
Abstract
Concepts or models of biological processes shape how we think about them, discuss them, and design experiments to test aspects of them. Because of the importance of our models of cell signaling by regulatory GTPases and the desire to extend those models to related signaling modules, I have throughout my career been fascinated by the similarities and differences between the modeling of heterotrimeric G protein and monomeric RAS superfamily GTPases. Recent discussions with colleagues led me to conclude that there is a growing divergence in how researchers model the activation and signaling processes of monomeric and trimeric GTPases and also a surprising lack of consensus within each camp. This series of articles arose in response to these discussions and is intended to spark new ones.
At last summer's FASEB meeting on ARFs/RABs I raised the question of whether there exists on membranes freely diffusible, activated GTPases, using a short history of (heterotrimeric) G proteins to contrast with my views of (monomeric) ARFs. This led to several interesting conversations, which continued after I returned to Emory. I am convinced that this is a fundamental question that shapes how we view GTPase signaling and design our experiments. As a result, I have asked a number of experts in the field of GTPase signaling to respond to the starting question, seen in my title. From Paul Liebman we have a historical perspective, which helps us appreciate the co-development of our models of biological membranes with those of G protein signaling, and also highlights the technical challenges encountered and lessons learned from the photoreceptor system. These issues are further refined and updated with newer technologies and data in the article from Arshavsky and Burns, which also focuses on rhodopsin—transducin—phosphodiesterase as the canonical GPCR/GEF—G protein—effector system that so strongly influenced early models and I think still colors our thinking in fundamental ways. Whether this canon can be appropriately extended to other or all GPCRs is explored in the articles by Ross and Hepler, which help us focus on existing data, key missing information, and well-reasoned suggestions as to how to obtain it. Articles from researchers focused on different families within the RAS superfamily were also solicited and currently include one from Martin Schwartz and Konstadinos Moissoglu, as a follow-up to their recent primary workCitation1 on RHO family activation and translocation onto membranes with a clear role for diffusion modeled into the process. We again see in this article the close relationship between our understanding of GTPase activation/signaling with membrane biology and lipid signaling. And Cathy Jackson adds some keen insights into the discussions by detailing and emphasizing the likely roles that “downstream” effectors play through interactions with GEFs and GTPases and pointing out that some GEFs can bind GTPases at two different sites, one catalytic and the other regulatory, that allow feed-forward signaling that is yet another type of signal amplifier. I offer a bit of background information below as both context for the other articles and to focus on some of what I believe are key questions in the fields today.
I remember some seminal papers from the 70s that used a mixture of mathematical modeling and wet bench data to argue that each GPCR can activate a large number of GTPases, which diffuse away to activate effectors. The simplest model for GPCR actions as guanine nucleotide exchange factors (GEFs) for heterotrimeric G proteins is shown in the left of , in which ligand binding leads to activation of a single Gα. I was lucky enough to have been present in the Gilman lab when others were developing the subunit dissociation model of G protein activation,Citation2,3 which posits that the activated (GTP-bound) Gα has reduced affinity for Gβγ, promoting dissociation of α from βγ and leading to the generation of 2 different activators of effectors per trimer. Not shown in the GPCR model in is the early, compelling, and paradigm promoting evidence that one activated GPCR can activate many G proteins, which diffuse on the surface of the membrane to encounter and activate effectors, thereby providing a biologically important amplification step to the process (see following articles for details). This has