
Abstract
Individual or combinations of somatic mutations found in genes from colorectal cancers can redirect the effects of chemotherapy and targeted agents on cancer cell survival and, consequently, on clinical outcome. Novel therapeutics with mechanisms of action that are independent of mutational status would therefore fulfill a current unmet clinical need. Here the CEA and CD3 bispecific single-chain antibody MEDI-565 (also known as MT111 and AMG 211) was evaluated for its ability to activate T cells both in vitro and in vivo and to kill human tumor cell lines harboring various somatic mutations commonly found in colorectal cancers. MEDI-565 specifically bound to normal and malignant tissues in a CEA-specific manner, and only killed CEA positive cells. The BiTE® antibody construct mediated T cell-directed killing of CEA positive tumor cells within 6 hours, at low effector-to-target ratios which were independent of high concentrations of soluble CEA. The potency of in vitro lysis was dependent on CEA antigen density but independent of the mutational status in cancer cell lines. Importantly, individual or combinations of mutated KRAS and BRAF oncogenes, activating PI3KCA mutations, loss of PTEN expression, and loss-of-function mutations in TP53 did not reduce the activity in vitro. MEDI-565 also prevented growth of human xenograft tumors which harbored various mutations. These findings suggest that MEDI-565 represents a potential treatment option for patients with CEA positive tumors of diverse origin, including those with individual or combinations of somatic mutations that may be less responsive to chemotherapy and other targeted agents.
Abbreviations
| BiTE® | = | bi-specific T cell engager |
| CEA | = | carcinoembryonic antigen |
| CEACAM5 | = | CEA-related cell adhesion molecule family member 5 |
| DHFR | = | dihydrofolate reductase |
| FFPE | = | formaldehye fixed paraffin embedded |
| EC50 | = | half maximal effective concentration |
| IV | = | intravenous |
| MEDI-565 | = | bispecific single-chain antibody specific for CEA and human CD3 |
| peripheral blood mononuclear cells | = | PBMC |
| SC | = | subcutaneous |
| scFv | = | single chain variable fragment |
| SEM | = | standard error of the mean |
| TMA | = | tissue microarray |
Introduction
Significant progress has been made during the past decade to treat patients with metastatic colorectal cancer (mCRC) following the approval of multiple new agents and treatment strategies. However, individual or combinations of somatic mutations in genes of mCRC can limit the effectiveness of standard chemotherapy and targeted therapies.Citation1 Thus, prognosis for patients with mCRC remains poor with 5-year survival rates <20%.Citation2 The development of novel agents that provide clinicians with therapies that work independently of the mutational status of the tumor may prove quite beneficial to enhance the overall survival of patients with mCRC.
Some of the most common somatic mutations found in genes of mCRC include mutated KRAS and BRAF oncogenes, activating PI3KCA mutations, loss of phosphatase and tensin homolog (PTEN) expression, and loss-of-function mutations in TP53.Citation3 Individually or in combination, these sets of mutations negatively impact the effects of chemotherapy and targeted agents on cancer cell survival and, in some cases, on clinical outcome.Citation4-9 Receptor-independent signaling of the epidermal growth factor receptor type 1 (EGFR-1) pathway by mutated KRAS, BRAF, or PI3KCA, or the loss of PTEN expression may eliminate the need of mCRC cells to express high levels of EGFR-1 Citation10 or activate intracellular signaling cascades through receptor-ligand interactions,Citation11 both factors that may limit the potential benefits of anti-EGFR-1 antibody therapy in patients with mCRC. For example, patients with tumors mutated in KRAS and BRAF do not benefit from an increased overall survival following administration of cetuximab (Erbitux®) or panitumumab (Vectibix®), monoclonal antibodies that block EGFR-1 signaling.Citation4,12-19 In addition, loss-of-function mutations in TP53 limits the ability of cancer cells to undergo p53-mediated apoptosis Citation20 which has been reported to reduce chemosensitivity to 5-fluorouracil (5-FU) Citation21,22 and in some cases of ovarian cancer limit clinical response following treatment with platinum-based chemotherapies. Citation23-30 Thus, mutational status has become an important biomarker to identify patients who are likely to benefit from a specific treatment, and has the potential to guide clinicians in prescribing a chemotherapy or targeted therapy.Citation31
Standard of care chemotherapy and targeting antibody therapies may initially reduce the tumor burden, but relapse for late-stage patients is common. The human immune response represents another opportunity to control neoplastic disease. Tumor antigen-specific T cells are readily found within tumors and in tumor-draining lymph nodes, but the clearance of established tumors by the immune system is rare.Citation32 Multiple mechanisms exist to subvert antigen-specific, T cell-mediated destruction of tumor cells thereby resulting in tumor immune evasion.Citation33 These include down-regulation of major histocompatibility (MHC) class I complexes on the surface of tumor cells, lack of tumor antigen processing and presentation, escape from T-cell induced destruction, as well as induction of T cell anergy, exhaustion, or apoptosis, and suppression of effector T cell function by co-inhibitory receptor pathway signaling or suppressor cell populations, in particular by regulatory T cells.Citation34
Bispecific T cell engager (BiTE®) antibody constructs were designed to transiently link effector T cells to tumor cells via concurrent binding to CD3 on peripheral and/or tumor-resident T cells and tumor-associated antigens on the surface of tumor cells.Citation35,36 This productive interaction results in the activation of T cells through CD3/T cell receptor complex signaling, and subsequent T cell-mediated lysis of targeted tumor cells. Consequently, BiTE® antibody constructs act independently of T cell receptor specificity, co-stimulatory or co-inhibitory signals, and peptide antigen presentation, thus bypassing several well-characterized mechanisms by which tumor cells evade immune recognition and destruction.Citation37 Some of the major functional characteristics of BiTE® antibody constructs that have been reported include a strict dependence on the presence of target cells for activation of T cells,Citation38 the creation of a functional cytolytic synapse,Citation39 serial T cell mediated lysis of target cells,Citation40 and highly potent redirected lysis of target cells.Citation41 In addition, neither antibody internalization nor T cell anergy were observed upon long-term treatment of tumor-bearing mice with a BiTE® antibody construct, suggesting that BiTE® antibody constructs themselves do not induce T cell anergy in the tumor microenvironment and can sustain an active T cell response during prolonged periods of T cell activation.Citation42
Currently, 4 different BiTE® antibody constructs are undergoing evaluation in clinical trials for the treatment of cancer, and these BiTE® antibody constructs target CD19 on B cell malignancies (blinatumomab; AMG 103; MT103), epithelial cell adhesion molecule (EpCAM; CD326) on adenocarcinomas (solitomab; AMG 110; MT110), prostate-specific membrane antigen (PSMA) on prostate adenocarcinomas (AMG 212; BAY2010112) and carcinoembryonic antigen (CEA/CEACAM5/CD66e) on gastrointestinal adenocarcinomas (MEDI-565; MT111; AMG 211). In studies of non-Hodgkin lymphoma and B-precursor acute lymphoblastic leukemia (B-ALL) patients, blinatumomab has been reported to induce a high rate of clinical benefit with an acceptable safety profile.Citation43-49 The blinatumomab data demonstrates clinical proof of principle for the platform technology. Clinical studies are ongoing in adult and pediatric patients with relapsed/refractory B-precursor ALL, adults with persistent minimal residual disease B-precursor ALL, and adults with relapsed diffuse large B cell lymphoma (DLBCL), (ClinicalTrials.gov identifiers NCT01471782, NCT01207388, NCT01466179, and NCT01741792). Solitomab targets EpCAM on both epithelial carcinomas as well as tumor-initiating cells,Citation50-52 and has entered a Phase I trial in patients with solid tumors that express EpCAM (ClinicalTrials.gov identifier NCT00635596). AMG 212 is being tested in a Phase I trial in patients with castration-resistant prostate adenocarcinoma (ClinicalTrials.gov identifier NCT01723475). MEDI-565 targets CEA and has entered Phase I clinical trials in patients with advanced gastrointestinal adenocarcinomas (ClinicalTrials.gov identifier NCT01284231).
CEA represents an attractive target for BiTE® antibody construct-mediated therapy. Historically, tumor-bound CEA has been used for cancer imaging and in vaccine and antibody-based therapeutic approaches for cancer treatment.Citation53-56 The antigen is limited to the apical surface and luminal portion of adult colonic epithelium under normal physiological conditions, but is over-expressed and loses its polarized distribution in CRC and other cancers resulting in expression of CEA over the entire cell surface.Citation57 This distribution in normal and neoplastic cells may result in specific targeting of CEA-positive tumor cells and sparing of normal colonic epithelium where T cell traffic is restricted to basolateral surfaces by the tight junctions separating apical and basolateral membranes. Interestingly, phospholipase cleavage of CEA from the surface of tumor cells Citation58 results in the accumulation of a soluble form in human blood.Citation57 Serum CEA levels are currently used clinically to monitor disease recurrence in patients after surgical resection of colorectal tumors,Citation59 and serves as a negative prognostic indicator in patients with gastrointestinal cancers.Citation60-62 However, soluble circulating CEA might pose a particular challenge because it could competitively inhibit the binding of CEA-specific targeted therapies and thereby interfere with its antitumor activity.
