
Abstract
Several hundred taxa of microorganisms—including bacteria, archaea and eukaryotes—inhabit the human body. What did it take for these species to become stable residents of humans? Recent reports illustrate how evolutionary changes in transcriptional circuits played a pivotal role in the adaptation of single-celled eukaryotes to colonize mammals.
An assembly of trillions of microorganisms comprising bacteria, archaea, and eukaryotes inhabit the human body. In the last few years, there has been a growing interest in understanding how this complex array of microbes functions, how it contributes to our health, and how imbalances in its composition (dysbiosis) can lead to disease. The most progress thus far has been made in inventorying our microbiota—albeit chiefly the bacterial component.Citation1 This cataloguing has shown that the collection of microbes that resides within us represents at most a few thousand taxa.Citation2-4 What did it take for these microorganisms to become stable residents of the human body?
The very same taxa that compose our microbiota—or at least closely related species of microbes—long predate higher eukaryotes. This means that these microorganisms were originally adapted to thrive in environments that likely offered little or no resemblance to the milieu in any mammalian host. Therefore, the species that ultimately were able to stably colonize our human ancestors likely underwent long adaptation processes. Several known molecular sources of evolutionary novelty—mutation, gene duplication, gene acquisition or loss, and de novo gene birth—likely played a role in this adaptation to a host-associated lifestyle. There is an extensive literature linking the acquisition of genes by horizontal transfer to the ability of microbes (particularly bacteria) to “infect” a host.Citation5 By contrast, examples that link this trait to other sources of phenotypic novelty are rather sparse. Here we highlight some recent work in eukaryotic microorganisms that illustrates the key role that evolutionary changes in transcriptional circuits and in gene regulation had in this process. Comprehensive reviews on the evolution of transcriptional regulatory networks in bacteria can be found elsewhere.Citation6–8
Linking Changes in Transcriptional Regulation to Phenotypic Novelty
The idea that evolutionary changes in gene regulation can generate phenotypic novelty is not new. Roy J. Britten and Eric H. Davidson in 1971,Citation9 as well as Mary-Claire King and Allan C. Wilson in 1975,Citation10 were among the first to discuss the importance of regulatory changes in evolution and proposed that differences among species could be attributed to this type of modification. The identification of meaningful “regulatory changes” among related species, however, requires some reasonable degree of knowledge of the regulatory circuitry of the organisms under study. Mapping such circuits is a task that requires much experimental work and that until recently was cumbersome even in the simplest single-celled organisms. Not surprisingly, it is only with the advent of large-scale DNA sequencing and functional genomics at the dawn of the 21st century that scientists have empirically started to examine whether, and how, “regulatory changes” influence phenotypic novelty at large. Indeed, in the last few years, researchers have documented how changes in transcriptional regulation underlie multiple morphological and physiological traits, particularly in animals and yeasts (for a recent review see ref. Eleven): For example, blonde hair color,Citation12 lactase persistenceCitation13 and the loss of androgen-dependent sensory vibrissae and penile spinesCitation14 in humans; alterations in insect wing morphology and coloring patterns;Citation15–16 and the incorporation of nutritional signals into the yeast mating process.Citation17
What do Evolutionary Changes in Transcriptional Regulation Entail?
In their simplest form, evolutionary changes to transcriptional circuits can entail one of 3 scenarios:Citation11 (1) Changes of regulon membership due to transcription regulator binding site turnover; (2) handover of a regulon from one transcription regulator to another; (3) recruiting a new transcription regulator to an existing regulon by the formation of new combinatorial interactions. These 3 mechanisms or variations thereof, alone or in combination, are at the heart of most examples involving the generation of phenotypic diversity due to changes in transcriptional regulation.
