
Abstract
The cation-independent mannose 6-phosphate (Man-6-P) receptor (CI-MPR) binds newly synthesized, Man-6-P-containing lysosomal acid hydrolases in the trans-Golgi network (TGN) for clathrin-mediated transport to endosomes. It has remained unresolved, however, whether acid hydrolase binding is required for exit of the CI-MPR from the TGN. To address this question we used a B cell line derived from a Mucolipidosis type II (MLII)/I-cell disease patient. In MLII patients, acid hydrolases do not acquire the Man-6-P recognition marker and as a consequence do not bind to the CI-MPR. This causes secretion of the majority of the acid hydrolases and a decreased lysosomal activity resulting in typical inclusion bodies. In agreement herewith, ultrastructural analysis of the MLII patient derived B cells showed numerous inclusion bodies with undigested material, which we defined as autolysosomes. By quantitative immuno-electron microscopy we then studied the distribution of the CI-MPR in these cells. We found that the level of co-localization of TGN-localized CI-MPR and clathrin was similar in MLII and control B cells. Moreover, the CI-MPR was readily found in endosomes of MLII cells and the TGN-to-early endosome ratio of CI-MPR labeling was unaltered. These data show that there is no block in TGN exit of the CI-MPR in the absence of Man-6-P-modified acid hydrolases. Notably, late endosomes and inclusion bodies in MLII B cells contained increased levels of the CI-MPR, which likely reflects the reduced degradative capacity of these compartments.
Introduction
Lysosomes mediate the degradation of a variety of macromolecules through the action of lysosomal acid hydrolases. The lysosomal targeting of most of the newly synthesized acid hydrolases is mediated by the mannose 6-phosphate receptors (MPRs), the 300 kD cation-independent MPR (CI-MPR) and the 46 kD cation-dependent MPR (CD-MPR).Citation1 After synthesis in the endoplasmic reticulum, the acid hydrolases acquire the mannose 6-phosphate (Man-6-P) recognition marker in the Golgi complex. The Man-6-P modification is a high affinity ligand for the MPRs and is generated in a 2-step process. The first step is mediated by UDP-GlcNAc: lysosomal enzyme N-acetylglucosamine-1-phosphotransferase (or phosphotransferase). Phosphotransferase is an α2β2γ2 heterohexameric enzyme that transfers GlcNAc-1-phosphate from UDP-GlcNAc to one or more high mannose residues on the acid hydrolases in the cis-Golgi complex.Citation2-5 Subsequently, GlcNAc-1-phosphodiester α-N-acetylglucosaminidase, also known as uncovering enzyme, generates the Man-6-P monoester by removing the GlcNAc.Citation6,7
In the TGN the MPRs are sorted into clathrin-coated vesicles for their transport to endosomes. This sorting step requires the heterotetrameric adaptor protein complex (AP)-1 and the Golgi-localized, γ-ear-containing, Arf-binding family of proteins (GGA), which recognize specific motifs in the cytosolic tail of the MPRs and mediate the recruitment of clathrin.Citation8,9 In the acidic environment of the endosomes the Man-6-P-containing acid hydrolases dissociate from the MPRs for delivery to lysosomes. The majority of the MPRs return to the TGN to mediate a new round of acid hydrolase transport. The retrieval of MPRs from endosomes is mediated by multiple retrograde transport machineries, which act from different stages of the endo-lysosomal system, i.e. early and late endosomes.Citation10
In addition to the TGN and endosomes, low levels (approximately 5–15%) of the MPRs are found at the plasma membrane.Citation11,12 Plasma membrane associated CI-MPR can mediate the endocytosis of secreted acid hydrolases and several other ligands, including insulin-like growth factor II (IGF-II), transforming growth factor-β1 and retinoic acid. The steady state subcellular distribution of the MPRs represents an equilibrium between the TGN, endosomes and plasma membrane. Internalization of the CI-MPR from the plasma membrane appears not affected by ligand depletion,Citation13,14 although the rate of internalization is increased by multivalent ligands.Citation15
It has remained unclear to what extent the trafficking of the CI-MPR is dependent on the availability of acid hydrolases in the TGN. Studies that used cycloheximide treatment to deplete the cells of Man-6-P-containing acid hydrolases, found no alterations in the endosomal level of the CI-MPR or in the exchange of the receptor between cell surface and intracellular membranes due to depletion of these ligands.Citation13,14 Since the subcellular distribution of the CI-MPR maintains an equilibrium between the various compartments, these data indirectly suggest that TGN exit is not impaired when ligands are absent. Several approaches were applied to directly evaluate the TGN exit in the absence of Man-6-P-containing ligands, but have resulted in conflicting data. In a first approach, the subcellular distribution of the CI-MPR was examined in fibroblasts from Mucolipidosis type II (MLII) patients.Citation16,17 MLII (or I-cell disease) is characterized by deficiency of the catalytic αβ subunits of phosphotransferase, which results in the generation of acid hydrolases that lack the Man-6-P recognition moiety. As a consequence, in most cell types of MLII patients, the majority of the acid hydrolases are no longer targeted to lysosomes, but secreted via the constitutive secretory pathway, resulting in elevated serum levels of acid hydrolases and decreased enzymatic activity in lysosomes.Citation18 Therefore, a hallmark of the disease is the accumulation of undegraded material in enlarged lysosomes, known as inclusion bodies.Citation18 Thus, since in MLII cells the hydrolases are no longer recognized by the MPRs in the TGN, these cells were used to study the MPR in the ligand unbound state. By electron microscopic immunocytochemistry of MLII fibroblasts, the CI-MPR was found to accumulate at the trans-side of the Golgi apparatus and in the TGN, suggesting a block in TGN exit.Citation16,17 Concomitantly, the CI-MPR level in endosomes was greatly reduced.Citation17 However, these conclusions were based on non-quantitative, cytochemical assays.