certainly shaped my thinking over the years and is why I was so struck by the fact that ARFs do not appear to behave as predicted by these models. This was highlighted, I thought quite simply, when we expressed different transmembrane protein “cargos” (e.g., mannose 6-phosphate receptor, furin, or amyloid precursor protein), each of which use ARFs to recruit a different set of coat proteins (e.g., adaptins, GGA1-3, COPI, or MINT3; one type of ARF effectors) required for their export from, or retention at, the Golgi. We found absolute specificity in pairing of cargo with adaptor for different cargos on the same membranes all apparently using the same set of ARF GTPases.Citation4 This observation alone seems sufficient to argue against the cargo-stimulated production of activated ARFs, which diffuse away from the GEF to encounter adaptors/effectors, as it provides no source of specificity in adaptor recruitment. Instead, there is more likely to be direct involvement of the cargo, ARF GEF, ARF, effector (coat proteins in this example) and even ARF GAPs into a protein complex that together dictate the specific outcome ( and ). In the model for cargo-dependent ARF activation shown in (right) it is important to realize what we do not know, as indicated by question marks. We do not know if there is a “ligand equivalent” that may bind and activate the transmembrane protein cargo, nor do we know how the presence of cargo at a membrane site leads to recruitment and/or activation of an ARF GEF (see ). I show the presence of a hypothetical “adapter” (not to be confused with coat proteins that are also often termed adaptors) to indicate the potential/likely homology to the RAS activation process, shown in the middle of . RAS activation by growth factor receptors, generically termed receptor tyrosine kinases (RTKs) in , involves recruitment to the membrane of the Grb2/SOS complex, the latter of which has RAS GEF activity, through binding of SH3 domains within Grb2 to specific phosphorylated motifs in the cytoplasmic tail of the RTK. Thus, while all regulatory GTPases are thought to require a GEF to activate them on the surface of the bilayer, G proteins use the heptahelical GPCRs that are intrinsic membrane proteins. In contrast, RASs and ARFs use GEFs that are recruited to membranes in a regulated fashion and may require distinct or concerted activation processes. Ligand binding to GPCRs leads to conformational changes that activate latent GEF activity. In contrast, ligand binding to RTKs promotes auto-phosphorylation that generates docking sites for the RAS GEF, Grb2/SOS. In addition, unknown ligands may bind to transmembrane protein cargos leading through unknown mechanisms to recruitment to the membrane of both ARF GEFs and the ARFs themselves to generate the activated ARFs and downstream signal.
Figure 1. Contrasting, simplified models of activation of G protein (left), RAS (middle), and ARF families of GTPases. GPCRs are a very large family (>800) of heptahelical membrane spanning proteins that bind ligands on the outside of the cell, leading to conformational changes that activate latent GEF activity for heterotrimeric G proteins on the cytoplasmic surface, promoting release of GDP and binding of the activating GTP. GPCRs can act catalytically to generate many activated Gα's per activated receptor, though may also retain the bound G protein subunits to act in more of a scaffolding role. One model of RAS activation (middle) is through the binding of a growth factor to its receptor on the outside of cells, resulting in auto-phosphorylation of the cytoplasmic tail of the receptor, and recruitment of the RAS GEF, Grb2/SOS, which activates the RAS already present on the plasma membrane. Thus, the GEF is recruited by the Receptor Tyrosine Kinase (RTK). Note that other RAS GEFs use different mechanisms (not shown). Less well understood is the role of transmembrane Cargos (e.g., mannose 6-phosphate receptor, amyloid precursor protein, etc) in recruiting or activating specific ARF GEFs (e.g., GBF1, BIG1/2, etc) at different sites inside cells. Both the ARF GEF and the ARF itself are recruited to the site of action. Roles for a ligand, binding to the cargo, or of an adaptor to physically couple the cargo to the ARF GEF are speculative and are included to highlight predicted functional homologies to the other GTPase systems.