Transcription Regulation Changes in Host-associated Microorganisms
We now know that changes in transcriptional regulation underlie a number of morphological and physiological traits in animals and other eukaryotes. Have similar regulatory changes played a significant role in the emergence of other traits, such as the ability of certain microbial species to colonize mammals? Despite the extensive effort dedicated to sequence microbial genomes and the inherent interest in understanding the biology of human-associated microorganisms, there are very few examples that directly connect changes in transcriptional circuits to the ability of any microbial taxon to reside in humans. In fact, most examples linking particular microbial loci to the ability to “infect” a host are instances of gene(s) acquisition by horizontal gene transfer.Citation5,6,18 The reason for this probably lies in the large differences that exist in gene content even among closely related microbes (particularly bacteria). Researchers interested in finding out the molecular basis of a host-related trait would be naturally inclined to look first into gene content differences. This inherent bias has left other potential sources of phenotypic novelty largely unexplored.
Transcriptional Regulation Changes in the Adaptation of the Yeast Candida albicans to its Mammalian Host
The yeast Candida albicans is the most prominent fungal species residing in humans. While it can thrive in multiple locales of the human body (e.g,. mouth, skin, gastrointestinal and genitourinary tracts), Candida's most common habitat is the human gut. Indeed, the majority, if not all, of healthy adults carry C. albicans as part of their normal gut microbiota.Citation19,20 In addition to C. albicans, other species in the Candida clade () are also associated with humans. By contrast, the related Saccharomyces clade () comprises mostly free-living, non-pathogenic yeasts. C. albicans and the model yeast S. cerevisiae last shared a common ancestor about 300 million years ago.Citation21 Despite the disparate niches that these 2 species occupy, C. albicans and S. cerevisiae share >80% of their genesCitation22 raising the question of what genetic changes contributed to the ability of Candida to reside in the human host. Perez et al.Citation23 have recently reported that the reshuffling of a portion of Candida's regulatory circuitry likely played a key role in the adaptation of this species to its host.
Figure 1. Transcriptional network expansion in the lineage leading to the human commensal and pathogenic yeast Candida albicans. The expansion and reshuffling of the Candida circuitry can be traced back to the successive duplications of a homolog of the S. cerevisiae transcription regulator LYS14. While LYS14 controls lysine biosynthesis in S. cerevisiae, each of the 4 resulting duplicated regulators in C. albicans has adopted a different role. A cladogram depicting the phylogenetic relationships among extant species of the Saccharomyces and Candida clades is shown to the left. The small orange circles in the cladogram represent the inferred gene duplication events that gave rise to the 4 C. albicans homologs. The thick arrows in the middle of the figure depict the genes encoding the LYS transcription regulators. Arrows of the same color represent orthologs based on phylogenetic reconstructions and synteny.Citation23 The right side of the figure shows a subset of each regulators’ target genes as determined by ChIP-chip.Citation23,27 Notice that the target genes included here are those that most likely contribute to the ability of C. albicans to colonize its mammalian host and cause disease.
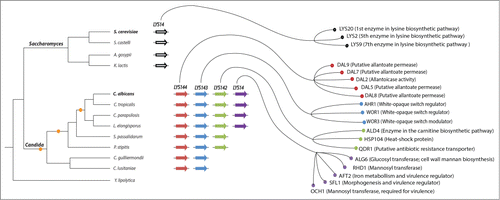
Perez et al. identified a group of transcription regulators that have undergone several successive duplications in the Candida clade (). By contrast, the free-living model yeast S. cerevisiae, as well as other species in the Saccharomyces clade, has a single copy. LYS14, as this gene is termed in S. cerevisiae, is a member of the fungal-specific zinc-cluster family of transcription regulators, and governs the synthesis of the amino acid lysine; that is, it confers upon S. cerevisiae the ability to grow in lysine-free medium.Citation24 The C. albicans genome, on the other hand, encodes 4 homologs of the S. cerevisiae LYS14 gene. Remarkably, none of these 4 genes is required for lysine biosynthesis in C. albicans,Citation25 suggesting that they regulate other aspects in the biology of this organism. Indeed, establishing the genome-wide in vivo binding locations (by chromatin immunoprecipitation) of each of the 4 proteins revealed some of the functions that they govern in C. albicans (): LYS143 feeds into the white-opaque regulatory circuit. This morphological switch between white and opaque cell types may be necessary for the fungus to adapt to different host niches.Citation26 LYS144 binds upstream of multiple genes encoding putative transporters and enzymes predicted to play a role in nutrient acquisition and metabolism;Citation27 these functions may be responsible for the impaired ability of the C. albicans lys144 mutant to colonize the mammalian gut.Citation27 LYS14 binds upstream of genes encoding enzymes predicted to modify the cell surface of the fungus, and thus it may play a role in immune evasion; consistent with this observation, the C. albicans lys14 mutant shows reduced virulence in a murine model of systemic infection.Citation27 Taken together, these findings indicate that each of the duplicated LYS regulators has a unique role in C. albicans biology. While the function of LYS142 remains to be uncovered, clearly the other 3 homologs control functions that are relevant within the host. Thus, while the single copy present in S. cerevisiae mediates lysine metabolism, the 4 copies in C. albicans have acquired completely different functions, at least some of which contribute to the ability of C. albicans to reside in the human host and cause disease.