MEDI-565 was developed from a set of BiTE® antibody constructs that redirected T cells in vitro to kill tumor cells expressing CEA and in vivo to inhibit growth of tumors cells.Citation63 A series of studies with MEDI-565 demonstrated that the BiTE® antibody construct could induce patient T cells to kill metastatic CRC specimens derived from patients previously treated with conventional chemotherapy.Citation64 Herein we extend these initial findings by characterizing the in vitro and in vivo pharmacology of MEDI-565. Key parameters assessed in this report include the BiTE® antibody construct's (a) bispecific binding capacity, (b) high potency of T cell killing of CEA-expressing cells, (c) functional effects on T cells, (d) mechanism of action of T cell killing, (e) ability to induce T cell killing of CEA-expressing cells independent of CEA density, of the presence of high levels of soluble CEA, or of the mutational status of tumor cell lines, and (f) capacity to inhibit growth of CEA-expressing tumors in xenograft mouse models. These results extend our understanding of the functional attributes of CEA-specific BiTE® antibody constructs and demonstrate the unique anti-tumoral properties of MEDI-565.
Results
Bispecific binding of MEDI-565 to CEA and human CD3
Flow cytometry binding studies were conducted to characterize the target antigens bound by the 2 binding domains of MEDI-565. MEDI-565 bound specifically to CHO cell lines stably expressing human (CHO/huCEA) and cynomolgus monkey CEA (CHO/cynoCEA) and not to the parental CHO cell line (Table S1). An antibody generated from the CEA binding domain of MEDI-565 bound only to cell lines expressing CEACAM5 and did not bind to cell lines expressing the other human CEACAM family members (Table S2). These results demonstrated that surface expression of CEA was required for binding of MEDI-565 to cells. In addition, MEDI-565 bound to CD3+ T cells from human peripheral blood mononuclear cells (PBMC) but not from cynomolgus monkey PBMCs (Table S1). Thus, MEDI-565 showed specificity and dual binding activity to both CEA-expressing cell lines and CD3-positive human T cells. Apparent equilibrium dissociation constant (Kd) values were determined for MEDI-565 binding to human CEA and CD3 from cell binding experiments. MEDI-565 bound to human CEA on CHO/huCEA cells with a Kd of 5.5 ± 2.2 nM and to CD3ϵ on primary human CD3 T cells with a Kd of 310 ± 67 nM (mean ± standard deviation).
MEDI-565 binds to malignant tissue of diverse origin
A targeted binding study was conducted to determine if MEDI-565 bound to normal epithelium and more broadly to cancerous tissue that typically express CEA. To accomplish this objective, MEDI-565 and a control BiTE® antibody construct were tested for their ability to bind cryosections of normal and cancerous human tissues. The control BiTE® antibody construct bound to an irrelevant antigen but shared the anti-CD3-binding arm with MEDI-565 and was used to determine the specificity of MEDI-565 in the tissue staining studies.
Membranous staining was observed with MEDI-565 in cancers included on 2 frozen human cancer tissue micro-arrays (TMAs-multi-tumor and pancreatic; ). The percentage of cancer specimens in these TMAs stained with MEDI-565 was as follows: 9 of 10 (90%) colon, 3 of 10 (30%) ovary, 5 of 10 (50%) breast, 7 of 10 (70%) lung, 1 of 10 (10%) prostate, and 9 of 10 (90%) pancreas. Of the 15 normal tissues (5 pancreas, 2 colon, 2 ovary, 2 breast, 2 lung and 3 prostate) present in these TMAs, staining with MEDI-565 was limited to colonic epithelium (2 of 2; 100%) (). Cytoplasmic and membranous staining with MEDI-565 was detected in 14 of 17 (82%) esophageal carcinomas representing 13 samples of squamous cell carcinoma (SCC), 2 samples of basal cell carcinoma (BCC) and 1 sample of adenocarcinoma. One section of normal squamous epithelium (1 of 1), and 2 hyperplastic squamous epithelium specimens (2 of 2) stained with both MEDI-565 and the control BiTE® antibody construct where the MEDI-565 staining predominated in the superficial layers while the 3 control BiTE® antibody construct treated samples stained in the basal layer of the epithelium. In the gastric cancer FFPE TMA (formaldehyde fixed paraffin embedded tissue micro-array), 61 of 68 (90%) gastric carcinomas representing 12 grade II, 42 grade III, and 14 grade IV demonstrated moderate to strong cytoplasmic and membranous staining in luminal epithelial cells with ascending tumor grade, whereas staining was not detected in the 15 normal gastric mucosae present in the same array. Cytoplasmic staining with MEDI-565 of hepatocytes in liver cancer TMA FFPE preparations was observed in 3 of 77 (4%) samples. In 2 of these samples, identical staining was also recorded using the control BiTE® antibody construct indicating that this interaction was not specific. These results indicate a very low expression of the antigen recognized by MEDI-565 in liver cancer.
Figure 1. Binding of MEDI-565 to normal and malignant human tissue sections. (A) Representative immunohistochemistry (IHC) images of normal human colon epithelium and normal human pancreas tissues stained with MEDI-565 or control BiTE® antibody construct. (B) Representative IHC images of human colorectal and pancreatic carcinoma tissues stained with MEDI-565 or control BiTE® antibody construct.
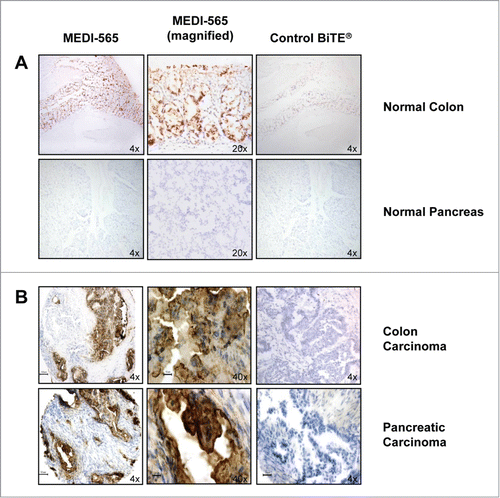
These results provided evidence that MEDI-565 bound specifically to epithelial cells within normal colon, but did not bind to epithelial cells or stromal elements within a variety of other normal tissues (). In contrast, MEDI-565 bound to a number of different tumors that included a majority of the tested samples originating from cancerous tissues of the colon, stomach, esophagus, pancreas, and lung (examples shown in ).
Characterization of the in vitro activity of MEDI-565
Similar to other described CEA-specific BiTE® antibody constructs,Citation63 the CEA- and CD3-binding arms of MEDI-565 were designed to redirect human T cells for the CEA-specific lysis of target cells. Parental CHO dihydrofolate reductase deficient (dhfr-) cells (CEA negative) and CHO cells stably expressing CEA were used in a flow cytometry-based assay to determine the cytotoxic activity of MEDI-565 against CEA-expressing cells with a non-mutated background. MEDI-565 induced T cells in a concentration-dependent manner to efficiently lyse CHO cells expressing the CEA antigen (), while the viability of the parental CHO cells remained unaltered, even when exposed to high concentrations of MEDI-565 (). A control BiTE® antibody construct did not induce T cell lysis of either the parental CHO cells or CHO cells expressing CEA (). These results demonstrated that MEDI-565-induced T cell killing required the expression of CEA on target cells.
Figure 2. Factors that influence MEDI-565-induced T cell killing of target cells: CEA expression on target cells, the presence of soluble CEA and effector:target ratios. (A) Activity of MEDI-565 (•) or control BiTE® antibody (□) to induce T cell killing of CEA positive CHO cells (CHO/huCEA) and CEA negative parental CHO cells (CHO dhfr-). (B) Effect of the indicated effector-to-target cell ratio on MEDI-565-induced T cell killing of CHO/huCEA cells over a range of MEDI-565 concentrations. In addition, the relationship between the maximum specific lysis and the EC50 values of lysis achieved in cytotoxicity assays are graphed versus E:T ratios. (C) Correlation (r2 = 0.77) of the estimated number of CEA binding sites for MEDI-565 on the surface of tumor cell lines with the potency (EC50 values) of redirected T cell lysis. The measured number of CEA molecules per cell for each of 6 tumor cell lines (ASPC-1, BxPC3, HPAC, HPAF II, H727 and LS174T) was plotted against the respective EC50 values of the percentage of tumor cells lysed from 3 individual donor T cells. P and r2 of the linear regression curve are listed on the graph. r2, coefficient of determination, calculated from Pearson's correlation coefficient (r). (D) Effect of the indicated concentrations of soluble CEA on MEDI-565 induced T cell killing of CHO/huCEA cells during a 72 hour assay. Data in the figure show representative results for assays using T cells from 3 different donors. All target cells were cultured for 72 hours with unstimulated human CD3+ T cells at an E:T ratio of 10:1 or as indicated in the figure. Error bars show the SEM in panels A, B and D.
