fedorova L, Imreh S, Beug H, Moustakas A. Sustained TGF b exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008; 27(9):1218-30; PMID:17724470; http://dx.doi.org/10.1038/sj.onc.1210741
- Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-b induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 1994; 127(6 Pt 2):2021-36; PMID:7806579; http://dx.doi.org/10.1083/jcb.127.6.2021
- Piek E, Moustakas A, Kurisaki A, Heldin C-H, ten Dijke P. TGF-b type I receptorALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 1999; 112 (Pt 24):4557-68; PMID:10574705
- Valcourt U, Kowanetz M, Niimi H, Heldin C-H, Moustakas A. TGF-b and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 2005; 16(4):1987-2002; PMID:15689496
- Janda E, Lehmann K, Killisch I, Jechlinger M, Herzig M, Downward J, Beug H, Grunert S. Ras and TGFb cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 2002; 156(2):299-313; PMID:11790801; http://dx.doi.org/10.1083/jcb.200109037
- Moustakas A, Heldin P. TGFb and matrix-regulated epithelial to mesenchymal transition. Biochim Biophys Acta 2014; 1840(8):2621-34; PMID:24561266; http://dx.doi.org/10.1016/j.bbagen.2014.02.004
- Mercado-Pimentel ME, Runyan RB. Multiple transforming growth factor-b isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 2007; 185(1-3):146-56; PMID:17587820; http://dx.doi.org/10.1159/000101315
- Azhar M, Runyan RB, Gard C, Sanford LP, Miller ML, Andringa A, Pawlowski S, Rajan S, Doetschman T. Ligand-specific function of transforming growth factor b in epithelial-mesenchymal transition in heart development. Dev Dyn 2009; 238(2):431-42; PMID:19161227; http://dx.doi.org/10.1002/dvdy.21854
- Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFb-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci 2008; 121(Pt 20):3317-24; PMID:18796538; http://dx.doi.org/10.1242/jcs.028282
- Luna-Zurita L, Prados B, Grego-Bessa J, Luxan G, del Monte G, Benguria A, Adams RH, Perez-Pomares JM, de la Pompa JL. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest 2010; 120(10):3493-507; PMID:20890042; http://dx.doi.org/10.1172/JCI42666
- Townsend TA, Robinson JY, How T, DeLaughter DM, Blobe GC, Barnett JV. Endocardial cell epithelial-mesenchymal transformation requires Type III TGFb receptor interaction with GIPC. Cell Signal 2012; 24(1):247-56; PMID:21945156; http://dx.doi.org/10.1016/j.cellsig.2011.09.006
- Stevens MV, Broka DM, Parker P, Rogowitz E, Vaillancourt RR, Camenisch TD. MEKK3 initiates transforming growth factor b 2-dependent epithelial-to-mesenchymal transition during endocardial cushion morphogenesis. Circ Res 2008; 103(12):1430-40; PMID:19008476; http://dx.doi.org/10.1161/CIRCRESAHA.108.180752
- Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through b-catenin-T-cell factor-4-dependent expression of transforming growth factor-b3. Mol Biol Cell 2008; 19(11):4875-87
- Jalali A, Zhu X, Liu C, Nawshad A. Induction of palate epithelial mesenchymal transition by transforming growth factor b3 signaling. Dev Growth Differ 2012; 54(6):633-48; PMID:22775504; http://dx.doi.org/10.1111/j.1440-169X.2012.01364.x
- Kaartinen V, Haataja L, Nagy A, Heisterkamp N, Groffen J. TGFb3-induced activation of RhoARho-kinase pathway is necessary but not sufficient for epithelio-mesenchymal transdifferentiation: implications for palatogenesis. Int J Mol Med 2002; 9(6):563-70; PMID:12011971