In a second approach, hepatocytes were treated with tunicamycin, which inhibits glycosylation and therefore impairs generation of the Man-6-P modification on acid hydrolases. This resulted in depletion of the CI-MPR from endosomes and accumulation in coated vesicles in the Golgi region,Citation19 suggesting that ligand binding is required to trigger CI-MPR exit from the TGN. Tunicamycin, however, is a general inhibitor that in addition to acid hydrolases affects many glycoproteins.
In a third approach, CI-MPR fusion proteins were made that lack the lumenal domain of the receptor and therefore mimic the receptor in its ligand-unbound state. These constructs were found to exit the TGN and localize to endosomes.Citation20,21 However, in one study the subcellular distribution shifted to the TGN,Citation20 suggesting that TGN exit was impaired. A drawback of this approach is that it requires overexpression of the fusion proteins, which may lead to a relative depletion of machinery proteins and altered trafficking.
In the present study we used a B cell line derived from an MLII patient to determine the subcellular distribution of the CI-MPR by an optimized quantitative immuno-electron microscopy (immuno-EM) approach.Citation22 Our results show that the receptor does not require the binding of acid hydrolases to exit the TGN. In addition, we found that the endo-lysosomal system in MLII B cells is affected, with a decrease in the number of late endosomes and lysosomes and the appearance of autolysosomes that accumulate undigested material. The autolysosomes contained elevated levels of the CI-MPR, which likely reflects the reduced degradative capacity of these compartments.
Results
Lysosomal acid hydrolases in MLII B cells do not acquire the Man-6-P recognition marker
To study CI-MPR trafficking in the absence of Man-6-P-containing acid hydrolases we used a B lymphocyte cell line from a patient diagnosed with MLII.Citation23 To confirm that the phosphotransferase activity was impaired in these cells, a mutation analysis was performed on GNPTAB, the gene encoding the catalytic αβ subunits of phosphotransferase, by cDNA sequencing. This revealed a 2-basepair deletion, 3665_3666delTC, in exon 19, which results in a frameshift and truncation of the αβ subunits at amino acid 1172. This mutation has been described in MLII patients and results in near complete loss of phosphotransferase activity in patients that are homozygous for this mutation.Citation24
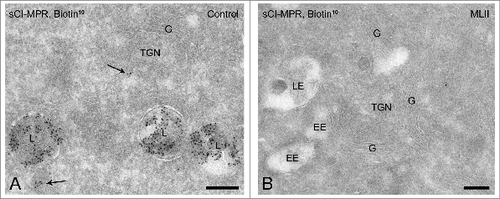
To test whether the activity of phosphotransferase was impaired in this B cell line, we investigated the presence of Man-6-P modifications in these cells. Ultrathin cryosections were incubated with biotinylated ligand-binding domain of the CI-MPR, which marks Man-6-P-containing acid hydrolases.Citation25 Subsequent immunogold labeling of the biotin moiety allows detection of Man-6-P-modified hydrolases by immuno-EM.Citation25 In control B cells Man-6-P-modified acid hydrolases were readily detectable, predominantly in late endosomes and lysosomes (). By contrast, the MLII B cells were completely devoid of label (). These results show that the acid hydrolases in the MLII B cells used in this study are not phosphorylated on their high mannose-type N-linked glycans.
Figure 1. Lysosomal acid hydrolases in MLII B cells do not acquire the Man-6-P modification. (A-B) Immunogold labeling for Man-6-P-modified acid hydrolases on ultrathin cryosections of control B cells (A) and MLII B cells (B). Sections were incubated with biotinylated ligand-binding domain of the CI-MPR (sCI-MPR) and subsequently immunogold labeled with anti-biotin antibodies and protein-A conjugated to 10 nm gold particles. In control B cells most labeling is present in late endosomes and lysosomes (A) and occasionally in transport vesicles (arrows in A). Labeling for Man-6-P-containing acid hydrolases is absent in MLII B cells (B). EE, early endosome; G, Golgi complex; L, lysosome; LE, late endosome. Bars, 200 nm.
CI-MPR association with clathrin-coated TGN membranes is unchanged in MLII B cells
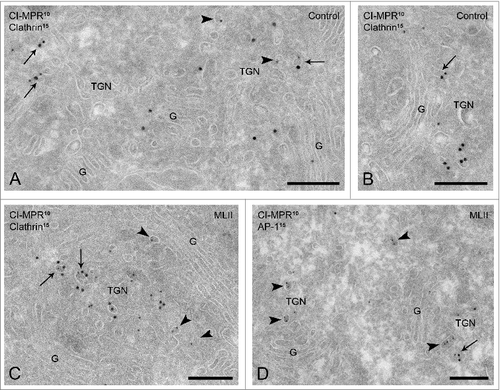
In the absence of the Man-6-P modification, acid hydrolases that arrive in the TGN can no longer bind to MPRs. To determine the effect of this cargo depletion on the trafficking of the CI-MPR, its distribution over the distinct TGN domains was evaluated in MLII and control B cells. In normal conditions, CI-MPRs are sorted into clathrin-coated TGN membranes that bud off to form carriers that deliver their cargo to endosomes. Therefore, the extent of co-localization of the CI-MPR and clathrin in TGN membranes of control and MLII B cells was quantified. The TGN was defined as those membranes at the trans-side of the Golgi complex that were within 500 nm distance of the trans-most cisterna.Citation11,26 In control B cells, low but consistent levels of clathrin labeling were obtained at the TGN, which occasionally co-localized with the CI-MPR (). In MLII B cells, the TGN exhibited similar ultrastructural characteristics and clathrin labeling. The CI-MPR was readily detectable in the clathrin-coated TGN membranes of MLII B cells (), where it also co-localized with the adaptor protein AP-1 (). Quantification of the co-localization of the CI-MPR and clathrin showed that in control B cells 19% of the total number of CI-MPR positive membranes also contained clathrin. This percentage was remarkably similar in MLII B cells, where 17% of the CI-MPR containing TGN membranes co-localized with clathrin.