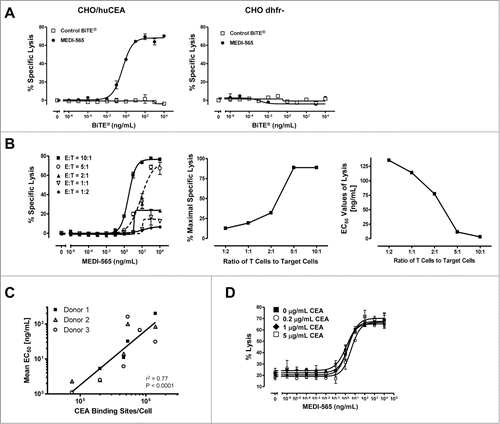
MEDI-565 was highly effective at the highest E:T cell ratios (10:1 and 5:1; ). The percentage of maximal lysis decreased rapidly between E:T cell ratios of 5:1 and 2:1 (middle panel, ) and remained low at E:T ratios of 1:1 and 1:2. Overall, the potency of MEDI-565 was inversely proportional to the E:T ratio, such that higher E:T cell ratios yielded lower EC50 values (right panel, ). Thus, these results suggest that MEDI-565 induced a concentration-dependent, polyclonal T cell response that was influenced by the number of T cells and resulted in the death of cells specifically expressing CEA.
Human tumor cell lines from diverse origins express various levels of CEA on their cell surface. To test whether CEA antigen density on the surface of cells correlated with MEDI-565 potency, we examined the ability of MEDI-565 to induce T cell killing of 6 selected tumor cell lines (ASPC-1, BxPC3, HPAC, HPAF II, H727 and LS174T) expressing varying levels of CEA density on their surface (). T cells from individual donors were combined with each tumor cell line and MEDI-565. Efficient T cell killing was observed for all tumor cell lines after 48 hours in culture, similar to data presented in . Interestingly, the surface density of CEA on the tumor cells did have an effect on the efficiency of MEDI-565 activity. We observed a significant inverse correlation (r2 = 0.77; P < 0.0001) in which the potency of MEDI-565 increased as the number of CEA binding sites on the tumor cells decreased (). Taken together, these results suggested that MEDI-565 can effectively induce human T cells to kill tumor cells expressing CEA, and the overall potency of MEDI-565 may depend upon the levels of CEA expressed by the target cells.
Table 1. Relationship between MEDI-565 directed cytotoxicity of cancer cell lines and their mutational status and CEA density. Results from various cytotoxicity assays are shown. Potency of redirected T cell lysis of human cancer cell lines is reported as EC50 values in ng/mL. Each assay used an unique set of donor T cells, an E:T ratio of 5:1 and an assay duration of 48 hours. Cytotoxicity measurements for ASPC1, MKN45 and PC3 cell assays utilized a flow cytometry-based readout; for LS174T, HT-29, BXPC3, PAN0813, HPAFII, HPAC, H727, A549 and BT474 cell assays a caspase 3 measurement was used. Somatic mutation status for the indicated genes for each cell line was compiled from the COSMIC database (http://www.sanger.ac.uk/cosmic). N, number of replicate lysis experiments; EC50, antibody concentration required for half-maximal effective cell lysis; CEA density, MEDI-565 binding sites per cell; ND, not determined
Figure 3. Kinetics of MEDI-565 mediated T cell cytotoxicity, activation and cytokine release. All data presented in this figure were collected from the same representative co-culture experiment; unstimulated human CD3+ T cells at an E:T ratio of 10:1 cultured with the indicated concentrations of MEDI-565 listed in panel A. Symbols listed in panel A are also used in panels B and C. (A) Percent specific lysis over time of DiO-labeled CHO/huCEA target cells incubated with CD3-enriched human T cells and the indicated concentrations of MEDI-565 or control BiTE® antibody construct. (B) Activation of CD8+ T cells over time as measured by upregulation of the T cell activation markers CD69 or CD25, at the indicated concentrations of MEDI-565 or control BiTE® antibody construct. (C) Secretion of cytokines, as indicated, into cell culture supernatants over time.
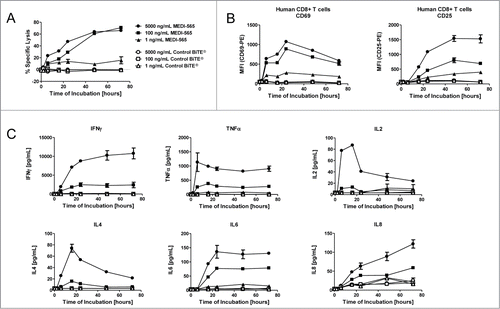
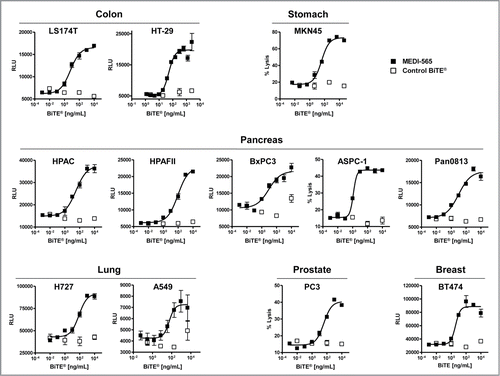
Figure 4. MEDI-565 induced T cell lysis of human cancer cell lines derived from various tissues. Activity of MEDI-565 (▪) or control BiTE® antibody (□) at the indicated concentrations to induce T cell killing of colon (LS174T, HT-29), stomach (MKN45) pancreatic (HPAC), and gastric (MKN45), pancreas (HPAC, HPAF II, BxPC3, ASPC-1, Pan08), lung (H727, A549), prostate (PC3) and breast (BT474) cancer cell lines after 42 hours in culture. RLU, Relative Light Units from caspase 3/7 assay;% lysis, lysis of tumor cells as determined by flow cytometry-based cytotoxicity assay. Error bars show the SEM.
CEA can be released by phospholipases from the cell surface,Citation58 accumulate in the blood,Citation57 and may pose a particular challenge to targeted therapies because it can compete with antibody binding and interfere with antitumor activity of a targeted therapy. Indeed, flow cytometry-based studies confirmed that a 1 hour pre-incubation of soluble CEA with MEDI-565 resulted in competitive inhibition of binding of MEDI-565 to cell surface CEA expressed on CHO/huCEA cells (data not shown). To determine the effect that soluble CEA may have on the activity of MEDI-565, CHO/huCEA cells and human CD3+ T cells were co-cultured with varying concentrations of MEDI-565 and soluble CEA for 72 hours. Target cell lysis was determined by flow cytometry as the percentage of target cells becoming PI-positive after 72 hours. shows the effect of 3 different concentrations of soluble CEA (sCEA) ranging from 0.2 to 5 μg/mL, with 5 μg/mL representing a level above that typically found in the serum of cancer patients with CEA positive tumors.Citation68,69 The concentrations of MEDI-565 for half-maximal lysis of target cells expressing CEA were 1.5 ng/mL for MEDI-565 alone, 4.0 ng/mL of MEDI-565 for 0.2 μg/mL of sCEA, 1.3 ng/mL of MEDI-565 for 1.0 μg/mL of sCEA and 2.1 ng/mL of MEDI-565 for 5.0 μg/mL of sCEA (). Thus, none of the selected concentrations of sCEA showed a substantial effect on the potency or magnitude of MEDI-565-mediated in vitro cytotoxicity.
We next wanted to test the kinetics of MEDI-565-mediated T cell killing of CEA positive tumor cells. In these studies, MEDI-565 activity was measured in co-culture assays on both T cells and target cells expressing CEA. T-cell killing of CHO/huCEA was dependent on the concentration of MEDI-565 and rapid; target cell death was detected within 6 hours of exposure and increased with time (measured up to 72 hours; ). T-cell-mediated killing of cells expressing CEA coincided with the de novo expression of the early T cell activation marker CD69 on resting peripheral T cells derived from human PBMC (). De novo expression of the late T cell activation marker CD25 was delayed as compared to CD69 expression, initially detected on resting peripheral T cells derived from human PBMC 16 hours after the initiation of the co-culture (). Maximal levels of CD69 and CD25 were reached at different times on the T cells (24 hours and 48 to 72 hours for CD69 and CD25, respectively). MEDI-565 activated T cells to produce cytokines concomitant with the first measurement of T cell activity (target cell killing and CD69 expression) at 6 hours (). The broad array of cytokines released by T cells and the majority of T cells (60–78% of CD69-positive T cells for cultures with 5 ng/mL of MEDI-565) becoming newly activated was consistent with the polyclonal nature of the T cell activation by MEDI-565.
To determine mechanistically how MEDI-565 induced in vitro T cell-mediated killing of tumor cell lines expressing CEA (), we assessed mechanisms consistent with standard cytotoxic T cells and seen with other BiTE® antibody constructs (Fig. S1).Citation70 Granzyme B and perforin were released by T cells in a CEA-specific manner and concurrently with poly adenosine ribose polymerase (PARP) cleavage (Fig. S1). T cells activated by MEDI-565 did not eliminate target cells in the presence of ethylenediaminetetraacetic acid (EDTA) demonstrating the need for extracellular calcium to allow the process to occur (data not shown). The addition of the pan-caspase inhibitors Z-VAD-FMK and Q-VD-OPh blocked pro-caspase cleavage in target cells (data not shown) suggesting that T‑cell killing mediated by MEDI-565 occurs through apoptosis.
