
Abstract
LRRK2 mutations are a frequent cause of familial Parkinson disease (PD) and are also found in a number of sporadic PD cases. PD-linked G2019S and I2020T mutations in the kinase domain of LRRK2 result in elevated kinase activity, which is required for the toxicity of these pathogenic variants in cell and animal models of PD. We recently reported that LRRK2 interacts with and phosphorylates a number of mammalian ribosomal proteins, several of which exhibit increased phosphorylation via both G2019S and I2020T LRRK2. Blocking the phosphorylation of ribosomal protein s15 through expression of phospho-deficient T136A s15 prevents age-associated locomotor deficits and dopamine neuron loss caused by G2019S LRRK2 expression in Drosophila indicating that s15 is a pathogenic LRRK2 substrate. We previously described that G2019S LRRK2 causes an induction of bulk mRNA translation that is blocked by T136A s15 or the protein synthesis inhibitor anisomycin. Here, we report the protective effects of the eIF4E/eIF4G interaction inhibitor 4EGI-1, in preventing neurodegenerative phenotypes in G2019S LRRK2 flies, and discuss how our findings and those of other groups provide a framework to begin investigating the mechanistic impact of LRRK2 on translation.
Introduction
LRRK2 is a large protein with several protein interaction domains and a central catalytic tri-domain consisting of GTPase Roc (Ras of complex proteins), COR (C-terminal of Roc) and kinase domains where several disease-segregating mutations are found.Citation1 A consensus exists that the most common LRRK2 mutation (G2019S) confers increased kinase activity and that in vivo G2019S LRRK2 toxicity can be effectively blocked by LRRK2 kinase inhibitors.Citation2-9 We and others have observed that a second PD-linked kinase domain mutation, I2020T, also results in elevated LRRK2 kinase activity, additionally highlighting the importance of LRRK2 kinase activity in PD etiology. Identification of LRRK2 substrates that contribute to its pathophysiological effects on the nervous system is necessary to elucidate the role of LRRK2 kinase activity in PD. Several candidate LRRK2 substrates have been reported, including endophilin A at the neuromuscular junction,Citation10 the ERM family proteins ezrin, radixin and moesin which link the actin cytoskeleton to plasma membrane proteins,Citation11 and eIF4E-BP1 (eukaryotic initiation factor 4E-binding protein 1)Citation12 which regulates cap-dependent translation. These substrates suggest a role for LRRK2 in synaptic vesicle endocytosis, cytoskeletal and mRNA translation regulation, respectively.
Increased Protein Synthesis Underlies Pathogenic LRRK2 Toxicity
We recently sought to identify LRRK2 kinase substrates through a multi-step process starting with a screen for LRRK2-interacting phospho-proteins by LRRK2 tandem affinity purification, IMAC enrichment of phospho-proteins and mass spectrometry in series. Interactors identified by this approach were subsequently screened via in vitro LRRK2 kinase assays to determine which are direct LRRK2 substrates. Using this method, we found that LRRK2 interacts with and phosphorylates several ribosomal proteins. Phosphorylation of the 40S ribosomal subunit protein s15 is increased by G2019S and I2020T LRRK2 and blocking s15 phosphorylation through expression of phospho-deficient T136A s15 rescues neurotoxicity in human dopamine neurons.Citation13 We next incorporated a Drosophila model of mutant LRRK2 toxicity in which flies expressing G2019S LRRK2 in dopamine neurons via the Ddc-gal4 driver exhibit accelerated age-dependent decline in locomotor function and a selective loss of dopamine neurons.Citation14 Ablation of dopamine signaling in the Drosophila nervous system results in pronounced locomotor deficits,Citation15,16 hence loss of locomotor activity concomitant with dopamine neuron death in this model is likely caused by an age-dependent loss of dopamine signaling. Phospho-deficient T136A s15 co-expression prevented these neurodegenerative phenotypes in G2019S LRRK2 flies and our studies also revealed that G2019S LRRK2 causes an increase in bulk protein synthesis which can be blocked by phospho-deficient s15, linking elevated protein synthesis to LRRK2 toxicity. Dietary treatment of flies with a low dose of the protein synthesis inhibitor anisomycin throughout adulthood also prevents neurodegenerative phenotypes from occurring in aged flies expressing G2019S LRRK2, providing independent verification of the link with aberrant protein synthesis.