Figure 2. The CI-MPR localizes to clathrin-coated/AP-1 positive TGN membranes in MLII B cells. Ultrathin cryosections of control B cells (A-B) and MLII B cells (C) double-immunogold labeled for the CI-MPR (10 nm gold particles) and clathrin (15 nm). In both control and MLII B cells, the CI-MPR was regularly observed in TGN membranes (arrowheads in A and C, respectively), in which it co-localized with clathrin (A-B and C, arrows). (D) MLII B cells double-labeled for the CI-MPR (10 nm, arrowheads) and AP-1 (15 nm) showing co-localization in a coated TGN vesicle (arrow). G, Golgi complex. Bars, 200 nm.
To extend these data to other cell types we also performed immuno-EM on MLII fibroblasts (Fig. S1) and osteoclasts (Fig. S2) derived from a mouse model of MLII (Gnptab−/− mice). The CI-MPR was readily detected in clathrin-coated TGN vesicles in both cell types. Together these data suggest that in the absence of Man-6-P-modified ligands in the TGN the CI-MPR is still sorted into clathrin-coated membranes.
MLII B cells accumulate inclusion bodies
The localization of the CI-MPR in clathrin-coated TGN membranes of MLII B cells suggests that the receptor traffics to endosomes in the absence of ligand. To be able to determine the endosomal localization of the CI-MPR in MLII B cells, we first needed to characterize the endo-lysosomal compartments in these cells. We therefore capitalized on a previous ultrastructural analysis, in which B cell endo-lysosomes were divided into 6 categories based on morphology, time dependent accessibility to endocytic tracers and the presence of molecular markers.Citation27 In summary, the following categories can be distinguished: (I) small, primary endocytic vesicles and tubules derived directly from the plasma membrane; (II and III) early endosomes with no or low label for the lysosomal membrane proteins LAMP-1 or LAMP-2; (IV) late endosomes with 9 or more intraluminal vesicles that represent the classical multivesicular bodies (MVBs), as shown in the human hepatoma cell line HepG2;Citation28 (V) late endosomes; (VI) multilaminar lysosomes. The MVBs (IV), late endosomes (V) and lysosomes (VI) are positive for LAMPs and major histocompatibility complex class II. The type V late endosomes are considered intermediates between MVBs (IV) and lysosomes (VI), containing both intraluminal vesicles and membrane sheets.
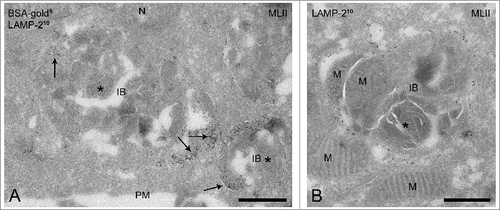
When a similar analysis was performed in the control and MLII B cells all 6 categories were observed. However, quantification of the number of compartments per cell profile () showed a striking difference in the occurrence of late endosomal compartments. The total number of late endosomes (type IV + V) was similar in both cell lines. However, in control B cells the majority of late endosomes represented the type V late endosomes, whereas the MLII B cells showed increased numbers of MVBs (type IV) and only few type V late endosomes (). In addition, a striking difference was observed in the number of lysosomes (VI). While in control B cells the lysosomes were the predominant compartments (36%) of all endo-lysosomal compartments (2.6 ± 0.55 per cell profile), in MLII B cells lysosomes were almost absent (0.3 ± 0.21 per cell profile). By contrast, MLII B cells contained numerous (3.3 ± 0.58 per cell profile) large vacuolar compartments () that were absent from control cells. These vacuoles were 500 nm - 3 μm in diameter and contained intraluminal vesicles, membrane sheets as well as amorphous electron dense material and cytoplasmic inclusions that could often be recognized as mitochondria (). The vacuoles were accessible to BSA-gold after 3 h of uptake (), indicating that they are compartments of endosomal origin. In addition, LAMP-1 (data not shown) and LAMP-2 were detected in their limiting membrane (), which positions them at a late stage of the endocytic pathway. We conclude that these enlarged endo-lysosomal compartments are the ultrastructural equivalent of the inclusion bodies that were previously described by light microscopy and conventional EM in different cell types of MLII patients.Citation29,30 The presence of BSA-gold and mitochondria indicates that these inclusion bodies receive material from both the endocytic and autophagic pathway, the cellular pathway to clear cytoplasmic components.Citation31 The presence of LAMP-1 and -2 in their limiting membranes further defines them as autolysosomes, which is in agreement with observations in MLII mouse models that show the presence of autolysosomes in exocrine secretory cells and compartments with similar morphology in various cell types.Citation32-34 In summary, the characterization of the endo-lysosomal system in MLII B cells shows that these cells have decreased numbers of type V late endosomes and lysosomes but instead accumulate inclusion bodies/autolyosomes with undigested material.
Table 1. MLII B cells accumulate inclusion bodies
Figure 3. MLII B cells accumulate inclusion bodies. (A-B) Ultrathin cryosections of MLII B cells showing the presence of inclusion bodies, which contain intraluminal vesicles, membrane sheets and undegraded material (indicated by asterisks). The endosomal nature of the inclusion bodies was confirmed by the presence of BSA-gold5 after 3 h of uptake (arrows in A) and the presence of the lysosomal membrane protein LAMP-2 (10 nm gold particles) in their limiting membrane. Occasionally mitochondria were recognized inside the inclusion bodies, as illustrated in (B). IB, inclusion body; M, mitochondrion; N, nucleus; PM, plasma membrane. Bars, 500 nm.