MEDI-565 mediates the T cell killing of CEA-positive tumor cells independently of mutational status
We next asked the question of whether individual or combinations of somatic mutations may affect the activity of MEDI-565 directed T cell killing of cancer cell lines. The cytotoxic activity of T cells directed by MEDI-565 against human cancer cell lines that were wild-type or mutant for key oncogenes and tumor suppressor genes was assessed by using a panel of 12 cancer cells lines that either lacked or possessed individual or combinations of somatic mutations in genes commonly found in colorectal cancer and known to mediate drug resistance (). For example, these tumor cell lines contain mutated KRAS and BRAF oncogenes, loss of PTEN expression and activating PI3KCA mutations that limit growth inhibition and clinical response by antagonist anti-EGFR antibodies including cetuximabCitation5,6,10 and mutated TP53 associated with reduced drug activityCitation9 and interference with apoptosis.Citation20 MEDI-565 induced T cell lysis of all cancer cell lines regardless of the gene mutation status (; Fig S2). The mean potency (EC50 concentrations) of MEDI-565 activity was 29 ± 20 ng/mL against the non-mutated MKN45 cell line and ranged from a mean of 3.5 ng/mL to 220 ng/mL against the panel of cancer cell lines with mutations in key oncogenes and tumor suppressors (). MEDI-565 was most active against ASPC-1, BT474, BXPC3 and LS174T cell lines; each harbored unique individual or combinations of mutated genes. MEDI-565 was least active against the HPAF II cell line (mean of 200 ng/mL) that possessed a KRAS and TP53 mutation. Overall, no trend or association was identified that related potency of MEDI-565 with an individual mutation or combinations of mutations commonly found in colorectal adenocarcinomas using this set of tumor cell lines. These results support the hypothesis that mutations in KRAS, BRAF, PTEN, PI3KCA, and TP53 may have limited impact on the ability of MEDI-565 to induce T cell killing of cancer cells in vitro.
Activity of MEDI-565 in KRAS pathway/TP53 non-mutated and mutated xenograft mouse models
The KRAS pathway and TP53 non-mutated gastric cancer line MKN45 and the mutated cell lines LS174T (colon), HT-29 (colon), HPAC (pancreas), HPAF II (pancreas) and H727 (lung) all express CEA and were used to establish xenograft tumor models in immunodeficient SCID mice. In addition, the HeyA8 (ovary) cell line that lacks expression of CEA was used as a control model in the mouse studies. Human T cells were administered to mice in the form of unstimulated PBMCs enriched for CD3+ T cells combined with the respective tumor cells at the indicated ratios before injection. Human PBMCs enriched for CD3+ T cells did not significantly affect the outgrowth of each tumor line in the absence of MEDI-565 (). Similarly, administration of the vehicle alone (PBS) or the control BiTE® antibody construct at 20 μg/dose/mouse for 5 d did not significantly inhibit growth of the human tumor cells in any of the mouse xenograft models. However, the control BiTE® antibody construct did produce noticeable (but not statistically significant) tumor growth reduction in the LS174T model (), and may represent a degree of non-specific activity of the anti-CD3 arm of the control BiTE® antibody construct in these experiments.
Figure 5. Antitumor activity of MEDI-565 in human tumor xenograft mouse models. (A) Mean tumor volume of Kras and PI3KCA mutant LS174T colon (CEA positive) or HeyA8 ovarian (CEA negative) tumor cells engrafted with human T cells in SCID mice and treated daily with PBS, MEDI-565 or control BiTE® antibody, as indicated (arrows) for 5 days. (B) Activity of MEDI-565 administered by the IV or SC routes of administration in mice engrafted with LS174T cells and human T cells. (C) Mean tumor volume of Kras, BRAF, PI3KCA and/or TP53 mutant tumors (HT-29, H727, HPAC, HPAF II) and wild-type (MKN45) tumors engrafted with human T cells. Error bars represent SEM. Arrows indicate study days when mice were administered test article. *, p < 0.05, Mann-Whitney rank sum test.
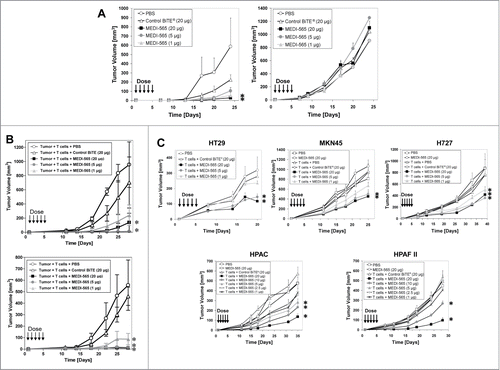
MEDI-565 was intravenously administered daily for 5 days to tumor-bearing mice and significantly inhibited the growth of the KRAS/PI3KCA mutant LS174T (CEA-positive; injected into the right hind flank) cancer cells by up to 95% as compared to the vehicle control group; in contrast, administration of MEDI-565 did not inhibit the growth of HeyA8 (lacks CEA expression; injected into the left hind flank) cancer cells (). All dose levels (20, 5 and 1 μg/dose/mouse) of MEDI-565 administered to the mice significantly inhibited growth of LS174T cells. Additionally, tumor growth in the LS174T xenograft mouse model was significantly inhibited up to 99% and 98% by MEDI-565 administered either IV (t1/2 of 4–6 hours) or SC (t1/2 of 4–6 hours), respectively, as compared to the vehicle control groups (). These results indicated that the in vivo activity of MEDI-565 required the expression of CEA by cancer cells and was independent of the route of administration.
In addition, IV administration of MEDI-565 in HPAC (KRAS mutant), HPAF II (KRAS/TP53 mutant), H727 (KRAS/TP53 mutant), HT29 (BRAF/PI3KCA/TP53 mutant) and MKN45 (wild-type) xenograft models inhibited tumor growth by as much as 72% (HPAC), 78% (HPAF II), 53% (H727), 58% (HT-29) and 52% (MKN45), compared to the control group (). Inhibition of tumor growth by MEDI-565 was dependent on the addition of T cells to the engraftment in the HPAC, HPAF II, HT29 and MKN45 models. Together, these in vivo studies demonstrate that the anti-cancer activity of MEDI-565 was dependent on the presence of T cells in the engraftment but independent of the mutational status of KRAS, BRAF, PTEN, PI3KCA and TP53 for the tumor cell lines tested.
Discussion
The non-polarized expression of CEA on human tumors represents an attractive target for the re-directed T-cell lysis of tumor cells mediated by BiTE® antibody constructs. We have shown that the CEA-specific BiTE® antibody MEDI-565 bound with relatively high frequency to primary human tumors that have a high prevalence of CEA-expression, including tumors of the colon, pancreas, stomach, esophagus, lung, and breast, as well as with a lower frequency to ovarian, prostate, and hepatocellular carcinomas. MEDI-565 demonstrated potent dose-dependent in vitro killing of tumor cell lines from a similarly diverse origin, including those derived from human colon, pancreatic, stomach, lung, breast, and prostate carcinomas. This killing occurred at relatively low effector-to-target cell ratios. Although immune cells frequently represent a minor percentage of a total tumor mass, localized infiltrates of T cells could provide high T cell-to-tumor cell ratios focally within a tumor.Citation71 These infiltrates, together with the rapid, potent, and serial killing mechanism mediated by BiTE® antibody constructs,Citation35 represent an effective means by which MEDI-565 may eliminate CEA-expressing tumors. Indeed, this is supported by our finding that human T cells from healthy donors can mediate CEA-dependent inhibition of tumor growth in immunocompromised mice at relatively low effector-to-target ratios.
CEA is enzymatically shed from tumor cells into the blood of patients with CEA-positive tumors,Citation57 and represents a potential sink for antibody therapies targeting CEA, including MEDI-565. Although sCEA competitively inhibited binding of MEDI-565 to surface CEA, MEDI-565 was still able to kill tumor cells in vitro in a manner that was insensitive to soluble CEA at concentrations (5 μg/mL) that are above those typically found in the plasma of cancer patients,Citation68,69 and in vivo in animals bearing human tumors expressing and shedding CEA. This suggests that CEA shed into the blood of cancer patients or into the local tumor microenvironment may not represent a significant pharmacological sink that would negatively affect the activity of MEDI-565. While the lack of inhibition of MEDI-565 activity in the presence of sCEA may be a particular advantage for targeting tumor cells expressing CEA, the basis for this observation is not yet understood and remains under investigation.
While diverse mutations of colorectal cancer did not seem to impact the activity of MEDI-565, increased surface density of CEA was associated with an increase in BiTE® antibody concentration required for lysis. In this study, no more than 6 cell lines were tested with a single T cell donor due to experimental constraints. However, the addition of more cell lines to this analysis could change the experimental results, and this is therefore an area of ongoing study. The findings contrast with the data for BiTE® antibody constructs directed against target antigens EphA2Citation72 or PSMA,Citation73 where the EC50 values decreased as target expression increased. The basis for this conundrum is currently not understood and requires future analysis. We consider it unlikely that an increased amount of shed CEA in higher expressing cells is neutralizing MEDI-565 activity as this BiTE® antibody construct was selected based on its insensitivity toward soluble CEA.Citation63 Another explanation could be that higher surface density of CEA may hinder synapse formation by cytotoxic T cells, which could provide an explanation why certain cancer cells retain high level expression. The limitation of surface antigen expression has been overcome with other BiTE® antibody constructs when longer duration (96 hour) experiments were evaluated.Citation74 While there may be some threshold level of expression that is required for pharmacologic activity, further pharmacodynamic evaluation of efficacy relationships may be quite variable, and its applicability to clinical activity is unknown.