The Protein Synthesis Inhibitor 4EGI-1 Blocks G2019S LRRK2 Toxicity
Translation proceeds through both cap-dependent and cap-independent mechanisms. Cap-dependent translation begins with the assembly of the eIF4F complex around the 5′ mRNA cap structure. eIF4F consists of the cap-binding protein eIF4E which binds directly to the 5′ m7G cap, the scaffold protein eIF4G and the helicase eIF4A, which unwinds secondary structure in the 5’-untranslated region (5′UTR) of the mRNACitation17 (). Other initiation factors and poly(A)-binding protein also bind to the eIF4F complex and are important in ribosome recruitment to the cap complex and translation initiation. Binding of the 43S pre-initiation complex to mRNA internal ribosome entry sites (IRES) initiates cap-independent translation which occurs without the need for eIF4E, or certain other initiation factors, depending on the type of IRES presentCitation18 (). Our reporter-based experiments in cell culture indicate that G2019S LRRK2 stimulates both cap-dependent and cap-independent translation. Since phospho-deficient s15 blocks the effects of G2019S LRRK2 on both modes of translation to a similar extent, insight into the relative contributions of increased cap-dependent and cap-independent translation to LRRK2 toxicity are not provided by phospho-deficient s15. A similar limitation applies to interpreting the protective effect of anisomycin treatment, as this inhibits the peptidyl transferase activity of the ribosome required for peptide bond formation and polypeptide release in both modes of translation. We additionally found that supplementing fly food with 10 μM 4EGI-1 throughout adulthood partially rescues the locomotor deficits and loss of dopamine neurons associated with G2019S LRRK2 expression (). 4EGI-1 specifically inhibits cap-dependent translation by interfering with the interaction between eIF4E and eIF4GCitation19 (). eIF4E is thought to be the limiting initiation factor required for eIF4F complex assembly.Citation20 Although the canonical model of cap-dependent translation holds that eIF4F complex is required for the translation of all mRNAs, numerous reports have demonstrated that manipulating levels of eIF4E directly or indirectly does not have a substantial impact on global mRNA translation, but predominantly affects translation of a specific subset of mRNAs, so-called ‘eIF4E-sensitive’ transcripts.Citation20 Hence, as a reduction of eIF4E activity by 4EGI-1 would likely have a similar effect as reducing eIF4E levels, these results suggest that cap-dependent translation of eIF4E-sensitive transcripts is important in pathogenic LRRK2 toxicity. Interestingly, treatment of control flies with the same dose of 4EGI-1 in food causes a pronounced impairment of locomotor activity in aged flies without affecting dopamine neuron numbers (). A lack of similar impairment of locomotor activity by 4EGI-1 in G2019S-LRRK2 flies is consistent with it being caused by a deficit in protein synthesis in control flies. This locomotor impairment occurs in the absence of outright death of dopamine neurons, suggesting that geotaxis deficits in control flies may preceed dopamine neuron death or possibly be independent of dopamine signaling. One apparent complication in the context of our previously published findings is that although disrupting the 4E/4G interaction with 4EGI-1 partially blocks pathogenic LRRK2 toxicity, flies expressing hypomorphic alleles of eIF4E do not exhibit protection from G2019S LRRK2.Citation13 This disparity may be related to a difference in the extent of eIF4E activity blockade, which may be more pronounced in eIF4E hypomorphs versus 4EGI-1 treatment. Alternatively, it could be caused by a differential effect of manipulating eIF4E only during adulthood in the case of 4EGI-1, vs. inhibition during embryonic and pupal stages for eIF4E mutant alleles tested. Notably, inhibiting cap-dependent translation with 4EGI-1 across a range of doses (not shown) yielded only partial rescue of LRRK2 neurotoxicity in G2019S LRRK2 transgenic flies that was maximal at 10 μM 4EGI-1 in fly food (). This is consistent with both modes of translation being important in the neurodegenerative phenotypes observed, and may warrant additional study of cap-independent translation on IRES-containing transcripts as specific mediators of LRRK2 toxicity. Another complication in the data is that while 4EGI-1 causes locomotor dysfunction in the control group, anisomycin does not. This, however, may be explained by the different mechanistic effects of the two compounds in their mode of translation inhibition. Specifically, anisomycin inhibits ribosomal translocation on all mRNAs while 4EGI-1 inhibits cap complex formation and is hypothesized to predominantly affect “eIF4E-sensitive” transcripts. Hence, the two drugs would likely have substantially different effects on translational profiles that may account for their divergent effects on control fly climbing ability.