- Paruchuri S, Yang JH, Aikawa E, Melero-Martin JM, Khan ZA, Loukogeorgakis S, Schoen FJ, Bischoff J. Human pulmonary valve progenitor cells exhibit endothelialmesenchymal plasticity in response to vascular endothelial growth factor-A and transforming growth factor-b2. Circ Res 2006; 99(8):861-9; PMID:16973908; http://dx.doi.org/10.1161/01.RES.0000245188.41002.2c
- Chen X, Xiao W, Liu X, Zeng M, Luo L, Wu M, Ye S, Liu Y. Blockade of JaggedNotch pathway abrogates transforming growth factor b2-induced epithelial-mesenchymal transition in human retinal pigment epithelium cells. Curr Mol Med 2014; 14(4):523-34; PMID:24694299; http://dx.doi.org/10.2174/1566524014666140331230411
- Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, Jechlinger M, Waerner T, Weith A, Beug H, et al. A crucial function of PDGF in TGF-b-mediated cancer progression of hepatocytes. Oncogene 2006; 25(22):3170-85; PMID:16607286; http://dx.doi.org/10.1038/sj.onc.1209083
- Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, Ruegg C, Hemmings BA. AktPKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3KAkt and TGF-b signaling axes. Cancer Discov 2012; 2(3):248-59; PMID:22585995; http://dx.doi.org/10.1158/2159-8290.CD-11-0270
- Wyatt L, Wadham C, Crocker LA, Lardelli M, Khew-Goodall Y. The protein tyrosine phosphatase Pez regulates TGFb, epithelial-mesenchymal transition, and organ development. J Cell Biol 2007; 178(7):1223-35; PMID:17893246; http://dx.doi.org/10.1083/jcb.200705035
- Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 2008; 9(6):582-9; PMID:18483486; http://dx.doi.org/10.1038/embor.2008.74
- Wang B, Koh P, Winbanks C, Coughlan MT, McClelland A, Watson A, Jandeleit-Dahm K, Burns WC, Thomas MC, Cooper ME, et al. miR-200a Prevents renal fibrogenesis through repression of TGF-b2 expression. Diabetes 2011; 60(1):280-7; PMID:20952520; http://dx.doi.org/10.2337/db10-0892
- Pickup M, Novitskiy S, Moses HL. The roles of TGFb in the tumour microenvironment. Nat Rev Cancer 2013; 13(11):788-99; PMID:24132110
- Massagué J. TGFb signalling in context. Nat Rev Mol Cell Biol 2012; 13(10):616-30; PMID:22992590; http://dx.doi.org/10.1038/nrm3434
- Moustakas A, Pardali K, Gaal A, Heldin C-H. Mechanisms of TGF-b signaling in regulation of cell growth and differentiation. Immunol Lett 2002; 82(1-2):85-91; PMID:12008039; http://dx.doi.org/10.1016/S0165-2478(02)00023-8
- Smith AP, Verrecchia A, Faga G, Doni M, Perna D, Martinato F, Guccione E, Amati B. A positive role for Myc in TGFb-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene 2009; 28(3):422-30; PMID:18978814; http://dx.doi.org/10.1038/onc.2008.395
- Yang L, Moses HL. Transforming growth factor b: tumor suppressor or promoter? Are host immune cells the answer? Cancer Res 2008; 68(22):9107-11; PMID:19010878; http://dx.doi.org/10.1158/0008-5472.CAN-08-2556
- Moustakas A, Heldin C-H. Dynamic control of TGF-b signaling and its links to the cytoskeleton. FEBS Lett 2008; 582(14):2051-65; PMID:18375206; http://dx.doi.org/10.1016/j.febslet.2008.03.027
- Deckers M, van Dinther M, Buijs J, Que I, Lowik C, van der Pluijm G, ten Dijke P. The tumor suppressor Smad4 is required for transforming growth factor b-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 2006; 66(4):2202-9; PMID:16489022; http://dx.doi.org/10.1158/0008-5472.CAN-05-3560
- Hesling C, Fattet L, Teyre G, Jury D, Gonzalo P, Lopez J, Vanbelle C, Morel AP, Gillet G, Mikaelian I, et al. Antagonistic regulation of EMT by TIF1g and Smad4 in mammary epithelial cells. EMBO Rep 2011; 12(7):665-72; PMID:21597466; http://dx.doi.org/10.1038/embor.2011.78