Factors other than the surface antigen density may also contribute to the differential sensitivity of tumor cells to MEDI-565 mediated killing. However, this is likely not mediated by constitutive activation of the PI3K/AKT axis, the RAS/RAF pathway or TP53, each having well characterized anti-apoptotic cellular effects.Citation9,75,76 For example, KRAS or BRAF mutant cell lines were killed with similar potency by MEDI-565 as compared to cell lines harboring wild-type KRAS or BRAF. Similarly findings with a BiTE® antibody construct targeting EGFR Citation77 suggest that the mechanism of action of BiTE® antibody constructs in general may be effective in eliminating tumors harboring mutations of the ras signal transduction pathway. This observation may have clinical significance for MEDI-565, especially in the treatment of patients with metastatic colorectal cancer where tumors bearing KRAS or BRAF mutations have been associated with reduced response rates to treatment with the EGFR-targeting monoclonal antibodies cetuximab and panitumumab. Citation4,5,12-14 Thus, although EGFR antibody-targeting therapy frequently encounters lower response rates in metastatic colorectal cancer patients harboring these mutations, the patients may respond to MEDI-565 treatment.
In addition to KRAS and BRAF mutations, MEDI-565 activates T cells to readily kill cancer cell lines harboring mutations in PI3KCA (gain of function) and PTEN (loss of function) that constitutively activate anti-apoptotic effects of the PI3K/AKT signaling pathway.Citation76 These mutations promote cancer cell growth and survival, and did not reduce the apoptotic signaling events and direct killing activity of MEDI-565 and T cells. The clinical significance of this observation in the context of colorectal cancer patients is unclear as conflicting reports suggest that patients with PIK3CA and PTEN mutations are likelyCitation78,79 or unlikelyCitation10,80 to respond to targeted therapy alone or with chemotherapy.
The mutational status of TP53 in a limited set of cancer cell lines did not affect the potency of MEDI-565 to induce T cell killing of cancer cell lines. However, the results of several studies suggest that the mutational status of TP53 may modulate the response of cancers to molecularly targeted agents, even if the agents are not designed to target the TP53 pathway directly.Citation22 For example, mutant TP53 is a predictor of better clinical outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab.Citation81 These observations suggest that molecular genotyping of tumors may yield molecular profiles that are useful for stratification and selection of patients who will benefit from targeted therapies, and may ultimately prove useful for selection of patients that receive BiTE® antibody therapies.
In summary, MEDI-565 represents a novel, potent therapeutic that appears to circumvent numerous tumor and immune escape mechanisms. These include mutation status in key tumor oncogenes and tumor suppressor genes as well as tumor immune evasion through MHC loss, peptide processing and presentation defects, and deactivation of T cell signaling through T cell exhaustion pathways. Loss of CEA expression by tumor cells remains a potential escape mechanism; loss of a tumor antigen remains a potential limitation for any therapy targeting tumor-associated proteins. As MEDI-565 has entered clinical testing in patients with gastrointestinal malignancies, the potential benefit of this T cell redirected therapy will now be evaluated directly in patients with advanced malignancies.
Materials and Methods
Construction of MEDI-565
The construction of BiTE® antibody constructs specific for CEA and CD3 have been described.Citation63 Briefly, a humanized CEACAM5-specific scFv fragment of A5B7Citation65 was fused via a short peptide linker to a de-immunized human CD3-specific scFvCitation66 derived from the murine monoclonal antibody L2K Citation67 in the orientation anti-human CEACAM5 VH-VL-glycine/serine linker-anti-human CD3 VH-VL. MEDI-565 was selected from a panel of these BiTE® antibody constructs, produced in CHO cells and affinity purified via a 6X histidine tag at the C-terminal end of the antibody.
Cell lines and reagents
All cell lines were obtained from the American Type Culture Collection and cultured in humidified chambers at 37°C and 5% CO2. All cells were cultured in media and fetal bovine serum (FBS, catalog #16140089) purchased from Life Technologies. BT474, H727, BxPC3, PAN0813, PC3, and MKN45 cells were cultured in RPMI 1640 (catalog #11875119) plus 10% FBS; LS174T and HPAF II in MEM L-glutamine (catalog #11095-080) plus 10% FBS; A549 and ASPC-1 in DMEM (catalog #11966025) plus 10% FBS; HPAC in a 50:50 mixture of DMEM and Ham's F12 (catalog #11320082) plus 10% FBS; HT-29 in McCoy's 5A modified medium (catalog #16600108) plus 10% FBS. Propidium iodide (PI) was purchased from Sigma-Aldrich (catalog #P4864). Soluble CEA derived from a human colon carcinoma cell line was obtained from Abcam (catalog #ab742). MEDI-565 and control BiTE® antibody construct (bispecific monoclonal antibody with the same C-terminal CD3 binding scFv as MEDI-565 but with an irrelevant N-terminal scFv that binds the herbicide mecroprop) were produced as described.Citation63
Flow cytometry-based binding studies
A total of 2.5 × 105 cells, including CHO cell lines or human peripheral blood mononuclear cells (PBMCs) enriched for CD3+ T cells, were suspended in phosphate-buffered saline, pH7.2 (PBS; Life Technologies, catalog #20012027) + 2% FBS and seeded into each well of 96‑well non-tissue culture treated plates (VWR, catalog #29445-154). Each cell type was incubated with MEDI-565 or control BiTE® antibody construct in PBS + 2% FBS buffer in triplicate for 1 hour at 4ºC in a total volume of 100 μL. After the incubation, the cells were washed 3-times with cold PBS + 2% FBS. Cells were subsequently incubated with 10 μg/mL PI (to exclude nonviable cells from analyses) and Alexa Fluor®488-conjugated mouse anti-penta-His antibodies (Qiagen, catalog #35310) for 1 hour at 4°C to detect each BiTE® antibody construct bound to cells. After 2 washes in cold PBS + 2% FBS, the cells were analyzed using a LSRII flow cytometer (Becton Dickinson) and FACSDiva software (Becton Dickinson). Fluorescence intensity of Alexa Fluor®488-conjugated mouse anti-penta-His antibodies bound to BiTE® antibodies on viable cells was determined by analysis of acquired data using FlowJo Software (TreeStar). Apparent dissociation equilibrium constant (Kd) values for MEDI-565 binding to human CEA (n = 3 experiments) and CD3ϵ (n = 3 experiments) were calculated from MEDI-565 binding curves in GraphPad Prism software for Windows, version 5.01, using non-linear regression analysis for single site binding.
Immunohistochemistry
Experiments were conducted at a dose of 5 μg/mL MEDI-565 in frozen multi-tumor human tissue micro arrays (TMA; TriStar, Inc.); one frozen pancreatic tumor TMA (TriStar, Inc.); 20 frozen esophageal specimens; one FFPE gastric cancer TMA; and one FFPE hepatocellular carcinoma TMA. Each TMA was prepared from individual tissue samples by AstraZeneca's Innovative Center China based in Shanghai, China. The panel of human cancerous tissues used as the test system included a 119 core multi-tumor TMA representative of 10 of the most frequent cancers (10 lung, 10 breast, 10 prostatic, 10 head and neck, 10 liver, 10 kidney, 10 pancreatic, 10 colonic, 9 ovarian, 10 melanoma and normal tissues consisting of 2 normal colon, 2 normal ovary, 2 normal breast, 2 normal lung, 2 normal prostate); a pancreatic tumor TMA that included 10 human pancreatic cancers and 5 normal pancreatic specimens; 17 esophageal carcinomas, esophageal mucosa from 3 normal donors; a gastric cancer FFPE TMA that includes 67 gastric carcinomas and gastric mucosa from 15 normal donors; and a liver cancer FFPE TMA that includes 76 hepatocellular carcinomas (HCC) and normal liver from 15 donors.
Tissue sections were stained using a Dako Autostainer (Dako North America). Briefly, after general protein blocking and blocking of endogenous peroxidases, sections were incubated with 1 μg/mL MEDI-565 or control BiTE® antibody construct. Bound primary antibody was detected using mouse anti-hexahistidine tag secondary antibody (Calbiochem/EMD Millipore Corporation, catalog #05-531) followed by Dako envision + dual link polymer and diaminobenzidine (DAB) colorimetric reagent (Dako, catalog #s K4061 and K400611). Tissues were counterstained with hematoxylin prior to mounting. Immunohistochemical staining was graded as: ± equivocal), 1+ (weak), 2+ (moderate), 3+ (strong), 4+ (intense), or negative by an experienced pathologist. Cells within a tissue section were considered positive for MEDI-565 binding if receiving a score of moderate, strong, or intense.
Isolation of human T cells
PBMC enriched for CD3+ T cells were isolated by the addition of 1 mL RosetteSep T-cell enrichment cocktail (Stem Cell Technologies, catalog #15061) per 20 mL of whole blood, followed by a 20-minute incubation, and subsequent T cell isolation by density gradient centrifugation using RosetteSep DM-L density medium (Stem Cell Technologies, catalog #15705). After centrifugation, the cells were washed 3 times with PBS containing 2% FBS and subsequently suspended in RPMI-1640 medium supplemented with 10% FBS.