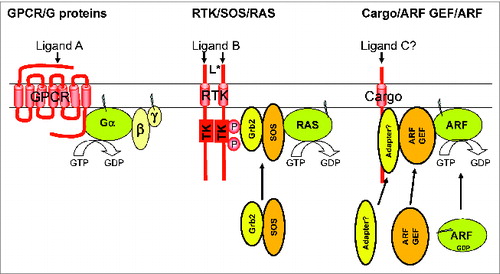
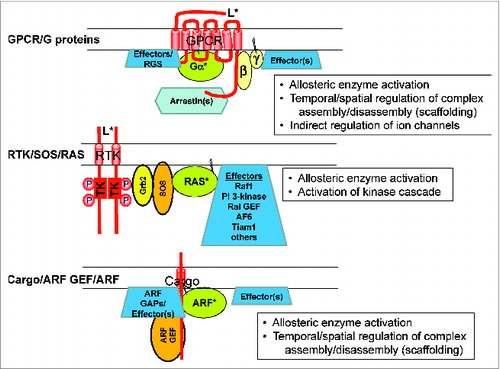
Figure 2. Modeling GTPase output as either/both allosteric regulation of enzymes and scaffolding to regulate the assembly of multi-subunit protein complexes. GPCRs (top) may be best known for their roles in activation of G proteins, leading to allosteric regulation of adenylyl cyclase, phospholipase Cβ, or Rho GEF, but are also increasingly appreciated to act as scaffolds for recruitment of effectors, RGS proteins, arrestins, and associated proteins that themselves may signal inside the cell or promote internalization of the complex. RAS protein signaling (middle) is best known for roles in oncogenesis through allosteric regulation of key pathways that include Raf1-MEK-ERK kinases, PI 3-kinase, RalGEF, and others. ARF signaling was earlier known for actions as an allosteric activator of the ADP-ribosyltransferase activity of cholera toxin from the human pathogen, Vibrio cholera, as well as the lipid modifying enzymes phospholipase D1 (PLD1), PI 4-kinase and PtdIns 4P 5-kinase (PI4P5K). But today it is perhaps best known for its role in recruiting coat proteins or complexes (COPI, GGA1-3, AP-1/3/4, MINT3) to specific membrane sites to coordinate nascent carrier biogenesis/coating. The extent to which lipid modifying and protein coating are integrated and work toward a common endpoint has been the source of much speculation. This is expected to be a common topic in the future for all GTPase families; i.e., the extent to which allosteric enzyme regulation and scaffolding synergize or antagonize the actions of each GTPase.
One example of why I think it is so important to discuss and compare our models of GTPase activation and action is the following. If signal amplification via one GEF generating multiple activated GTPases with lateral diffusion on the surface of the bilayer is off the table, then instead our thinking should focus on temporal control or the proofreading aspect of GTPase signaling. That is, if the lifetime of the activated GTPase (G*) controls the magnitude of the output, then we have great opportunity for signal amplification. But if the output is stoichiometric, i.e., 1 GTPase:1 complex/effector, then the lifetime of G* is more likely controlling the fidelity of the complex during assembly, and also imposing directionality to the assembly/disassembly processes through regulated GTP hydrolysis.
Another conclusion that emerged from my thinking on these issues is that signal amplification elicits different world views between the G protein and RAS superfamily camps. In the former, we see that the second messenger hypothesis was central to early models for G protein signaling and the importance of signal amplification. Binding of one ligand molecule on the cell surface generating huge changes in cAMP or CaCitation2+ in cytosol, came from those observations. In contrast, what is clearly emerging from studies of RAB biologies are “GTPase cascades” in which one activated RAB recruits to a membrane a GEF for the next GTPase which in turn recruits the GAP for the former one, and so on. With each activated RAB also recruiting or acting on distinct sets of effectors. This concatenation of activated GTPases results in signal amplification in the number of effectors affected by the sequential list of activated RABs. In the case of the RABs, whose functions are closely linked to the regulation of vesicular traffic, this cascade is closely tied to (perhaps even determines) vesicle maturation. Although several labs have contributed in important ways to this model it is the subject of a recent review from Mizuno-Yamasaki, et al.Citation5 that does a far better job explaining the details than I can here. I find the G protein signal amplifier and RAB GTPase cascades to be useful models with divergent roles for each of the different components, despite the conservation of overall biochemical properties (GTP binding, GEF or GAP activities, etc). There are likely to be related or important variants of these models emerging from studies of these or other GTPase families and I encourage those of you with such views to share them by adding to this series.