How did the 4 regulators differentiate from one another in the Candida lineage? Perez et al. combined large-scale biochemical measurements and full-genome molecular biology methods to show that a combination of 3 mechanisms accounts for the diversification of the recently-duplicated transcription regulators: (1) Slightly different intrinsic, monomer DNA binding specificities (i.e., variations in a core motif); (2) different preferences for half-site arrangements (direct vs. inverted repeats; preferred distances between repeats); and (3) interaction with co-factors. These 3 differences add up to give each regulator the specificity to govern the expression of a separate set of target genes (for a recent review see ref.Citation28).
Changes in Gene Expression Regulation Played a Role in the Evolution of Pathogenicity in Some Yeast Lineages
The yeast S. cerevisiae is ubiquitous in nature and can be found under a variety of ecological niches. It is also the most commonly used fungal species for industrial production purposes, for example in brewing and baking. While generally innocuous, an increasing number of S. cerevisiae infections have been reported, particularly in immunocompromised individuals.Citation29 No specific virulence factors have been described yet, but the abundant genetic and genomic resources associated with this species render it an ideal organism to investigate the genetic changes associated with the transition to a pathogenic lifestyle.Citation30
To determine whether changes in gene expression regulation played a role in the evolution of pathogenicity in this yeast, Fraser et al.Citation30 sought evidence for lineage-specific natural selection acting on the cis-regulation of entire sets of genes. For this, they employed genome-wide allele-specific expression (ASE) data from a hybrid between 2 diverged S. cerevisiae lineages: a common laboratory strain and a pathogenic strain. Genes were then grouped in categories based on functional annotation or physical interaction, and the genes in each category were tested for biased ASE directionality (that is, the number of genes with increased expression from alleles of the pathogenic strain, as opposed to alleles from the common laboratory strain) compared to the genome as a whole. This analysis revealed a complex of 17 genes, 10 of which showed lower expression of the alleles coming from the pathogenic strain. Further population-genetic tests established that the observed expression patterns of this group of genes were likely due to positive selection rather than to relaxed negative selection. Thus, most of these genes underwent positive selection for cis-acting downregulation in an ancestor of the pathogenic strain. This complex of 17 genes is enriched for proteins involved in the early stages of endocytosis. While it remains to be determined the mechanism(s) whereby these genes contribute to virulence, it is clear that their downregulation confers a growth advantage within the mammalian host. Thus, modifying the patterns of expression of this group of genes contributed to the ability of some S. cerevisiae lineages to infect and proliferate in humans.