The CI-MPR is transported to early endosomes in MLII B cells
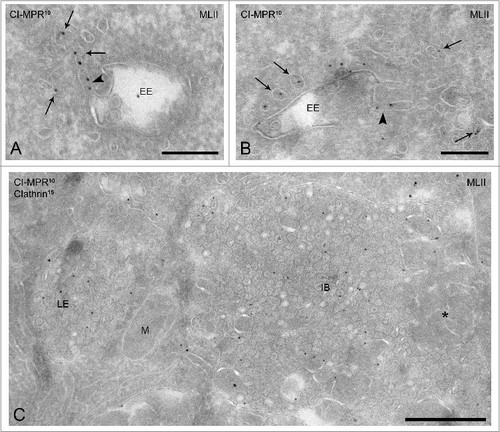
Following the categorization described in the previous paragraph, we next determined the endosomal delivery of the CI-MPR in MLII B cells. Although the CI-MPR is believed to deliver its cargo to both early and late endosomes, early endosomes are generally regarded as the main target compartments.Citation21,35 In agreement with this, the CI-MPR was readily found in early endosomes (type II + III) of MLII B cells, as well as in early endosome-associated tubules and vesicles (, arrows), which likely represent a mixture of TGN derived vesicles that deliver the CI-MPR to endosomes and recycling vesicles emerging from endosomes. Endosomal compartments in MLII fibroblasts (Fig. S1) and Gnptab−/− osteoclasts (Fig. S2) also significantly labeled for the CI-MPR, which indicates that endosomal delivery of the CI-MPR is not impaired in the absence of acid hydrolase binding.
Figure 4. The CI-MPR accumulates in late endosomes and inclusion bodies in MLII B cells. (A-C) Immunogold labeling for the CI-MPR (10 nm gold particles) in MLII B cells shows a normal localization to early endosomes (A-B) and associated vesicles (arrows). Moreover, the CI-MPR is present in typical recycling tubules emanating from the early endosomal vacuole (arrowheads in A and B). (C) Late endosomes and inclusion bodies (undegraded material indicated by the asterisk) show increased lumenal labeling for the CI-MPR. EE, early endosome; IB, inclusion body; LE, late endosome; M, mitochondrion. Bars A-B, 200 nm; bar in C, 500 nm.
In order to compare the relative distribution of the CI-MPR in control and MLII B cells, sections were randomly screened in the electron microscope. All CI-MPR representing gold particles were counted and the percentage of total labeling associated with a specific compartment was calculated (). In control B cells, the major pools of CI-MPR labeling were the TGN, endosomes and plasma membrane (), which is in agreement with previous studies in other cell types.Citation11 In MLII B cells the relative portion of CI-MPR label associated with the TGN and early endosomes was unaltered, whereas more CI-MPR was found in late endosomes. The plasma membrane labeling was slightly reduced (5.1 ± 1.42% vs 11.5 ± 0.67% for control cells), although this difference did not exceed the natural fluctuations observed in other cell types.Citation11,12, The labeling in cytoplasmic vesicles and tubules was reduced in MLII cells from 51.4 ± 1.94% to 30.2 ± 1.32%. Although this is a significant decrease, it is difficult to draw a conclusion form these data since this pool likely reflects a mixture of transport carriers.
Table 2. Subcellular distribution of the CI-MPR in MLII B cells compared to control B cells
Since the CI-MPR cycles between the TGN and endosomes, a block in exit from the TGN would have shifted the steady-state distribution accordingly. To address this, we compared the TGN-to-endosome ratios of the CI-MPR in control and MLII B cells. Only the early endosomes were taken into account for this quantification, because these are regarded as the main target compartments of TGN-derived CI-MPR Citation21,35 and because degradation is not yet a factor of importance in these early compartments but compromised in later endocytic compartments (see below). We found that the TGN-to-early endosome ratio in both cells was strikingly similar; in both control and MLII cells ∼0.9. Moreover, also in MLII cells the CI-MPR was found in typical recycling tubules emanating from early endosomes, illustrating that recycling proceeds from earlier stage endosomes (, arrowhead). Together, these results suggest that there is no block in CI-MPR cycling between the TGN and early endosomes in the absence of Man-6-P-containing ligands. In addition, the localization of the CI-MPR was evaluated in later endosomal compartments (). The MLII cells showed a striking increase in CI-MPR labeling in late endosomes, especially type IV MVBs, and inclusion bodies (20.3 ± 1.83% and 16.4 ± 1.63%, respectively) (). This increase is most likely due to the reduced degradative capacity of these compartments caused by the lack of acid hydrolases in these cells.
Discussion
Although the protein machinery that is involved in CI-MPR trafficking is relatively well understood, it has remained controversial whether binding to acid hydrolases is required for the trafficking of the CI-MPR from the TGN to endosomes.Citation13,14,16,17,19 In this paper we used quantitative immuno-EM to study the subcellular distribution of endogenous CI-MPR in the presence and absence of Man-6-P-containing acid hydrolases. Our data support the model that the CI-MPR is a constitutively cycling receptor that does not require ligand binding to exit the TGN.