Although key aspects of models for G proteins vs. RAS superfamily members has been quite divergent, recent data suggest more commonality than previously appreciated. While once we thought of G proteins solely as allosteric activators or inhibitors of enzymes (e.g., adenylyl cyclase, phospholipase Cβ, cGMP phosphodiesterase, Rho GEF) at the plasma membrane, it is now widely appreciated that GPCRs often act as signaling centers or platforms (the term scaffold is often used in such contexts) that coordinate interactions of a large and growing list of proteins involved in a variety of aspects of cell signaling.Citation6-8 In addition to G proteins and their effectors there is evidence of direct binding to Regulators of G protein Signaling (RGS) proteins, which possess GAP activity for G proteins, and arrestins to GPCRs. Signaling by GPCRs, G proteins and their various interactors is also understood today to traffic throughout the cell and need not be limited to the cell surface. This is increasingly similar to a central function of the ARFs, which are best known for their role in regulating vesicular traffic at the Golgi, endosomes, and cell surface through direct binding to a number of different coat proteins or complexes (see ) and also the RABs. Thus, perhaps all families of GTPases act in different ways, to allosterically regulate both specific enzymes and the assembly of multi-subunit protein complexes. Which of these types of output is viewed as the most important for any one GTPase or GTPase family will obviously depend upon the context. But the fact that it is common for both to be occurring, perhaps on the same membrane, and that there is a finite pool of GTPases makes questions about specificity and diffusion of GTPase signaling only more important to address and model.
Although the question posed relates specifically to GTPase activation, I cannot leave this introductory article without some comparisons between the different models for termination of GTPase signaling. Though most GTPases have intrinsic GTPase activity that spontaneously hydrolyzes bound GTP, these are typically quite low and it is commonly assumed that GTPase signaling in the cell is terminated as a result of hydrolysis promoted by a GTPase activating protein (GAP) for RAS superfamily members and termed RGS for G proteins. GAP/RGS proteins can increase the rates of GTP hydrolysis by as much as five orders of magnitude and thus promptly silence the signal output from any substrate GTPase. But it is clearly a mistake to think of GAP/RGS proteins solely as terminators of signaling. This is particularly true with the ARF family as I believe that probably all ARF GAPs are effectors.Citation9 This idea was first emphasized to me when we cloned all known ARF GAPs in the yeast, S. cerevisiae, as high copy suppressors of the loss of ARF activityCitation10,11; an activity inconsistent with pure ARF signal terminators. Another way to consider roles for GAP/RGS proteins may be to compare to the two modes of signaling described above. If the function studied involves the GTPase acting as allosteric activator (or inhibitor) of a specific enzymatic activity, then it seems likely that the GAP/RGS will act in a silencing mode. However, if the GTPase instead is acting in a proofreading mode for regulating assembly and maturation of a protein complex, it is important for the GTPase to be activated to recruit components but then silenced to allow further maturation of the complex (e.g., see above discussion of RABs). Thus, it is very likely cycling of an ARF through GTP and GDP bound states during vesicle biogenesis that explains its function (and need for GTP hydrolysis) and absence from the mature vesicular carrier. Such a model makes it unlikely that GTP hydrolysis by a GTPase involved in vesicle biogenesis is also involved in the same vesicle's uncoating or fusion. This need for cycling to do its job in the cell can explain some confusing, or I believe mis-interpreted, data from the use of non-hydrolyzable GTP analogs (e.g., GTPγS) or expression of dominant mutants of GTPases. This model of GAPs acting as both effectors and terminators of GTPase signaling has been seen with G proteinsCitation12-16 and I expect in all GTPase families, but I am less familiar with others.