Figure 1. Schematic representation of eukaryotic cap-dependent and cap-independent translation and protein synthesis inhibitor sites of action. LRRK2 phosphorylates the 40S ribosomal subunit protein s15 which is increased via G2019S LRRK2 to stimulate both cap-dependent and cap-independent translation in bicistronic reporter assays.Citation13 In cap-dependent translation, the 40S ribosomal subunit binds several initiation factors (eIFs 1,1A, 2, 3, 5 not shown) to become the 43S pre-initiation complex which binds to the eIF4F cap complex (comprised of eIFs 4A, 4E, 4G plus other associated factors) assembled at the m7G mRNA cap. 4EGI-1 interacts with eIF4E and blocks cap complex formation thereby inhibiting cap-dependent translation. In cap-independent translation, the 43S pre-initiation complex binds to internal ribosome entry sites (IRES) on mRNA to initiate scanning along the 5’UTR and eventual start codon recognition. Other initiation factors except eIF4E may be involved in ribosomal recruitment to IRES (not shown). Following translation initiation, progression of the elongating ribosome along mRNA can be halted by the peptidyl transferase inhibitor anisomycin. Both 4EGI-1 and anisomycin are reversible inhibitors of protein synthesis.
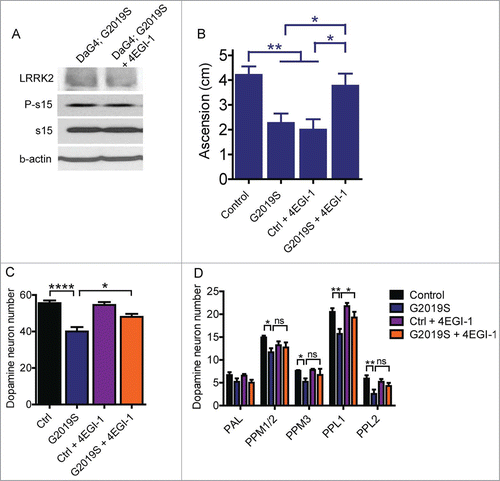
Figure 2. 4EGI-1 partially blocks G2019S LRRK2 toxicity in 6-week-old Drosophila. (A) Treatment of fly food with 10 μM 4EGI-1 does not affect P-s15 levels in flies expressing G2019S LRRK2 via Da-Gal4. (B) Climbing deficits in aged G2019S LRRK2 flies is rescued by 10 μM 4EGI-1 (ANOVA, Bonferroni's post-test * p < 0.05, ** p < 0.01, n = 15–24 flies per genotype). (C) Total dopamine neuron number is significantly rescued by 10 μM 4EGI-1 (ANOVA, Bonferroni's post-test, * p < 0.05, **** p < 0.0001, n = 7–11 flies per genotype). (D) Of the major dopamine neuron clusters, only PPL1 shows a significant rescue effect in G2019S LRRK2 transgenics following 4EGI-1 treatment (individual ANOVAs, Bonferroni's post-test, * p < 0.05, ** p < 0.01, n = 7–11 flies per genotype). Data from control and G2019S LRRK2 groups in (D) were previously published.Citation13
LRRK2 was previously reported to phosphorylate 4E-BP1 at several phospho-sites that are subject to direct mTORC1 phosphorylation.Citation12 Phosphorylation of 4E-BP1 prevents its binding to eIF4E, thereby increasing levels of free eIF4E available for cap complex formation.Citation17 If LRRK2 modulated translation through 4E-BP1 phosphorylation in addition to its effects on s15, it would also be consistent with the protective effect of 4EGI-1 found here. However, whether 4E-BP1 is a direct LRRK2 substrate is unclear as several groups including ours have been unable to detect an increase in phospho-4E-BP1 levels in transgenic LRRK2 models.Citation13,21,22 Hence, the involvement of 4E-BP1 phosphorylation in LRRK2-mediated translational effects remains to be clarified.