We provide several lines of evidence to support the conclusion that ligand binding is not required for exit of the CI-MPR from the TGN. First, we found that the level of co-localization of the CI-MPR and clathrin in the TGN of MLII and control B cells was similar (17% and 19% co-localization, respectively). This result suggests that also in the absence of Man-6-P-containing acid hydrolases the CI-MPR is sorted into clathrin-coated vesicles. Since exit of the CI-MPR from the TGN requires its AP-1 and GGA-dependent sorting into clathrin-coated vesicles Citation8,9 this is a first indication that the exit is not impaired. Second, CI-MPR label was readily found in early and late endosomes in MLII B cells, showing that transport to the endo-lysosomal system occurs. Thirdly, quantification of the steady-state subcellular distribution of the CI-MPR in control and MLII B cells showed that the TGN-to-early endosome ratio was unchanged in the cargo depleted cells. Moreover, the CI-MPR was often found in typical recycling tubules emerging from endosomes (). Early endosomes are generally regarded as the main target compartments for the delivery of clathrin-coated CI-MPR vesicles from the TGN,Citation21,35 but also as the compartments for retromer-dependent recycling of CI-MPR to the TGN.Citation26,28,36,37 The balance between TGN exit and retrograde recycling determines the steady state distribution of the CI-MPR. Our data are therefore consistent with the model that both the exit of the CI-MPR from the TGN and retrograde transport from early endosomes continues in the absence of Man-6-P-containing ligands. Since the CI-MPR mediates both TGN-to-endosome transport of acid hydrolases as well as the uptake of ligands from the plasma membrane, its constitutive cycling may be required to keep the receptor available in the different pools and in appropriate equilibrium.Citation38 Our data, however, do not provide any information on the kinetics of cycling. The rate of CI-MPR internalization from the plasma membrane has been reported to increase upon binding of multivalent ligands.Citation15 Therefore, it is conceivable that the rate of CI-MPR cycling is altered in MLII cells.
Our conclusions are contradictory to previous studies in MLII fibroblasts,Citation16,17 which showed by EM immunocytochemistry that the CI-MPR accumulates in the TGN and no longer travels to endosomes in the absence of Man-6-P-modified ligands. However, in these studies the CI-MPR was still found on the plasma membrane of the MLII fibroblasts,Citation17 suggesting that at least a portion of the CI-MPR was able to exit the TGN. To investigate whether the differences between these and our studies might be cell type dependent, we used our cryosectioning and immunogold labeling approach to localize the CI-MPR in MLII fibroblasts (GM02013). In contrast to the previous studies we could detect the CI-MPR on the early endosomal limiting membrane and associated tubules and vesicles of MLII fibroblasts (Fig. S1), which makes it unlikely that the differences in our findings are explained by the different cell types used. Furthermore, in a parallel study of a mouse model of MLII (Gnptab−/− mice) we assessed the distribution of the CI-MPR in various tissues and osteoclasts and found no obvious differences in the subcellular distribution by immuno-EM (Fig. S2). Therefore, the discrepancy between our data and the previously published work is likely explained by the techniques used. Brown et al. applied EM immunocytochemistry, which requires permeabilization of the cells prior to primary antibody incubation and subsequent incubation with horseradish peroxidase-conjugated secondary antibody.Citation17 The use of detergents for permeabilization may result in local differences in penetration of the antibody and the loss of peripheral proteins, which may explain why they observed low endosomal CI-MPR levels.Citation39 Furthermore, EM immunocytochemistry cannot be used to obtain quantitative information.
MLII / I-cell disease is characterized by the presence of ‘inclusion bodies’ that were initially identified by light microscopy and conventional EM.Citation29,30 The accumulation of inclusion bodies is caused by impaired degradation of lysosomal contents due to the missorting of acid hydrolases. Our ultrastructural analysis of the endo-lysosomal compartments in control and MLII B cells showed that the latter were depleted of type V late endosomes and lysosomes. Instead they contained numerous enlarged, LAMP-1/2-positive vacuoles with endocytic (internalized BSA-gold) and undigested cytoplasmic material (e.g., mitochondrial remnants), defining them as autolysosomes.Citation32 The decrease in the number of type V late endosomes and lysosomes likely reflects a defect in endosomal maturation due to the reduced delivery of acid hydrolases to the endosomal system. In addition, these data are consistent with a report showing that inhibition of autolysosomal protein degradation abrogates lysosome reformation.Citation40 The autolysosomes are the likely ultrastructural counterparts of the inclusion bodies, since they were absent from control cells. In agreement with this finding, our recent immuno-EM studies in MLII mice showed a massive accumulation of autolysosomes in exocrine secretory glands.Citation32 The presence of inclusion bodies/autolysosomes in MLII B cells indicates that the transport of lysosomal hydrolases in these cells is strongly dependent on the MPRs.
Materials and Methods
Antibodies and reagents
Chemicals were obtained from Sigma-Aldrich unless otherwise specified. The CI-MPR was detected with the rabbit polyclonal antibody GVII, a kind gift from Dr K von Figura (Göttingen University, Germany), which was previously used in immuno-EM.Citation41 Mouse monoclonal antibodies against LAMP-1, LAMP-2, clathrin heavy chain and AP-1 were purchased from BD Biosciences. Rabbit polyclonal antibody against biotin was purchased from Rockland Immunochemicals, Inc. Man-6-P-modified lysosomal acid hydrolases were detected with biotinylated sCI-MPR, which was a generous gift from Dr. P. Lobel (Rutgers University, Piscataway, NJ). Lyophilized sCI-MPR was reconstituted to a final concentration of 10 μg/ml in PBS + 1% wt/vol bovine serum albumin (BSA), containing 5 mM β-glycerol phosphate disodium salt pentahydrate and 0.02% wt/vol sodium azide (Merck). Rabbit polyclonal antibody against IgM was obtained from Sanquin. Rabbit polyclonal antibody against mouse immunoglobulins (DAKO) was used as a bridging antibody between mouse monoclonal antibodies and protein A-gold (Cell Microscopy Center, UMC Utrecht, the Netherlands).
Cell lines and culture
The EBV-transformed MLII B cell line, derived from a patient clinically diagnosed with MLII (described in Citation42), and a control human B cell line (JY) were kindly provided by Dr. G. Griffith (University of Cambridge, UK). Cells were cultured in RPMI 1640 medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin (Life Technologies). Cells were kept at 37°C in a 5% CO2 humidified incubator until they reached a density of 3–5 × 105 cells/ml, upon which they were harvested by centrifugation.