- Sundqvist A, Zieba A, Vasilaki E, Herrera Hidalgo C, Soderberg O, Koinuma D, Miyazono K, Heldin C-H, Landegren U, ten Dijke P, et al. Specific interactions between Smad proteins and AP-1 components determine TGFb-induced breast cancer cell invasion. Oncogene 2013; 32(31):3606-15; PMID:22926518; http://dx.doi.org/10.1038/onc.2012.370
- Termén S, Tan EJ, Heldin C-H, Moustakas A. p53 regulates epithelial-mesenchymal transition induced by transforming growth factor b. J Cell Physiol 2013; 228(4):801-13; PMID:23018556; http://dx.doi.org/10.1002/jcp.24229
- Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, Yu WH, Rehman SK, Hsu JL, Lee HH, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol 2011; 13(3):317-23; PMID:21336307; http://dx.doi.org/10.1038/ncb2173
- Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, et al. A p53miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol 2011; 195(3):417-33; PMID:22024162; http://dx.doi.org/10.1083/jcb.201103097
- Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D, Knezevic J, Greene SB, Darr D, Troester MA, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Acad Sci U S A 2012; 109(8):2778-83; PMID:21633010; http://dx.doi.org/10.1073/pnas.1018862108
- Morishita A, Zaidi MR, Mitoro A, Sankarasharma D, Szabolcs M, Okada Y, D’Armiento J, Chada K. HMGA2 is a driver of tumor metastasis. Cancer Res 2013; 73(14):4289-99; PMID:23722545; http://dx.doi.org/10.1158/0008-5472.CAN-12-3848
- Pegoraro S, Ros G, Piazza S, Sommaggio R, Ciani Y, Rosato A, Sgarra R, Del Sal G, Manfioletti G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013; 4(8):1293-308; PMID:23945276
- Peinado H, Quintanilla M, Cano A. Transforming growth factor b-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem 2003; 278(23):21113-23; PMID:12665527; http://dx.doi.org/10.1074/jbc.M211304200
- Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL, et al. A SNAIL1-SMAD34 transcriptional repressor complex promotes TGF-b mediated epithelial-mesenchymal transition. Nat Cell Biol 2009; 11(8):943-50; PMID:19597490; http://dx.doi.org/10.1038/ncb1905
- Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by dEF1 proteins in epithelial mesenchymal transition induced by TGF-b. Mol Biol Cell 2007; 18(9):3533-44; PMID:17615296
- Verschueren K, Remacle JE, Collart C, Kraft H, Baker BS, Tylzanowski P, Nelles L, Wuytens G, Su MT, Bodmer R, et al. SIP1, a novel zinc fingerhomeodomain repressor, interacts with Smad proteins and binds to 5’-CACCT sequences in candidate target genes. J Biol Chem 1999; 274(29):20489-98; PMID:10400677; http://dx.doi.org/10.1074/jbc.274.29.20489
- Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor b-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 2000; 275(47):36803-10; PMID:10969078; http://dx.doi.org/10.1074/jbc.M005912200
- Galliher AJ, Schiemann WP. Src phosphorylates Tyr284 in TGF-b type II receptor and regulates TGF-b stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res 2007; 67(8):3752-8; PMID:17440088; http://dx.doi.org/10.1158/0008-5472.CAN-06-3851
- Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. TGF-b activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J 2007; 26(17):3957-67; PMID:17673906; http://dx.doi.org/10.1038/sj.emboj.7601818
- Mu Y, Sundar R, Thakur N, Ekman M, Gudey SK, Yakymovych M, Hermansson A, Dimitriou H, Bengoechea-Alonso MT, Ericsson J, et al. TRAF6 ubiquitinates TGFb type I receptor to promote its cleavage and nuclear translocation in cancer. Nat Commun 2011; 2:330; PMID:21629263; http://dx.doi.org/10.1038/ncomms1332
- Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFb receptors controls epithelial cell plasticity. Science 2005; 307(5715):1603-9; PMID:15761148; http://dx.doi.org/10.1126/science.1105718
- Dillon RL, Muller WJ. Distinct biological roles for the akt family in mammary tumor progression. Cancer Res 2010; 70(11):4260-4; PMID:20424120; http://dx.doi.org/10.1158/0008-5472.CAN-10-0266
- Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, Danino M, Karlan BY, Slamon DJ. Overexpression of AKT2protein kinase Bb leads to up-regulation of b1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res 2003; 63(1):196-206; PMID:12517798
- Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, Muller WJ. Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res 2009; 69(12):5057-64; PMID:19491266; http://dx.doi.org/10.1158/0008-5472.CAN-08-4287
- Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD, Wang LH. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 2007; 67(5):1979-87; PMID:17332325; http://dx.doi.org/10.1158/0008-5472.CAN-06-1479
- Villagrasa P, Diaz VM, Vinas-Castells R, Peiro S, Del Valle-Perez B, Dave N, Rodriguez-Asiain A, Casal JI, Lizcano JM, Dunach M, et al. Akt2 interacts with Snail1 in the E-cadherin promoter. Oncogene 2012; 31(36):4022-33; PMID:22158034; http://dx.doi.org/10.1038/onc.2011.562
- Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal 2009; 2(92):ra62; PMID:19825827; http://dx.doi.org/10.1126/scisignal.2000356
- Lamouille S, Derynck R. Cell size and invasion in TGF-b-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 2007; 178(3):437-51; PMID:17646396; http://dx.doi.org/10.1083/jcb.200611146
- Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. TGF-b-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci 2012; 125(Pt 5):1259-73; PMID:22399812; http://dx.doi.org/10.1242/jcs.095299
- Serrano I, McDonald PC, Lock FE, Dedhar S. Role of the integrin-linked kinase (ILK)Rictor complex in TGFb-1-induced epithelial-mesenchymal transition (EMT). Oncogene 2013; 32(1):50-60; PMID:22310280; http://dx.doi.org/10.1038/onc.2012.30
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 2009; 10(5):295-304; PMID:19308066; http://dx.doi.org/10.1038/nrg2540
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res 2011; 21(3):381-95; PMID:21321607; http://dx.doi.org/10.1038/cr.2011.22
- Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 2013; 19(11):1438-49; PMID:24202396; http://dx.doi.org/10.1038/nm.3336
- Wu CY, Tsai YP, Wu MZ, Teng SC, Wu KJ. Epigenetic reprogramming and post-transcriptional regulation during the epithelial-mesenchymal transition. Trends Genet 2012; 28(9):454-63; PMID:22717049; http://dx.doi.org/10.1016/j.tig.2012.05.005
- Malouf GG, Taube JH, Lu Y, Roysarkar T, Panjarian S, Estecio MR, Jelinek J, Yamazaki J, Raynal NJ, Long H, et al. Architecture of epigenetic reprogramming following Twist1 mediated epithelial-mesenchymal transition. Genome Biol 2013; 14(12):R144; PMID:24367927; http://dx.doi.org/10.1186/gb-2013-14-12-r144
- McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nat Struct Mol Biol 2011; 18(8):867-74; PMID:21725293; http://dx.doi.org/10.1038/nsmb.2084
- Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, Evers BM, Zhou BP. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest 2012; 122(4):1469-86; PMID:22406531; http://dx.doi.org/10.1172/JCI57349
- Ramadoss S, Chen X, Wang CY. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. J Biol Chem 2012; 287(53):44508-17; PMID:23152497; http://dx.doi.org/10.1074/jbc.M112.424903
- Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3Ahistone deacetylase 1 (HDAC1)HDAC2 complex. Mol Cell Biol 2004; 24(1):306-19; PMID:14673164; http://dx.doi.org/10.1128/MCB.24.1.306-319.2004
- Dong C, Wu Y, Wang Y, Wang C, Kang T, Rychahou PG, Chi YI, Evers BM, Zhou BP. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 2013; 32(11):1351-62; PMID:22562246; http://dx.doi.org/10.1038/onc.2012.169
- Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, Escriva M, Hernandez-Munoz I, Di Croce L, Helin K, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol 2008; 28(15):4772-81; PMID:18519590; http://dx.doi.org/10.1128/MCB.00323-08
- Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010; 29(35):4896-904; PMID:20562920; http://dx.doi.org/10.1038/onc.2010.234
- Peinado H, Del Carmen Iglesias-de la Cruz M, Olmeda D, Csiszar K, Fong KS, Vega S, Nieto MA, Cano A, Portillo F. A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. EMBO J 2005; 24(19):3446-58; PMID:16096638; http://dx.doi.org/10.1038/sj.emboj.7600781
- Lin Y, Wu Y, Li J, Dong C, Ye X, Chi YI, Evers BM, Zhou BP. The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J 2010; 29(11):1803-16; PMID:20389281; http://dx.doi.org/10.1038/emboj.2010.63
- Millanes-Romero A, Herranz N, Perrera V, Iturbide A, Loubat-Casanovas J, Gil J, Jenuwein T, Garcia de Herreros A, Peiro S. Regulation of heterochromatin transcription by Snail1LOXL2 during epithelial-to-mesenchymal transition. Mol Cell 2013; 52(5):746-57; PMID:24239292
- Javaid S, Zhang J, Anderssen E, Black JC, Wittner BS, Tajima K, Ting DT, Smolen GA, Zubrowski M, Desai R, et al. Dynamic chromatin modification sustains epithelial-mesenchymal transition following inducible expression of Snail-1. Cell Rep 2013; 5(6):1679-89; PMID:24360956; http://dx.doi.org/10.1016/j.celrep.2013.11.034
- Yang F, Sun L, Li Q, Han X, Lei L, Zhang H, Shang Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J 2012; 31(1):110-23; PMID:21983900; http://dx.doi.org/10.1038/emboj.2011.364
- Stadler SC, Vincent CT, Fedorov VD, Patsialou A, Cherrington BD, Wakshlag JJ, Mohanan S, Zee BM, Zhang X, Garcia BA, et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3b activates TGF-b signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc Natl Acad Sci U S A 2013; 110(29):11851-6; PMID:23818587; http://dx.doi.org/10.1073/pnas.1308362110
- Dumont N, Wilson MB, Crawford YG, Reynolds PA, Sigaroudinia M, Tlsty TD. Sustained induction of epithelial to mesenchymal transition activates DNA methylation of genes silenced in basal-like breast cancers. Proc Natl Acad Sci U S A 2008; 105(39):14867-72; PMID:18806226; http://dx.doi.org/10.1073/pnas.0807146105
- Lombaerts M, van Wezel T, Philippo K, Dierssen JW, Zimmerman RM, Oosting J, van Eijk R, Eilers PH, van de Water B, Cornelisse CJ, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J Cancer 2006; 94(5):661-71; PMID:16495925
- Papageorgis P, Lambert AW, Ozturk S, Gao F, Pan H, Manne U, Alekseyev YO, Thiagalingam A, Abdolmaleky HM, Lenburg M, et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res 2010; 70(3):968-78; PMID:20086175; http://dx.doi.org/10.1158/0008-5472.CAN-09-1872
- Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol 2009; 11(3):228-34; PMID:19255566; http://dx.doi.org/10.1038/ncb0309-228
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10(5):593-601; PMID:18376396; http://dx.doi.org/10.1038/ncb1722
- Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 2008; 283(22):14910-4; PMID:18411277; http://dx.doi.org/10.1074/jbc.C800074200
- Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 2008; 68(19):7846-54; PMID:18829540; http://dx.doi.org/10.1158/0008-5472.CAN-08-1942
- Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, Morris M, Wyatt L, Farshid G, Lim YY, et al. An autocrine TGF-bZEBmiR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell 2011; 22(10):1686-98; PMID:21411626; http://dx.doi.org/10.1091/mbc.E11-02-0103
- Samavarchi-Tehrani P, Golipour A, David L, Sung HK, Beyer TA, Datti A, Woltjen K, Nagy A, Wrana JL. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010; 7(1):64-77; PMID:20621051; http://dx.doi.org/10.1016/j.stem.2010.04.015
- Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, Hermeking H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011; 10(24):4256-71; PMID:22134354; http://dx.doi.org/10.4161/cc.10.24.18552
- Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor bSmad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol 2008; 28(22):6773-84; PMID:18794355; http://dx.doi.org/10.1128/MCB.00941-08
- Johansson J, Berg T, Kurzejamska E, Pang MF, Tabor V, Jansson M, Roswall P, Pietras K, Sund M, Religa P, et al. MiR-155-mediated loss of CEBPb shifts the TGF-b response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene 2013; 32(50):5614-24; PMID:23955085; http://dx.doi.org/10.1038/onc.2013.322
- Li CH, Chen Y. Targeting long non-coding RNAs in cancers: progress and prospects. Int J Biochem Cell Biol 2013; 45(8):1895-910; PMID:23748105; http://dx.doi.org/10.1016/j.biocel.2013.05.030
- Padua Alves C, Fonseca AS, Muys BR, de Barros ELBR, Burger MC, de Souza JE, Valente V, Zago MA, Silva WA Jr. Brief report: The lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells 2013; 31(12):2827-32; PMID:24022994; http://dx.doi.org/10.1002/stem.1547
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464(7291):1071-6; PMID:20393566; http://dx.doi.org/10.1038/nature08975
- Matouk IJ, Raveh E, Abu-lail R, Mezan S, Gilon M, Gershtain E, Birman T, Gallula J, Schneider T, Barkali M, et al. Oncofetal H19 RNA promotes tumor metastasis. Biochim Biophys Acta 2014; 1843(7):1414-26
- Fan Y, Shen B, Tan M, Mu X, Qin Y, Zhang F, Liu Y. TGF-b-Induced Upregulation of malat1 promotes bladder cancer metastasis by associating with suz12. Clin Cancer Res 2014; 20(6):1531-41; PMID:24449823; http://dx.doi.org/10.1158/1078-0432.CCR-13-1455
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010; 7(1):51-63; PMID:20621050; http://dx.doi.org/10.1016/j.stem.2010.04.014
- Liu X, Sun H, Qi J, Wang L, He S, Liu J, Feng C, Chen C, Li W, Guo Y, et al. Sequential introduction of reprogramming factors reveals a time-sensitive requirement for individual factors and a sequential EMT-MET mechanism for optimal reprogramming. Nat Cell Biol 2013; 15(7):829-38; PMID:23708003; http://dx.doi.org/10.1038/ncb2765
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100(7):3983-8; PMID:12629218; http://dx.doi.org/10.1073/pnas.0530291100
- Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature 2010; 465(7299):704-12; PMID:20535199; http://dx.doi.org/10.1038/nature09229
- Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res 2013; 73(6):1981-92; PMID:23378344; http://dx.doi.org/10.1158/0008-5472.CAN-12-2962
- Smith CL, Baek ST, Sung CY, Tallquist MD. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res 2011; 108(12):e15-26; PMID:21512159; http://dx.doi.org/10.1161/CIRCRESAHA.110.235531