The Link Between Increased Protein Synthesis and Neuronal Toxicity
An important question arising from our findings is how elevated translation ultimately leads to neuronal degeneration in the context of Parkinson disease. One possibility is that increased expression of one or more key proteins results in toxicity either directly or through triggering a toxic cascade of events leading to neuronal demise. Alternatively, stimulating widespread anabolic protein production may place an excessive burden on cellular energy stores or protein quality control mechanisms leading to dysfunction in a neuronal-specific manner. Protein synthesis is tightly regulated and coordinated based on environmental conditions and available cell resources. The translation of certain mRNAs is highly dependent on these translational control mechanisms which is important for cell homeostasis and may be perturbed by mutant LRRK2. The role of wild type LRRK2 in translation is unclear, but if pathogenic mutations such as G2019S result in a loss of translational control, then this may be central to PD pathogenesis. Changes in neuronal translation profiles would be undesirable under basal conditions but are likely to be more detrimental under conditions unfavorable to anabolic protein production, e.g. during cell stress, in a manner that perturbs homeostasis and therefore renders neurons more vulnerable to harm. In the context of PD neurodegeneration, the hypothesis that mutant LRRK2 causes dysregulated translation that enhances neuronal susceptibility to stress is consistent with a combinatorial influence of genetics, aging and environmental influences on disease development.
Conclusions
There is accumulating evidence that LRRK2 interacts with components of the translation machinery and that altered mRNA translation is important in mutant LRRK2 toxicity. We found that LRRK2 interacts with ribosomal proteins and that phosphorylation of the 40S subunit protein s15 is key to the increased bulk protein synthesis, age-related dopamine neuron loss and climbing defects observed in G2019S LRRK2 transgenic Drosophila. Anisomycin treatment completely blocks LRRK2 toxicity in G2019S LRRK2 flies whereas 4EGI-1 partially rescues toxic phenotypes in these flies. The partial rescue effects of 4EGI-1, which blocks only cap-dependent translation is consistent with a role for both cap-dependent and cap-independent translation in LRRK2 toxicity. From these findings, several hypotheses can be generated as to how enhanced translation results in neuronal toxicity such as loss of translational control, loss of protein quality control, depletion of cell energy stores, or upregulation of specific (and currently unknown) proteins that lead to neuronal death.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the NIH/NINDS NS38377, the JPB Foundation, a New York Stem Cell Foundation-Druckenmiller Fellowship to I.M. L.A-A is supported by a La Caixa Foundation grant. T.MD is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation and the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation's Parkinson Disease Programs.
References
- Cookson M R. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson disease. Nat Rev Neurosci 2010; 11:791-7.; PMID:21088684; http://dx.doi.org/10.1038/nrn2935
- Anand V S, Reichling L J, Lipinski K, Stochaj W, Duan W, Kelleher K, Pungaliya P, Brown E L, Reinhart P H, Somberg R, et al. Investigation of leucine-rich repeat kinase 2 : enzymological properties and novel assays. The FEBS journal 2009; 276;466-78; PMID:19076219; http://dx.doi.org/10.1111/j.1742-4658.2008.06789.x
- Covy J P, Giasson, B I. Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem Biophysical Res Commun 2009; 378:473-7; PMID:19027715; http://dx.doi.org/10.1016/j.bbrc.2008.11.048
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug M P, Beilina A, Blackinton J, Thomas K J, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis 2006; 23:329-41; PMID:16750377
- Lee BD, Shin J H, VanKampen J, Petrucelli L, West A B, Ko H S, Lee Y I, Maguire-Zeiss K A, Bowers W J, Federoff H J, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson disease. Nat Med 2010; 16:998-1000; PMID:20729864; http://dx.doi.org/10.1038/nm.2199