Cytotoxicity assays
T cells (PBMC enriched for CD3+ cells) and target (CHO or tumor) cell lines were incubated with MEDI-565 in RPMI-1640 plus 10% FBS, and T cell activation and killing of target cells was subsequently determined by either flow cytometry or by the measurement of caspase 3 released into cell culture media. To distinguish target cells from effector CD3+ T cells during flow cytometry, the green fluorescent membrane dye 3,3′-dioctadecyloxacarbocyanine (Vybrant DiO; Life Technologies, catalog #V22889) was incubated with target cells for 5 minutes at 37°C for integration of the dye into the plasma membrane. Dyed target cells were mixed with effector cells at an effector-to-target (E:T) ratio of 5:1 (or as indicated in the text), seeded into non-tissue culture treated 96-well plates and then cultured at 37°C in an atmosphere of 5% CO2 for various periods of time. T cell activation was measured by flow cytometry by monitoring the up-regulation of CD25 (IL2 receptor α chain) or CD69. CD8-positive T cells were incubated with a PECy7-conjugated mouse anti-human CD8 antibody (BD Biosciences, catalog #557750) combined with an APC-conjugated anti-human CD25 monoclonal antibody (eBioscience, catalog #555434) or a PE-conjugated anti-human CD69 monoclonal antibody (BD Biosciences, catalog #557050). The release of cytokines into cell culture supernatants was measured using a cytokine-specific electrochemiluminescent-based assay (Mesoscale Discovery, catalog #K15010A-4). Cell viability was measured during flow cytometry by the cellular uptake of PI by non-viable cells. The specific activity of MEDI-565 was defined as the percentage of PI-positive cells among total DiO labeled cells, subtracted for background levels of non-viable cells among cells incubated with T cells in the absence of MEDI-565. FACSDiva software (Becton Dickinson) was used to acquire flow cytometry events and FlowJo software (Treestar, Inc.) for flow cytometry data analysis. Alternatively, the viability of non-labeled cells was determined by measurement of total caspase 3 released into cell culture media by lysed cells using an electrochemiluminescence-based assay (MesoScale Discovery, catalog #K151CFD-3). Total caspase 3 levels in the culture medium were represented as relative light units (RLU). Graphical and statistical analyses of experimental data were performed using GraphPad Prism software, version 5.01 (Graphpad Software, Inc.). Sigmoidal dose-response curves typically had r2 values > 0.90, and calculated half-maximal effective concentrations (EC50) were used for comparison of bioactivity. Parallel studies using the same T cells with targets cells amenable to both flow cytometry and total caspase 3 readouts, demonstrated that EC50 values were comparable but the percent specific lysis was not (data not shown). Therefore, RLU values were not converted to a percent specific lysis.
Determination of CEA antigen binding sites on human tumor cell lines
QIFIKIT® (Dako North America, Inc., catalog #K0078) was used to quantitatively determine the number of CEA determinants on the surface of cells. CEA surface density was determined using linear regression analysis of a flow-cytometry based readout of an anti-CEA antibody (IgG1 antibody consisting of 2 CEA-specific scFv domains derived from the CEA-specific scFv used to construct MEDI-565) bound to the cell surface of cancer cell lines and to control beads for determination of a standard curve.
Measurement of granzyme B and perforin in cell culture supernatants and cleaved poly-adenosine diphosphate ribose polymerase (PARP) in cell lysates
Human PBMC enriched for CD3+ T cells isolated from healthy donors and CHO/huCEA target cells were incubated at an E:T ratio of 10:1 with increasing concentrations of MEDI-565 or control BiTE® antibody construct. The presence of granzyme B and perforin in supernatants and cleaved PARP protein in target cell lysates after 72 hours of incubation was determined by ELISA for granzyme B (Diaclone SAS, catalog #850.790.096) and perforin (Diaclone SAS, catalog #850.860.096), and by the BD OptEIA set for human cleaved PARP (BD Biosciences, catalog #552592).
Xenograft tumor models in SCID mice
Four to 6 week old female CB-17 severe combined immunodeficient (SCID) mice (Taconic Farms, Inc.) used for efficacy models were housed in an Association for Animal Accreditation of Laboratory Animal Care-accredited and U.S. Department of Agriculture-licensed facility under sterile and standardized environmental conditions (20 ± 1°C room temperature, 50 ±10% relative humidity, and 12 hour light-dark cycle). Mice received autoclaved food and bedding, and acidified drinking water ad libitum. SCID mice were treated weekly with intraperitoneally administered rabbit anti-sialo GM1 antibody to deplete NK cells. Human tumor cell lines were harvested from cell culture by trypsinization, washed once with PBS, resuspended in PBS, and mixed with freshly isolated human PBMC enriched for CD3+ T cells from healthy donors. Cell suspensions of 1 to 2.5 × 106 tumor cells admixed with human T cells were injected in a total volume of 0.2 mL/mouse. The ratios of implanted effector T cells to tumor cells were as follows: LS174T and HeyA8, 1:2 (); LS174T 1:2 (); HT-29; MKN, 1.7:1; H727, 1.5:1; HPAC, 1:1; HPAFII, 1:1 (). MEDI-565, control BiTE® antibody construct, or PBS test articles at the indicated doses were administered intravenously (IV) or subcutaneously (SC) one hour after tumor cell/PBMC injection, and subsequently each day over the following 4 d for a total of 5 treatments. Tumor growth was measured with calipers in 2 perpendicular dimensions, and tumor volume (mm3) was calculated using the formula (width2 × length)/2. All data points were reported as the arithmetic mean ± SEM for each group. Anti-cancer effect, expressed as a percent tumor growth inhibition (TGI), was calculated using the formula (1 - [mean tumor volume of treatment group] ÷ [mean tumor volume of control group]) × 100.
Statistical analyses
The Mann-Whitney rank sum test was used to determine statistically significant differences between treatment groups for in vivo efficacy models. The coefficient of determination (r2), calculated from Pearson's correlation coefficient (r), was used to assess the correlation between MEDI-565 binding sites and EC50 values for cytotoxicity assays using 6 cell lines with the same 3 independent T cell donors.
Disclosure of Potential Conflicts of Interest
The authors would like to disclose the following conflict of interest: This study was supported by MedImmune, LLC, Gaithersburg, MD and Amgen Research (Munich) GmbH, Munich, Germany. The authors of this publication are employed by MedImmune LLC, Amgen, and Roche Glycart, as indicated in the manuscript.
Supplementary_Material.zip
Download Zip (367.1 KB)Funding
Study supported by MedImmune, LLC, Gaithersburg, MD and Amgen Research (Munich) GmbH, Munich, Germany.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M, Silver J, Ogino S, Hooshmand S, Kwak E, et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 2009; 101:465-72; PMID:19603024; http://dx.doi.org/10.1038/sj.bjc.6605164
- AmericanCancerSociety. Colorectal Cancer Facts & Figures 2011-2013. Atlanta, GA: American Cancer Society; 2011; 1–30.