Mutation analysis
To perform a mutation analysis on the cDNA of GNPTAB, total RNA was first isolated from MLII B cells with the high pure RNA isolation kit (Roche applied science) according to the manufacturer's protocol. RT-PCR was performed on total RNA with oligo(dT)15 primers in the presence of 5 mM MgCl2 (Promega). cDNA of GNPTAB was amplified with the primers 5′ – GGA GGG GTG ATG CTG TTC AA – 3′ and 5′ – TCA GAG CTG CTC AAC ACA CA – 3′. PCR was performed with Phusion high-fidelity PCR master mix (Finnzymes) in the presence of 3% dimethylsulfoxide (Merck). Parameters were as follows: 35 cycles, denaturation at 98°C for 10s, annealing at 60°C for 30s, extension at 72°C for 30s. The PCR product was gel purified and sequenced with the Big Dye terminator v1.1 cycle sequencing kit (Applied Biosystems) and the following primers: 5′ – CTC AGG CTC CTC GGG GCG T – 3′, 5′ – GCA TCA CAT CAC AAA GAC ATC TC – 3′, 5′ – TGG TCA ATG TTG TCA TTC AGG C – 3′, 5′ – ACT GCA TAT CCT CAG AAT GGC – 3′, 5′ – ATC TTG CTG TGG TCT CCA AGT – 3′, 5′ – TCT GGG CTG TAG AAT TCA G – 3′.
Immuno-electron microscopy
Cells were cultured as described and fixed for immuno-EM upon reaching a density of 3–5 × 105 cells/ml. Fixation was achieved by adding 0.4% wt/vol glutaraldehyde (GA; Polysciences Europe GmbH) + 4% wt/vol paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB; pH = 7.4) to an equal volume of culture medium for 15 minutes at room temperature (RT). Subsequent post-fixation with 0.2% wt/vol GA + 2% wt/vol PFA in 0.1 M PB was for >2 h at RT. Embedding for immuno-EM, preparation of ultrathin cryosections and immunogold labeling was performed as previously described.Citation22 Sections were analyzed in a Jeol 1200 EX electron microscope (Tokyo, Japan) at 80 kV. To detect Man-6-P-modified acid hydrolases, ultrathin cryosections were first blocked with PBS + 1% wt/vol BSA for 2 h at 4°C, followed by overnight incubation with biotinylated sCI-MPR at 4°C. Subsequent immunogold labeling with anti-biotin was performed following the standard procedure.Citation22 To identify endo-lysosomal compartments, cells were incubated with BSA conjugated to 5 nm gold particles (BSA-gold5; Cell Microscopy Center, UMC Utrecht, the Netherlands). To this end, cells were transferred to RPMI 1640 medium containing 1% FBS. BSA-gold5 was added at a final OD of 5 and cells were incubated in a 5% CO2 humidified incubator at 37°C for 3 h. The cells were then washed in cold, serum-free medium, fixed in 0.2% wt/vol GA + 2% wt/vol PFA in 0.1 M PB and processed for cryo-immunogold labeling.
Quantifications
The number of endo-lysosomal compartments in control B cells and MLII B cells was determined by analyzing 30 cell profiles (>200 compartments) of ultrathin cryosections immunogold labeled for either LAMP-1 or LAMP-2. The different categories were according to a previous morphological study in B cells.Citation27 Early endosomes (type II and III) contained up to 8 intraluminal vesicles and MVBs (type IV) contained 9 or more intraluminal vesicles.Citation28 All statistical calculations were performed in Microsoft Excel. Values are expressed as the average number of compartments per cell profile ± the standard error of the mean (SEM), n = 30. P-values were determined by Student's t test. The percentage of CI-MPR and clathrin labeled TGN membranes from the total number of CI-MPR labeled TGN membranes was analyzed from images taken at a magnification of 30,000× in the electron microscope. All membranes located in an area within 500 nm from the most trans-Golgi cisterna were designated as TGN. At least 20 TGNs were assessed for each cell line. To determine the subcellular localization of the CI-MPR, sections double immunogold labeled for the CI-MPR and IgM were assessed. Cells with high IgM labeling were excluded from the counting, to avoid variations due to differences in the activation state of the cells. Quantifications were performed by randomly screening through ultrathin cryosections containing cell profiles with a good overall ultrastructure. All gold particles within a distance of 15 nm from a membrane were regarded as positive label. Endo-lysosomal compartments were defined as described.Citation27,28 For each condition 2 × 300 gold particles were counted, from sections located on different grids. Numbers are expressed as percentages of the total number of gold particles ± standard deviations.
Supplemental Materials and Methods
The fibroblast lines GM02013 (MLII, compound heterozygote with 3395_3398dupCTAC and 3665_3666delTC, phosphotransferase activity undetectable) and GM02014 (control) were obtained from the Coriell Institute for Medical Research. The cells were maintained in Dulbecco's Modified Eagle Medium (Life Technologies) supplemented with 15% FBS, 2 mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin (Life Technologies). Bone marrow-derived macrophages from Gnptab−/− and wild-type mice were differentiated into osteoclasts as described.Citation43 The cells were fixed in 2% wt/vol PFA + 0.2% wt/vol GA in 0.1 M PB pH = 7.4 and prepared for cryosectioning and immunogold labeling as described in the main text. To detect the CI-MPR and clathrin, the sections were incubated with the rabbit anti-human CI-MPR antibody GVII, a kind gift from Dr K von Figura (Göttingen University, Germany) or rabbit anti-bovine CI-MPR antibody (generous gift from Dr S Kornfeld, Washington University in St Louis, St Louis, MO), both previously used in immuno-EM41,43 and a mouse monoclonal antibody to the clathrin heavy chain (BD Biosciences). Antibodies were marked by 10 or 15 nm protein A-gold particles (Cell Microscopy Center, UMC Utrecht, the Netherlands).