Modeling of GAP/RGS actions took on even greater significance for me when I read in the review from Ross and Wilkie on RGS/GAP proteinsCitation14: “If a receptor regulates multiple G proteins, however, then selective modulation of one Gα by an RGS (GAP) protein can qualitatively change the nature of output from a single receptor.” That is, with different Gα's being locally activated by one receptor, if those Gα-GTP's are then exposed to different RGS/GAPs that themselves have different specificities the nature of the output and effectors activated can be determined in large part by the GAPs (!). I had not previously considered these issues as likely sources of specificity in G protein signaling. I trust this idea has not escaped the attention of the pharmaceutical industry or anyone working on the design of modulators or inhibitors of GPCR signaling pathways with high specificity. I have for a long time been convinced that the field sorely needs more and better detailed biochemical characterizations of the affinities and specificities of each GTPase for each GEF, GAP, and effector, using the full length proteins and not just the isolated GEF (e.g., SEC7), GAP, or GTPase-binding domains. While such “simple” assays will certainly miss some biologically important regulatory factors (e.g., lipids, other proteins) they should form the basis for better models of GTPase pathways and the specificities required in biology.
I conclude my thoughts with the observation that the G protein and RAS superfamily fields began in different ways that I think have colored their evolutions. G proteins were first predicted and then identified and purified as essential regulatory components in hormone stimulated (notably β-adrenergic agonists like adrenaline and isoproterenol) adenylyl cyclase activity (apologies to the photoreceptor people). Those purifications, and later cloning and homology searching, identified the family of GTP-binding α subunits whose primary functions are coupling of GPCRs to effectors. In contrast, the RAS superfamily began in the middle, with the GTPases (RAS itself, ARF, etc) at a time when cloning and homology searching was more common and led to the rapid realization of larger families of paralogs. In many cases we didn't have a biology associated with the GTPase under study, and we have been identifying components in the pathways ever since. I can point to over 20 effectors of ARF GTPases and there are likely to be a similar number for RAS and other members of the superfamily. But I cannot yet describe for most of those effectors how one is activated to the exclusion of the others. Thus, what seems to me to be lagging behind in the RAS superfamily fields are not only the sources of specificity in determining which effector(s) is in play in specific cases, but also how the GEFs are activated. We know many of the GEFs, the functional homologs of the GPCRs, but what are the “ligand equivalents”? What regulates the GEFs? I think in the case of ARFs, the GTPase regulated pathways are much more likely to be constitutive in nature, tunable by mass action (e.g., changes in the levels of cargo at specific membrane sites or possibly of a lipid stimulator) rather than the acute changes in ligand concentration at the cell surface that require the rapid generation of an amplified signal that changes a biological outcome. This appears to be the situation with the RHO/RAC model described in the article from Schwartz and Moissoglu, as they point out the lack of point mutants but instead changes in GTPase expression levels showing correlations to diseases states. My goal here is simply to spark discussions that hopefully generate fruitful experimentation that continue the evolution of more comprehensive and specific models for GTPase signaling. It is a shame that as these fields have each gotten so large, we no longer have meetings that include G protein, RAS, RAB, ARF, RAN, RHO, etc researchers all together. I think we still have much to learn from one another, both in the commonalities and in the differences in the varied and critical cellular actions of our favorite signal transducers.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I thank the many colleagues who have discussed these topics with me over the years and in the generation of this Reasoned Debate series, too numerous to list. I readily admit to many sins of omission in models and citations in this article and figures as they are intended as summaries for discussion and are nowhere close to exhaustive. Many key findings and pathways were not included either due to my own ignorance or the need to stay focused on a few general points. There are many closely related topics that are interesting and extend or contradict statements made above. Not the least are novel roles for non-canonical GEFs for G proteins (e.g., Ric-8), membrane traffic of GTPases and their actions at different sites, and cross-talk between signaling by GTPases in different families (ARF-RAB, ARF-RHO, ARF-G proteins, etc).