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108-13; PMID:17932254; http://dx.doi.org/10.1126/science.1145720
- Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757-65; PMID:18946061; http://dx.doi.org/10.1056/NEJMoa0804385
- De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010; 11:753-62; PMID:20619739; http://dx.doi.org/10.1016/S1470-2045(10)70130-3
- Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 2009; 27:5924-30; PMID:19884556; http://dx.doi.org/10.1200/JCO.2008.21.6796
- Hawkins DS, Demers GW, Galloway DA. Inactivation of p53 enhances sensitivity to multiple chemotherapeutic agents. Cancer Res 1996; 56:892-8; PMID:8631030
- Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol 2005; 23:7518-28; PMID:16172461; http://dx.doi.org/10.1200/JCO.2005.00.471
- Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9:701-13; PMID:19693097
- Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS, Nasser S, Arango D, Shin J, Klampfer L, et al. PIK3CA mutationPTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res 2008; 68:1953-61; PMID:18339877; http://dx.doi.org/10.1158/0008-5472.CAN-07-5659
- Chen J, Huang XF, Katsifis A. Activation of signal pathways and the resistance to anti-EGFR treatment in colorectal cancer. J Cell Biochem 2010; 111:1082-6; PMID:21053280; http://dx.doi.org/10.1002/jcb.22905
- Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008; 26:5705-12; PMID:19001320; http://dx.doi.org/10.1200/JCO.2008.18.0786
- Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol 2009; 6:519-27; PMID:19636327; http://dx.doi.org/10.1038/nrclinonc.2009.111
- Prenen H, Tejpar S, Van Cutsem E. New strategies for treatment of KRAS mutant metastatic colorectal cancer. Clin Cancer Res 2010; 16:2921-6; PMID:20460490; http://dx.doi.org/10.1158/1078-0432.CCR-09-2029
- Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:1626-34; PMID:18316791; http://dx.doi.org/10.1200/JCO.2007.14.7116
- Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A. Oncogenic activation of the RASRAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 2007; 67:2643-8; PMID:17363584; http://dx.doi.org/10.1158/0008-5472.CAN-06-4158
- Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 2007; 97:1139-45; PMID:17940504; http://dx.doi.org/10.1038/sj.bjc.6604009
- Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouche O, Landi B, Louvet C, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26:374-9; PMID:18202412; http://dx.doi.org/10.1200/JCO.2007.12.5906
- Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C, Andreola S, Bajetta E, et al. PI3KCAPTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 2009; 20:84-90; PMID:18669866; http://dx.doi.org/10.1093/annonc/mdn541
- Li R SP, Schwartz D, Matas D, Almog N, Wolkowicz R, Goldfinger N, Pei H, Prokocimer M, Rotter V. Mutant p53 protein expression interferes with p53-independent apoptotic pathways. Oncogene 1998; 16:3269-77; PMID:9681825; http://dx.doi.org/10.1038/sj.onc.1201867
- Longley DB, Boyer J, Allen WL, Latif T, Ferguson PR, Maxwell PJ, McDermott U, Lynch M, Harkin DP, Johnston PG. The role of thymidylate synthase induction in modulating p53-regulated gene expression in response to 5-fluorouracil and antifolates. Cancer Res 2002; 62:2644-9; PMID:11980662
- Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 1999; 104:263-9; PMID:10430607; http://dx.doi.org/10.1172/JCI6863
- Goff BA, Ries JA, Els LP, Coltrera MD, Gown AM. Immunophenotype of ovarian cancer as predictor of clinical outcome: evaluation at primary surgery and second-look procedure. Gynecol Oncol 1998; 70:378-85; PMID:9790791; http://dx.doi.org/10.1006/gyno.1998.5094
- Righetti SC, Della Torre G, Pilotti S, Menard S, Ottone F, Colnaghi MI, Pierotti MA, Lavarino C, Cornarotti M, Oriana S, et al. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res 1996; 56:689-93; PMID:8630996
- Marx D, Meden H, Ziemek T, Lenthe T, Kuhn W, Schauer A. Expression of the p53 tumour suppressor gene as a prognostic marker in platinum-treated patients with ovarian cancer. Eur J Cancer 1998; 34:845-50; PMID:9797696; http://dx.doi.org/10.1016/S0959-8049(97)10169-1
- Mayr D, Pannekamp U, Baretton GB, Gropp M, Meier W, Flens MJ, Scheper R, Diebold J. Immunohistochemical analysis of drug resistance-associated proteins in ovarian carcinomas. Pathol Res Pract 2000; 196:469-75; PMID:10926324; http://dx.doi.org/10.1016/S0344-0338(00)80048-5
- Gadducci A, Di Cristofano C, Zavaglia M, Giusti L, Menicagli M, Cosio S, Naccarato AG, Genazzani AR, Bevilacqua G, Cavazzana AO. P53 gene status in patients with advanced serous epithelial ovarian cancer in relation to response to paclitaxel- plus platinum-based chemotherapy and long-term clinical outcome. Anticancer Res 2006; 26:687-93; PMID:16739339
- Lavarino C, Delia D, Di Palma S, Zunino F, Pilotti S. p53 in drug resistance in ovarian cancer. Lancet 1997; 349:1556; PMID:9167490; http://dx.doi.org/10.1016/S0140-6736(05)62140-X
- Buttitta F, Marchetti A, Gadducci A, Pellegrini S, Morganti M, Carnicelli V, Cosio S, Gagetti O, Genazzani AR, Bevilacqua G. p53 alterations are predictive of chemoresistance and aggressiveness in ovarian carcinomas: a molecular and immunohistochemical study. Br J Cancer 1997; 75:230-5; PMID:9010031; http://dx.doi.org/10.1038/bjc.1997.38
- Lavarino C, Pilotti S, Oggionni M, Gatti L, Perego P, Bresciani G, Pierotti MA, Scambia G, Ferrandina G, Fagotti A, et al. p53 gene status and response to platinumpaclitaxel-based chemotherapy in advanced ovarian carcinoma. J Clin Oncol 2000; 18:3936-45; PMID:11099323
- Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American society of clinical oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009; 27:2091-6; PMID:19188670; http://dx.doi.org/10.1200/JCO.2009.21.9170
- Talmadge JE, Donkor M, Scholar E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev 2007; 26:373-400; PMID:17717638; http://dx.doi.org/10.1007/s10555-007-9072-0
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:21436444; http://dx.doi.org/10.1126/science.1203486
- Whiteside TL. Inhibiting the inhibitors: evaluating agents targeting cancer immunosuppression. Expert Opin Biol Ther 2010; 10:1019-35; PMID:20415597; http://dx.doi.org/10.1517/14712598.2010.482207
- Baeuerle PA, Reinhardt C. Bispecific T-cell engaging antibodies for cancer therapy. Cancer Res 2009; 69:4941-4; PMID:19509221; http://dx.doi.org/10.1158/0008-5472.CAN-09-0547
- Frankel SR, Baeuerle PA. Targeting T cells to tumor cells using bispecific antibodies. Curr Opin Chem Biol 2013; 17:385-92; PMID:23623807; http://dx.doi.org/10.1016/j.cbpa.2013.03.029
- Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today 2005; 10:1237-44; PMID:16213416; http://dx.doi.org/10.1016/S1359-6446(05)03554-3
- Brischwein K, Parr L, Pflanz S, Volkland J, Lumsden J, Klinger M, Locher M, Hammond SA, Kiener P, Kufer P, et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J Immunother 2007; 30:798-807; PMID:18049331; http://dx.doi.org/10.1097/CJI.0b013e318156750c
- Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol 2006; 43:763-71; PMID:16360021; http://dx.doi.org/10.1016/j.molimm.2005.03.007
- Hoffmann P, Hofmeister R, Brischwein K, Brandl C, Crommer S, Bargou R, Itin C, Prang N, Baeuerle PA. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-CD3-bispecific single-chain antibody construct. Int J Cancer 2005; 115:98-104; PMID:15688411; http://dx.doi.org/10.1002/ijc.20908
- Molhoj M, Crommer S, Brischwein K, Rau D, Sriskandarajah M, Hoffmann P, Kufer P, Hofmeister R, Baeuerle PA. CD19-CD3-bispecific antibody of the BiTE class is far superior to tandem diabody with respect to redirected tumor cell lysis. Mol Immunol 2007; 44:1935-43; PMID:17083975; http://dx.doi.org/10.1016/j.molimm.2006.09.032
- Amann M, D’Argouges S, Lorenczewski G, Brischwein K, Kischel R, Lutterbuese R, Mangold S, Rau D, Volkland J, Pflanz S, et al. Antitumor activity of an EpCAMCD3-bispecific BiTE antibody during long-term treatment of mice in the absence of T-cell anergy and sustained cytokine release. J Immunother 2009; 32:452-64; PMID:19609237; http://dx.doi.org/10.1097/CJI.0b013e3181a1c097
- Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, Noppeney R, Viardot A, Hess G, Schuler M, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008; 321:974-7; PMID:18703743; http://dx.doi.org/10.1126/science.1158545
- Handgretinger R, Zugmaier G, Henze G, Kreyenberg H, Lang P, von Stackelberg A. Complete remission after blinatumomab-induced donor T-cell activation in three pediatric patients with post-transplant relapsed acute lymphoblastic leukemia. Leukemia 2011; 25:181-4; PMID:20944674; http://dx.doi.org/10.1038/leu.2010.239
- Goebeler M, Viardot A, Noppeney R, Krause S, Mackensen A, Rupertus K, Kanz L, Knop S, Topp M, Scheele J, et al. Blinatumomab (CD3CD19 BiTE antibody) results in a high response rate in patients with relapsed non-Hodgkin lymphoma (NHL) including MCL and DLBCL. Ann Oncol 2011; 22 (Supplement 4):iv104-iv5
- Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, Gokbuget N, Neumann S, Goebeler M, Viardot A, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19CD3-bispecific BiTE antibody blinatumomab. Blood 2012; 119:6226-33; PMID:22592608; http://dx.doi.org/10.1182/blood-2012-01-400515