Supplemental data for this article can be accessed on the publisher's website.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
954441_Supplementary_Materials.zip
Download Zip (9.8 MB)Acknowledgments
We thank the members of the Department of Cell Biology for helpful discussions and in particular Marc van Peski and René Scriwanek for their help with preparation of the electron micrographs, Dr Gillian Griffiths, Dr Peter Lobel and Dr Kurt von Figura for kindly providing the B cell lines, biotinylated sCI-MPR and CI-MPR antibody, respectively.
Additional information
Funding
References
- Ghosh P, Dahms NM, Kornfeld S. Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 2003; 4:202-12; PMID:12612639; http://dx.doi.org/10.1038/nrm1050
- Bao M, Booth JL, Elmendorf BJ, Canfield WM. Bovine UDP-N-acetylglucosamine:lysosomal-enzyme N-acetylglucosamine-1-phosphotransferase. I. Purification and subunit structure. J Biol Chem 1996; 271:31437-45; PMID:8940155; http://dx.doi.org/10.1074/jbc.271.49.31437
- Reitman ML, Kornfeld S. UDP-N-acetylglucosamine:glycoprotein N-acetylglucosamine-1-phosphotransferase. Proposed enzyme for the phosphorylation of the high mannose oligosaccharide units of lysosomal enzymes. J Biol Chem 1981; 256:4275-81; PMID:6452459
- Reitman ML, Kornfeld S. Lysosomal enzyme targeting. N-Acetylglucosaminylphosphotransferase selectively phosphorylates native lysosomal enzymes. J Biol Chem 1981; 256:11977-80; PMID:6457829
- Waheed A, Hasilik A, von Figura K. UDP-N-acetylglucosamine:lysosomal enzyme precursor N-acetylglucosamine-1-phosphotransferase. Partial purification and characterization of the rat liver Golgi enzyme. J Biol Chem 1982; 257:12322-31; PMID:6288715
- Varki A, Kornfeld S. Purification and characterization of rat liver alpha-N-acetylglucosaminyl phosphodiesterase. J Biol Chem 1981; 256:9937-43; PMID:6268636
- Waheed A, Hasilik A, von Figura K. Processing of the phosphorylated recognition marker in lysosomal enzymes. Characterization and partial purification of a microsomal alpha-N-acetylglucosaminyl phosphodiesterase. J Biol Chem 1981; 256:5717-21; PMID:6263889
- Ghosh P, Kornfeld S. The GGA proteins: key players in protein sorting at the trans-Golgi network. Eur J Cell Biol 2004; 83:257-62; PMID:15511083; http://dx.doi.org/10.1078/0171-9335-00374
- Ghosh P, Kornfeld S. The cytoplasmic tail of the cation-independent mannose 6-phosphate receptor contains four binding sites for AP-1. Arch Biochem Biophys 2004; 426:225-30; PMID:15158672; http://dx.doi.org/10.1016/j.abb.2004.02.011
- Pfeffer SR. Entry at the trans-face of the Golgi. Cold Spring Harbor Perspect Biol 2011; 3; PMID:21421921; http://dx.doi.org/10.1101/cshperspect.a005272
- Klumperman J, Hille A, Veenendaal T, Oorschot V, Stoorvogel W, von Figura K, Geuze HJ. Differences in the endosomal distributions of the two mannose 6-phosphate receptors. J Cell Biol 1993; 121:997-1010; PMID:8099077; http://dx.doi.org/10.1083/jcb.121.5.997
- Braulke T, Mach L, Hoflack B, Glossl J. Biosynthesis and endocytosis of lysosomal enzymes in human colon carcinoma SW 1116 cells: impaired internalization of plasma membrane-associated cation-independent mannose 6-phosphate receptor. Arch Biochem Biophys 1992; 298:176-81; PMID:1326252; http://dx.doi.org/10.1016/0003-9861(92)90109-A
- Braulke T, Gartung C, Hasilik A, von Figura K. Is movement of mannose 6-phosphate-specific receptor triggered by binding of lysosomal enzymes? J Cell Biol 1987; 104:1735-42; PMID:2953746; http://dx.doi.org/10.1083/jcb.104.6.1735
- Pfeffer SR. The endosomal concentration of a mannose 6-phosphate receptor is unchanged in the absence of ligand synthesis. J Cell Biol 1987; 105:229-34; PMID:3038925; http://dx.doi.org/10.1083/jcb.105.1.229
- York SJ, Arneson LS, Gregory WT, Dahms NM, Kornfeld S. The rate of internalization of the mannose 6-phosphate/insulin-like growth factor II receptor is enhanced by multivalent ligand binding. J Biol Chem 1999; 274:1164-71; PMID:9873065; http://dx.doi.org/10.1074/jbc.274.2.1164
- Brown WJ. Cation-independent mannose 6-phosphate receptors are concentrated in trans Golgi elements in normal human and I-cell disease fibroblasts. Eur J Cell Biol 1990; 51:201-10; PMID:2161763
- Brown WJ, Farquhar MG. Accumulation of coated vesicles bearing mannose 6-phosphate receptors for lysosomal enzymes in the Golgi region of I-cell fibroblasts. Proc Natl Acad Sci U S A 1984; 81:5135-9; PMID:6147848; http://dx.doi.org/10.1073/pnas.81.16.5135
- Braulke T, Raas-Rothschild A, Kornfeld S. I-cell disease and pseudo-Hurler polydystrophy: disorders of lysosomal enzyme phosphorylation and localization. Online Metab Mol Bases Inherit Dis 2013; Chapter 138; http://dx.doi.org/10.1036/ommbid.167
- Brown WJ, Constantinescu E, Farquhar MG. Redistribution of mannose-6-phosphate receptors induced by tunicamycin and chloroquine. J Cell Biol 1984; 99:320-6; PMID:6330128; http://dx.doi.org/10.1083/jcb.99.1.320