Additional information
Funding
References
- Moissoglu K, Kiessling V, Wan C, Hoffman BD, Norambuena A, Tamm LK, et al. Regulation of Rac1 translocation and activation by membrane domains and their boundaries. J Cell Sci 2014; 127(pt 11):2565-76
- Northup JK, Smigel MD, Sternweis PC, Gilman AG. The subunits of the stimulatory regulatory component of adenylate cyclase. Resolution of the activated 45,000-dalton (alpha) subunit. J Biol Chem 1983; 258(18):11369-76; PMID: 6309844
- Northup JK, Sternweis PC, Gilman AG. The subunits of the stimulatory regulatory component of adenylate cyclase. Resolution, activity, and properties of the 35,000-dalton (beta) subunit. J Biol Chem 1983; 258(18):11361-8; PMID: 6309843
- Caster AH, Sztul E, Kahn RA. A role for cargo in Arf-dependent adaptor recruitment. J Biol Chem 2013; 288(21):14788-804; PMID: 23572528; http://dx.doi.org/10.1074/jbc.M113.453621
- Mizuno-Yamasaki E, Rivera-Molina F, Novick P. GTPase networks in membrane traffic. Annu Rev Biochem 2012; 81:637-59; PMID: 22463690; http://dx.doi.org/10.1146/annurev-biochem-052810-093700
- Ferre S, Casado V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 2014; 66(2):413-34; PMID: 24515647; http://dx.doi.org/10.1124/pr.113.008052
- Rebois RV, Hebert TE. Protein complexes involved in heptahelical receptor-mediated signal transduction. Receptor Channel 2003; 9(3):169-94; PMID: 12775338
- Malbon CC, Tao J, Wang HY. AKAPs (A-kinase anchoring proteins) and molecules that compose their G-protein-coupled receptor signalling complexes. Biochem J 2004; 379(Pt 1):1-9; PMID: 14715081; http://dx.doi.org/10.1042/BJ20031648
- East MP, Kahn RA. Models for the functions of Arf GAPs. Semin Cell Dev Biol 2011; 22(1):3-9; PMID: 20637885; http://dx.doi.org/10.1016/j.semcdb.2010.07.002
- Zhang CJ, Bowzard JB, Anido A, Kahn RA. Four ARF GAPs in Saccharomyces cerevisiae have both overlapping and distinct functions. Yeast 2003; 20(4):315-30; PMID: 12627398; http://dx.doi.org/10.1002/yea.966
- Zhang CJ, Cavenagh MM, Kahn RA. A family of Arf effectors defined as suppressors of the loss of Arf function in the yeast Saccharomyces cerevisiae. J Biol Chem 1998; 273(31):19792-6; PMID: 9677411; http://dx.doi.org/10.1074/jbc.273.31.19792
- Chen Z, Singer WD, Sternweis PC, Sprang SR. Structure of the p115RhoGEF rgRGS domain-Galpha13/i1 chimera complex suggests convergent evolution of a GTPase activator. Nat Struct Mol Biol 2005; 12(2):191-7; PMID: 15665872; http://dx.doi.org/10.1038/nsmb888
- Kozasa T. Regulation of G protein-mediated signal transduction by RGS proteins. Life Sci 2001; 68(19-20):2309-17; PMID: 11358341
- Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 2000; 69:795-827; PMID: 10966476; http://dx.doi.org/10.1146/annurev.biochem.69.1.795
- Shankaranarayanan A, Thal DM, Tesmer VM, Roman DL, Neubig RR, Kozasa T, Tesmer JJ. Assembly of high order G alpha q-effector complexes with RGS proteins. J Biol Chem 2008; 283(50):34923-34; PMID: 18936096; http://dx.doi.org/10.1074/jbc.M805860200
- Wells CD, Liu MY, Jackson M, Gutowski S, Sternweis PM, Rothstein JD, Kozasa T, Sternweis PC. Mechanisms for reversible regulation between G13 and Rho exchange factors. J Biol Chem 2002; 277(2):1174-81; PMID: 11698392; http://dx.doi.org/10.1074/jbc.M105274200