- Nagorsen D, Baeuerle PA. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp Cell Res 2011; 317:1255-60; PMID:21419116; http://dx.doi.org/10.1016/j.yexcr.2011.03.010
- Topp MS, Gokbuget N, Zugmaier G, Degenhard E, Goebeler ME, Klinger M, Neumann SA, Horst HA, Raff T, Viardot A, et al. Long-term follow-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in B-lineage ALL. Blood 2012; 120:5185-7; PMID:23024237; http://dx.doi.org/10.1182/blood-2012-07-441030
- Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, Horst HA, Raff T, Viardot A, Schmid M, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. J Clin Oncol 2011; 29:2493-8; PMID:21576633; http://dx.doi.org/10.1200/JCO.2010.32.7270
- Haas C, Krinner E, Brischwein K, Hoffmann P, Lutterbuse R, Schlereth B, Kufer P, Baeuerle PA. Mode of cytotoxic action of T cell-engaging BiTE antibody MT110. Immunobiology 2009; 214:441-53; PMID:19157637; http://dx.doi.org/10.1016/j.imbio.2008.11.014
- Witthauer J, Schlereth B, Brischwein K, Winter H, Funke I, Jauch KW, Baeuerle P, Mayer B. Lysis of cancer cells by autologous T cells in breast cancer pleural effusates treated with anti-EpCAM BiTE antibody MT110. Breast Cancer Res Treat 2009; 117:471-81; PMID:18819003; http://dx.doi.org/10.1007/s10549-008-0185-0
- Herrmann I, Baeuerle PA, Friedrich M, Murr A, Filusch S, Ruttinger D, Majdoub MW, Sharma S, Kufer P, Raum T, et al. Highly efficient elimination of colorectal tumor-initiating cells by an EpCAMCD3-bispecific antibody engaging human T cells. PLoS One 2010; 5:e13474; PMID:20976159; http://dx.doi.org/10.1371/journal.pone.0013474
- Behr TM, Liersch T, Greiner-Bechert L, Griesinger F, Behe M, Markus PM, Gratz S, Angerstein C, Brittinger G, Becker H, et al. Radioimmunotherapy of small-volume disease of metastatic colorectal cancer. Cancer 2002; 94:1373-81; PMID:11877768; http://dx.doi.org/10.1002/cncr.10308
- Granowska M, Britton KE, Mather SJ, Morris G, Ellison D, Soobramoney S, Talbot IC, Northover JM. Radioimmunoscintigraphy with technetium-99m labelled monoclonal antibody, 1A3, in colorectal cancer. Eur J Nucl Med 1993; 20:690-8; PMID:8404956; http://dx.doi.org/10.1007/BF00181760
- Mayer A, Tsiompanou E, O’Malley D, Boxer GM, Bhatia J, Flynn AA, Chester KA, Davidson BR, Lewis AA, Winslet MC, et al. Radioimmunoguided surgery in colorectal cancer using a genetically engineered anti-CEA single-chain Fv antibody. Clin Cancer Res 2000; 6:1711-9; PMID:10815889
- Francis RJ, Sharma SK, Springer C, Green AJ, Hope-Stone LD, Sena L, Martin J, Adamson KL, Robbins A, Gumbrell L, et al. A phase I trial of antibody directed enzyme prodrug therapy (ADEPT) in patients with advanced colorectal carcinoma or other CEA producing tumours. Br J Cancer 2002; 87:600-7; PMID:12237768; http://dx.doi.org/10.1038/sj.bjc.6600517
- Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 1999; 9:67-81; PMID:10202129; http://dx.doi.org/10.1006/scbi.1998.0119
- Sack TL, Gum JR, Low MG, Kim YS. Release of carcinoembryonic antigen from human colon cancer cells by phosphatidylinositol-specific phospholipase C. J Clin Invest 1988; 82:586-93; PMID:3042807; http://dx.doi.org/10.1172/JCI113636
- Graham RA, Wang S, Catalano PJ, Haller DG. Postsurgical surveillance of colon cancer: preliminary cost analysis of physician examination, carcinoembryonic antigen testing, chest x-ray, and colonoscopy. Ann Surg 1998; 228:59-63; PMID:9671067; http://dx.doi.org/10.1097/00000658-199807000-00009
- Duffy MJ. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem 2001; 47:624-30; PMID:11274010
- Locker GY, Hamilton S, Harris J, Jessup JM, Kemeny N, Macdonald JS, Somerfield MR, Hayes DF, Bast RC Jr. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. J Clin Oncol 2006; 24:5313-27; PMID:17060676; http://dx.doi.org/10.1200/JCO.2006.08.2644
- Rother M. Carcinoembryonic antigen monitoring for early detection of asymptomatic incurable metastatic colorectal cancer. J Clin Oncol 2007; 25:1293-4; author reply 4; PMID:17401025; http://dx.doi.org/10.1200/JCO.2006.10.0974
- Lutterbuese R, Raum T, Kischel R, Lutterbuese P, Schlereth B, Schaller E, Mangold S, Rau D, Meier P, Kiener PA, et al. Potent control of tumor growth by CEACD3-bispecific single-chain antibody constructs that are not competitively inhibited by soluble CEA. J Immunother 2009; 32:341-52; PMID:19342971; http://dx.doi.org/10.1097/CJI.0b013e31819b7c70
- Osada T, Hsu D, Hammond S, Hobeika A, Devi G, Clay TM, Lyerly HK, Morse MA. Metastatic colorectal cancer cells from patients previously treated with chemotherapy are sensitive to T-cell killing mediated by CEACD3-bispecific T-cell-engaging BiTE antibody. Br J Cancer 2010; 102:124-33; PMID:19953093; http://dx.doi.org/10.1038/sj.bjc.6605364
- Chester KA, Robson L, Keep PA, Pedley RB, Boden JA, Boxer GM, Hawkins RE, Begent RH. Production and tumour-binding characterization of a chimeric anti-CEA Fab expressed in Escherichia coli. Int J Cancer 1994; 57:67-72; PMID:8150543; http://dx.doi.org/10.1002/ijc.2910570113
- Jones TD, Hanlon M, Smith BJ, Heise CT, Nayee PD, Sanders DA, Hamilton A, Sweet C, Unitt E, Alexander G, et al. The development of a modified human IFN-alpha2b linked to the Fc portion of human IgG1 as a novel potential therapeutic for the treatment of hepatitis C virus infection. J Interferon Cytokine Res 2004; 24:560-72; PMID:15450132; http://dx.doi.org/10.1089/jir.2004.24.560
- Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, Offner S, Locher M, Urbig T, Raum T, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol 2006; 43:1129-43; PMID:16139892; http://dx.doi.org/10.1016/j.molimm.2005.07.034
- Bhatnagar J, Tewari HB, Bhatnagar M, Austin GE. Comparison of carcinoembryonic antigen in tissue and serum with grade and stage of colon cancer. Anticancer Res 1999; 19:2181-7; PMID:10472328
- Flamini E, Mercatali L, Nanni O, Calistri D, Nunziatini R, Zoli W, Rosetti P, Gardini N, Lattuneddu A, Verdecchia GM, et al. Free DNA and carcinoembryonic antigen serum levels: an important combination for diagnosis of colorectal cancer. Clin Cancer Res 2006; 12:6985-8; PMID:17145818; http://dx.doi.org/10.1158/1078-0432.CCR-06-1931
- Baeuerle PA, Reinhardt C, Kufer P. BITE: a new class of antibodies that recruit T-cells. Drugs of the Future 2008; 33:137-47; http://dx.doi.org/10.1358/dof.2008.033.02.1172578
- Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 2010; 29:1093-102; PMID:19946335; http://dx.doi.org/10.1038/onc.2009.416
- Hammond SA, Lutterbuese R, Roff S, Lutterbuese P, Schlereth B, Bruckheimer E, Kinch MS, Coats S, Baeuerle PA, Kufer P, et al. Selective targeting and potent control of tumor growth using an EphA2CD3-Bispecific single-chain antibody construct. Cancer Res 2007; 67:3927-35; PMID:17440108; http://dx.doi.org/10.1158/0008-5472.CAN-06-2760
- Friedrich M, Raum T, Lutterbuese R, Voelkel M, Deegen P, Rau D, Kischel R, Hoffmann P, Brandl C, Schuhmacher J, et al. Regression of human prostate cancer xenografts in mice by AMG 212BAY2010112, a novel PSMACD3-Bispecific BiTE antibody cross-reactive with non-human primate antigens. Mol Cancer Ther 2012; 11:2664-73; PMID:23041545; http://dx.doi.org/10.1158/1535-7163.MCT-12-0042
- Krupka C, Kufer P, Kischel R, Zugmaier G, Bogeholz J, Kohnke T, Lichtenegger FS, Schneider S, Metzeler KH, Fiegl M, et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell-engaging antibody AMG 330. Blood 2013; 123:356-65; PMID:24300852; http://dx.doi.org/10.1182/blood-2013-08-523548
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 2007; 7:295-308; PMID:17384584; http://dx.doi.org/10.1038/nrc2109
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9:550-62; PMID:19629070; http://dx.doi.org/10.1038/nrc2664
- Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B, Friedrich M, Thomas O, Lorenczewski G, Rau D, et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A 2010; 107:12605-10; PMID:20616015; http://dx.doi.org/10.1073/pnas.1000976107
- Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, Lambrechts D, Van Cutsem E, Tejpar S. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res 2009; 15:3184-8; PMID:19366826; http://dx.doi.org/10.1158/1078-0432.CCR-08-2961
- Park JH, Han SW, Oh DY, Im SA, Jeong SY, Park KJ, Kim TY, Bang YJ, Park JG. Analysis of KRAS, BRAF, PTEN, IGF1R, EGFR intron 1 CA status in both primary tumors and paired metastases in determining benefit from cetuximab therapy in colon cancer. Cancer Chemother Pharmacol 2011; 68:1045-55; PMID:21340604; http://dx.doi.org/10.1007/s00280-011-1586-z
- Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 2009; 69:1851-7; PMID:19223544; http://dx.doi.org/10.1158/0008-5472.CAN-08-2466
- Oden-Gangloff A, Di Fiore F, Bibeau F, Lamy A, Bougeard G, Charbonnier F, Blanchard F, Tougeron D, Ychou M, Boissiere F, et al. TP53 mutations predict disease control in metastatic colorectal cancer treated with cetuximab-based chemotherapy. Br J Cancer 2009; 100:1330-5; PMID:19367287; http://dx.doi.org/10.1038/sj.bjc.6605008