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A (), et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011; 147(4):759-72; PMID:22078877; http://dx.doi.org/10.1016/j.cell.2011.09.048
- Morel AP, Hinkal GW, Thomas C, Fauvet F, Courtois-Cox S, Wierinckx A, Devouassoux-Shisheboran M, Treilleux I, Tissier A, Gras B, et al. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet 2012; 8(5):e1002723; PMID:22654675; http://dx.doi.org/10.1371/journal.pgen.1002723
- Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci 2013; 126(Pt 1):21-9; PMID:23516327; http://dx.doi.org/10.1242/jcs.120907
- Xing F, Okuda H, Watabe M, Kobayashi A, Pai SK, Liu W, Pandey PR, Fukuda K, Hirota S, Sugai T, et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011; 30(39):4075-86; PMID:21499308; http://dx.doi.org/10.1038/onc.2011.122
- Louie E, Nik S, Chen JS, Schmidt M, Song B, Pacson C, Chen XF, Park S, Ju J, Chen EI. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res 2010; 12(6):R94; PMID:21067584; http://dx.doi.org/10.1186/bcr2773
- Lock FE, McDonald PC, Lou Y, Serrano I, Chafe SC, Ostlund C, Aparicio S, Winum JY, Supuran CT, Dedhar S. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 2013; 32(44):5210-9; PMID:23208505; http://dx.doi.org/10.1038/onc.2012.550
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013; 339(6119):580-4; PMID:23372014; http://dx.doi.org/10.1126/science.1228522
- Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-b in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem 1983; 258(11):7155-60; PMID:6602130
- Lip GY, Chin BS, Blann AD. Cancer and the prothrombotic state. Lancet Oncol 2002; 3(1):27-34; PMID:11908507; http://dx.doi.org/10.1016/S1470-2045(01)00619-2
- Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011; 20(5):576-90; PMID:22094253; http://dx.doi.org/10.1016/j.ccr.2011.09.009
- Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer 2011; 11(2):123-34; PMID:21258396; http://dx.doi.org/10.1038/nrc3004
- Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 2012; 13(5):518-27; PMID:22440112; http://dx.doi.org/10.1016/S1470-2045(12)70112-2
- Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 2012; 379(9826):1591-601; PMID:22440947; http://dx.doi.org/10.1016/S0140-6736(12)60209-8
- Kondo M, Cubillo E, Tobiume K, Shirakihara T, Fukuda N, Suzuki H, Shimizu K, Takehara K, Cano A, Saitoh M, et al. A role for Id in the regulation of TGF-b-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ 2004; 11(10):1092-101; PMID:15181457; http://dx.doi.org/10.1038/sj.cdd.4401467
- Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor b and bone morphogenetic protein. Mol Cell Biol 2004; 24(10):4241-54; PMID:15121845; http://dx.doi.org/10.1128/MCB.24.10.4241-4254.2004
- Buijs JT, Henriquez NV, van Overveld PG, van der Horst G, ten Dijke P, van der Pluijm G. TGF-b and BMP7 interactions in tumour progression and bone metastasis. Clin Exp Metastasis 2007; 24(8):609-17; PMID:18008174; http://dx.doi.org/10.1007/s10585-007-9118-2
- Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massagué J, Benezra R. TGF-b-Id1 signaling opposes twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep 2013; 5(5):1228-42; PMID:24332369; http://dx.doi.org/10.1016/j.celrep.2013.11.014
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012; 22(6):725-36; PMID:23201165; http://dx.doi.org/10.1016/j.ccr.2012.09.022
- Akhurst RJ, Hata A. Targeting the TGFb signalling pathway in disease. Nat Rev Drug Discov 2012; 11(10):790-811; PMID:23000686; http://dx.doi.org/10.1038/nrd3810