Conclusion
Human mucosal surfaces are laden with trillions of microorganisms encompassing all 3 domains of life. Yet this vast array of microbes represents only a few thousand different taxa—a small fraction of the tens of millions of microbial species estimated to exist on our planet. The microorganisms that ultimately were able to stably colonize our human ancestors likely underwent long adaptation processes, which plausibly involved numerous modifications to their genomes. To date, most examples linking particular microbial loci to the ability to “infect” a host are instances of gene acquisition by horizontal gene transfer, leaving other potential sources of phenotypic novelty largely unexplored. Recent work in the yeasts Candida albicans and Saccharomyces cerevisiae have revealed how changes in transcriptional circuits played a role in the adaption of these species to colonize and cause disease in humans. These changes have been uncovered by genome-wide experimental and computational studies that mapped the gene regulatory networks of microorganisms that are evolutionarily related but that differ in their host-colonizing capabilities. We posit that evolutionary changes in transcriptional circuits were pivotal in endowing microorganisms with the ability to inhabit the human host. The prevalence of these kinds of changes can soon be empirically determined as similar experimental approaches are extended to other members of the human microbiota.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Research on Candida albicans in the Pérez laboratory is supported by the Interdisziplinäres Zentrum für Klinische Forschung der Universität Würzburg (Projekt A-296).
References
- Pérez JC, Johnson AD. Regulatory circuits that enable proliferation of the fungus Candida albicans in a mammalian host. PLoS Pathog 2013; 9: e1003780; http://dx.doi.org/10.1371/journal.ppat.1003780
- Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, Creasy HH, Earl AM, FitzGerald MG, Fulton RS. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486: 207-214; PMID:22699609; http://dx.doi.org/10.1038/nature11234.
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464: 59-65; PMID:20203603; http://dx.doi.org/10.1038/nature08821
- Li J, Huijue J, Cai X, Zhong H, Feng Qiang, Sunagawa S, Arumugam M, Kultima JR, Prifiti E, Nielsen T, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 2014; 32: 834-841; PMID:24997786; http://dx.doi.org/10.1038/nbt.2942
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature 2000; 405: 299-304; PMID:10830951; http://dx.doi.org/10.1038/35012500
- Perez JC, Groisman EA. Evolution of transcriptional regulatory circuits in bacteria. Cell 2009; 138: 233-244; PMID:19632175; http://dx.doi.org/10.1016/j.cell.2009.07.002
- Capra EJ, Laub MT. Evolution of two-component signal transduction systems. Annu Rev Microbiol 2012; 66: 325-347; PMID:22746333; http://dx.doi.org/10.1146/annurev-micro-092611-150039
- Chalancon G, Babu MM. Structure and evolution of transcriptional regulatory networks. Babu MM, editor. Bacterial Gene Regulation and Transcriptional Networks. Norfolk, UK. Caister Academic Press. 2013. p. 121-138
- Britten RJ, Davidson EH. Repetitive and non-repetitie DNA sequences and a speculation on the origins of evolutionary novelty. Quart Rev Biol 1971; 46: 111-138; PMID:5160087; http://dx.doi.org/10.1086/406830
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science 1975; 188: 107-116; PMID:1090005; http://dx.doi.org/10.1126/science.1090005
- Li H, Johnson AD. Evolution of transcription networks - Lessons from yeasts. Curr Biol 2010; 20: R746-R753; PMID:20833319; http://dx.doi.org/10.1016/j.cub.2010.06.056