- Conibear E, Pearse BM. A chimera of the cytoplasmic tail of the mannose 6-phosphate/IGF-II receptor and lysozyme localizes to the TGN rather than prelysosomes where the bulk of the endogenous receptor is found. J Cell Sci 1994; 107 (Pt 4):923-32; PMID:8056846
- Waguri S, Dewitte F, Le Borgne R, Rouille Y, Uchiyama Y, Dubremetz JF, Hoflack B. Visualization of TGN to endosome trafficking through fluorescently labeled MPR and AP-1 in living cells. Mol Biol Cell 2003; 14:142-55; PMID:12529433; http://dx.doi.org/10.1091/mbc.E02-06-0338
- Slot JW, Geuze HJ. Cryosectioning and immunolabeling. Nat Protoc 2007; 2:2480-91; PMID:17947990; http://dx.doi.org/10.1038/nprot.2007.365
- Griffiths GM, Isaaz S. Granzymes A and B are targeted to the lytic granules of lymphocytes by the mannose-6-phosphate receptor. J Cell Biol 1993; 120:885-96; PMID:8432729; http://dx.doi.org/10.1083/jcb.120.4.885
- Kudo M, Brem MS, Canfield WM. Mucolipidosis II (I-cell disease) and mucolipidosis IIIA (classical pseudo-hurler polydystrophy) are caused by mutations in the GlcNAc-phosphotransferase alpha/beta -subunits precursor gene. Am J Hum Genet 2006; 78:451-63; PMID:16465621; http://dx.doi.org/10.1086/500849
- Valenzano KJ, Kallay LM, Lobel P. An assay to detect glycoproteins that contain mannose 6-phosphate. Anal Biochem 1993; 209:156-62; PMID:8465950; http://dx.doi.org/10.1006/abio.1993.1096
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol 2004; 164:1065-76; PMID:15051738; http://dx.doi.org/10.1083/jcb.200311064
- Kleijmeer MJ, Morkowski S, Griffith JM, Rudensky AY, Geuze HJ. Major histocompatibility complex class II compartments in human and mouse B lymphoblasts represent conventional endocytic compartments. J Cell Biol 1997; 139:639-49; PMID:9348281; http://dx.doi.org/10.1083/jcb.139.3.639
- Mari M, Bujny MV, Zeuschner D, Geerts WJ, Griffith J, Petersen CM, Cullen PJ, Klumperman J, Geuze HJ. SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic 2008; 9:380-93; PMID:18088323; http://dx.doi.org/10.1111/j.1600-0854.2007.00686.x
- Leroy JG, Demars RI. Mutant Enzymatic and cytological phenotypes in cultured human fibroblasts. Science 1967; 157:804-6; PMID:17842782; http://dx.doi.org/10.1126/science.157.3790.804
- Rapola J, Autio S, Aula P, Nanto V. Lymphotic inclusions in I-cell disease. J Pediat 1974; 85:88-90; PMID:4368882; http://dx.doi.org/10.1016/S0022-3476(74)80294-5
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 2009; 335:1-32; PMID:19802558
- Boonen M, van Meel E, Oorschot V, Klumperman J, Kornfeld S. Vacuolization of mucolipidosis type II mouse exocrine gland cells represents accumulation of autolysosomes. Mol Biol Cell 2011; 22:1135-47; PMID:21325625; http://dx.doi.org/10.1091/mbc.E10-07-0584
- Kollmann K, Damme M, Markmann S, Morelle W, Schweizer M, Hermans-Borgmeyer I, Rochert AK, Pohl S, Lubke T, Michalski JC, et al. Lysosomal dysfunction causes neurodegeneration in mucolipidosis II ‘knock-in’ mice. Brain: A J Neurol 2012; 135:2661-75; PMID:22961545; http://dx.doi.org/10.1093/brain/aws209
- Kollmann K, Pestka JM, Kuhn SC, Schone E, Schweizer M, Karkmann K, Otomo T, Catala-Lehnen P, Failla AV, Marshall RP, et al. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol Med 2013; 5:1871-86; PMID:24127423; http://dx.doi.org/10.1002/emmm.201302979
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 2003; 72:395-447; PMID:12651740; http://dx.doi.org/10.1146/annurev.biochem.72.121801.161800
- Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, van der Sluijs P, Bonifacino JS. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J Cell Biol 2008; 183:513-26; PMID:18981234; http://dx.doi.org/10.1083/jcb.200804048
- van Weering JR, Verkade P, Cullen PJ. SNX-BAR-mediated endosome tubulation is co-ordinated with endosome maturation. Traffic 2012; 13:94-107; PMID:21973056; http://dx.doi.org/10.1111/j.1600-0854.2011.01297.x
- Royle SJ, Murrell-Lagnado RD. Constitutive cycling: a general mechanism to regulate cell surface proteins. BioEssays: News Rev Mol, Cell Dev Biol 2003; 25:39-46
- Griffiths G. Fine Structure Immunocytochemistry. Springer-Verlag, 1993; http://dx.doi.org/10.1007/978-3-642-77095-1
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465:942-6; PMID:20526321; http://dx.doi.org/10.1038/nature09076
- Geuze HJ, Slot JW, Strous GJ, Hasilik A, von Figura K. Possible pathways for lysosomal enzyme delivery. J Cell Biol 1985; 101:2253-62; PMID:2933416; http://dx.doi.org/10.1083/jcb.101.6.2253
- Layton GT, Harris SJ, Gearing AJ, Hill-Perkins M, Cole JS, Griffiths JC, Burns NR, Kingsman AJ, Adams SE. Induction of HIV-specific cytotoxic T lymphocytes in vivo with hybrid HIV-1 V3:Ty-virus-like particles. J Immunol 1993; 151:1097-107; PMID:8335892
- van Meel E, Boonen M, Zhao H, Oorschot V, Ross FP, Kornfeld S, Klumperman J. Disruption of the Man-6-P targeting pathway in mice impairs osteoclast secretory lysosome biogenesis. Traffic 2011; 12:912-24.