- Liu Z, Hamamichi S, Lee B D, Yang D, Ray A, Caldwell G A, Caldwell K A, Dawson T M, Smith W W, Dawson V L. Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson disease models. Hum Mol Genet 2011; 20:3933-42; PMID:21768216; http://dx.doi.org/10.1093/hmg/ddr312
- Luzon-Toro B, Rubio de la Torre E, Delgado A, Perez-Tur J, Hilfiker S. Mechanistic insight into the dominant mode of the Parkinson disease-associated G2019S LRRK2 mutation. Hum Mol Genet 2007; 16:2031-9; PMID:17584768; http://dx.doi.org/10.1093/hmg/ddm151
- Smith W W, Pei Z, Jiang H, Dawson V L, Dawson T M, Ross C A. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 2006; 9:1231-3; PMID:16980962; http://dx.doi.org/10.1038/nn1776
- West A B, Moore D J, Choi C, Andrabi S A, Li X, Dikeman D, Biskup S, Zhang Z, Lim K L, Dawson V L, et al. Parkinson disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet 2007; 16:223-2; PMID:17200152; http://dx.doi.org/10.1093/hmg/ddl471
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock, P J, Morais V A, Vilain S, Haddad D, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 2012; 75:1008-21; PMID:22998870; http://dx.doi.org/10.1016/j.neuron.2012.08.022
- Parisiadou L, Xie C, Cho H J, Lin X, Gu X L, Long C X, Lobbestael E, Baekelandt V, Taymans J M, Sun L, et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci 2009; 29:13971-80; PMID:19890007; http://dx.doi.org/10.1523/JNEUROSCI.3799-09.2009
- Imai, Y., Gehrke, S., Wang, H.Q., Takahashi, R., Hasegawa, K., Oota, E., and Lu, B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J 2008; 27:2432-43.; PMID:18701920; http://dx.doi.org/10.1038/emboj.2008.163
- Martin I, Kim J W, Lee B D, Kang H C, Xu J C, Jia H, Stankowski J, Kim M S, Zhong J, Kumar M, et al. Ribosomal protein s15 phosphorylation mediates LRRK2 neurodegeneration in Parkinson disease. Cell 2014; 157:472-85; PMID:24725412; http://dx.doi.org/10.1016/j.cell.2014.01.064
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, et al. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A 2008; 105:2693-8; PMID:18258746; http://dx.doi.org/10.1073/pnas.0708452105
- Friggi-Grelin F, Coulom H, Meller M, Gomez D, Hirsh J, Birman S. Targeted gene expression in drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J Neurobiol 2003; 54:618-27; PMID:12555273
- Riemensperger T, Isabel G, Coulom H, Neuser K, Seugnet L, Kume K, Iche-Torres M, Cassar M, Strauss R, Preat T, et al. Behavioral consequences of dopamine deficiency in the Drosophila central nervous system. Proc Natl Acad Sci U S A 2011; 108:834-9; PMID:21187381; http://dx.doi.org/10.1073/pnas.1010930108
- Hershey J W, Sonenberg N, Mathews M B. Principles of translational control: an overview. Cold Spring Harb Perspect Biol 2012; 4; PMID:23209153; http://dx.doi.org/10.1101/cshperspect.a011528
- Jackson R J, Hellen, C U, Pestova T V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 2010; 11:113-27; PMID:20094052; http://dx.doi.org/10.1038/nrm2838
- Hoeffer C A, Cowansage K K, Arnold E C, Banko J L, Moerke N J, Rodriguez R, Schmidt E K, Klosi E, Chorev M, Lloyd R E, et al. Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proc Natl Acad Sci U S A 2011; 108:3383-8; PMID:21289279; http://dx.doi.org/10.1073/pnas.1013063108
- Roux P P, Topisirovic I. Regulation of mRNA translation by signaling pathways. Cold Spring Harb Perspects Biol 2012; 4:1-23; PMID:22888049; http://dx.doi.org/10.1101/cshperspect.a012252
- Kumar A, Greggio E, Beilina A, Kaganovich A, Chan D, Taymans J M, Wolozin B, Cookson M R . The Parkinson disease associated LRRK2 exhibits weaker in vitro phosphorylation of 4E-BP compared to autophosphorylation. PloS one 2010; 5:e8730; PMID:20090955; http://dx.doi.org/10.1371/journal.pone.0008730
- Trancikova A, Mamais A, Webber P J, Stafa K, Tsika E, Glauser L, West A B, Bandopadhyay R, Moore D J. Phosphorylation of 4E-BP1 in the mammalian brain is not altered by LRRK2 expression or pathogenic mutations. PloS one 2012; 7, e47784; PMID:23082216; http://dx.doi.org/10.1371/journal.pone.0047784