- Guenther CA, Tasic B, Luo L, Bedell MA, Kingsley DM. A molecular basis for blond hair color in Europeans. Nat Genet 2014; 46: 748-752; PMID:24880339; http://dx.doi.org/10.1038/ng.2991
- Tishkoff SA, Reed FA, Ranciaro A, Voight BF, Babbitt CC, Silverman JS, Powell K, Mortensen HM, Hirbo JB, Osman M, et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat Genet 2007; 39: 31-40; PMID:17159977; http://dx.doi.org/10.1038/ng1946
- McLean CY, Reno PL, Pollen AA, Bassan AI, Capellini TD, Guenther C, Indjeian VB, Lim X, Menke DB, Schaar BT, et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 2011; 471: 216-9; PMID:21390129; http://dx.doi.org/10.1038/nature09774
- Gompel N, Prud’homme B, Wittkopp PJ, Kassner VA, Carroll SB. Chance caught on wings: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature 2005; 433: 481-487; PMID:15690032; http://dx.doi.org/10.1038/nature03235
- Werner T, Koshikawa S, Williams TM, Carroll SB. Generation of novel wing colour pattern by the Wingless morphogen. Nature 2010; 464: 1143-8; PMID:20376004; http://dx.doi.org/10.1038/nature08896
- Booth LN, Tuch BB, Johnson AD. Intercalation of a new tier of transcription regulation into an ancient circuit. Nature 2010; 486: 959-63; http://dx.doi.org/10.1038/nature09560
- Groisman E, Ochman H. Pathogenicity Island: Bacterial evolution in quantum leaps. Cell 1996; 87: 791-4; PMID:8945505; http://dx.doi.org/10.1016/S0092-8674(00)81985-6
- Odds FC. Candida infections: an overview. Crit Rev Microbiol 1987; 15: 1-5; PMID:3319417
- Calderone RA, Gow NA. Host recognition by Candida species. Calderone RA, editor. Candida and Candidiasis. Washington (D.C.). ASM Press. 2002. p. 107-122.
- Taylor JW, Berbee ML. Dating divergences in the fungal tree of life: review and new analyses. Mycologia 2006; 98: 838-49; PMID:17486961; http://dx.doi.org/10.3852/mycologia.98.6.838
- Braun BR, van Het Hoog M, d’Enfert C, Martchenko M, Dungan J, Kuo A, Inglis DO, Uhl MA, Hogues H, Berriman M, et al. A human curated annotation of the Candida albicans genome. PLoS Genet 2005; 1: 36-57; PMID:16103911; http://dx.doi.org/10.1371/journal.pgen.0010001
- Pérez JC, Fordyce PM, Lohse MB, Hanson-Smith V, DeRisi JL, Johnson AD. How duplicated transcription regulators can diversify to govern the expression of nonoverlapping sets of genes. Genes Dev 2014; 28: 1272-7; http://dx.doi.org/10.1101/gad.242271.114
- Ramos F, Dubois E, Pierard A. Control of enzyme synthesis in the lysine biosynthetic pathway of Saccharomyces cerevisiae. Evidence for a regulatory role of gene LYS14. Eur J Biochem 1998; 171: 171-6; http://dx.doi.org/10.1111/j.1432-1033.1988.tb13773.x
- Homann OR, Dea J, Noble SM, Johnson AD. A phenotypic profile of the Candida albicans regulatory network. PLoS Genet. 2009; 5: e1000783; PMID:20041210; http://dx.doi.org/10.1371/journal.pgen.1000783
- Pande K, Chen C, Noble SM. Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet 2003; 45: 1088-91; http://dx.doi.org/10.1038/ng.2710
- Pérez JC, Kumamoto CA, Johnson AD. Candida albicans commensalism and pathogenicity are intertwined traits directed by a tightly knit transcriptional regulatory circuit. PLoS Biol 2013; 11: e1001510; http://dx.doi.org/10.1371/journal.pbio.1001510
- Blake VM, Barolo S. Genome evolution: How sister genes grow apart. Curr Biol 2014; 24: R695-7; PMID:25093562; http://dx.doi.org/10.1016/j.cub.2014.06.049
- Enache-Angoulvant A and Hannequin C. Invasive Saccharomyces invasion: A comprehensive review. Clin Infect Dis 2005; 41: 1559-68; PMID:16267727; http://dx.doi.org/10.1086/497832
- Fraser HB, Levy S, Chavan A, Shah HB, Pérez JC, Zhou Y, Siegal ML, Sinha H. Polygenic cis-regulatory adaptation in the evolution of yeast pathogenicity. Genome Res 2012; 22: 1930-9; PMID:22645260; http://dx.doi.org/10.1101/gr.134080.111