
Abstract
Obesity is often associated with systemic insulin resistance, and the decline of insulin sensitivity marks the progression of obesity into a disease state. We recently generated a mouse with adipose-specific ablation of the p110α phosphoinositide 3-kinase (PI3K) catalytic subunit to model insulin resistance in this organ. The phenotypes of this animal revealed novel roles of adipose PI3K signaling in regulating body weight and systemic glucose and lipid homeostasis. Loss of p110α in the brown adipose tissue resulted in reduced expression of mitochondrial-associated genes and decreased respiration in brown adipocytes. Reduced activity of the brown adipose tissue in p110α-null mice lowered their energy expenditure, which promoted obesity and systemic metabolic dysfunction with increased lipid deposition in the liver. Loss of PI3K activity did not affect adiposity until sexual maturation, suggesting that the effect of adipose PI3K on obesity might be linked to the development of puberty. Elevated leptin in the p110α knockout mice might interfere with the reproductive axis to delay pubertal development. The increase in adiposity induced by adipose-specific loss of p110α provides a link between insulin resistance and obesity onset and may also provide deeper insight into changes in prepubescent insulin sensitivity that can affect metabolism later in life.
Introduction
Obesity is caused by an energy imbalance resulting from caloric intake that exceeds caloric expenditure. Consumption of calories that are not burned off by exercise or other increases in metabolic activity produces weight gain. Obesity was long thought to be simply a by-product of persistent overeating and an unhealthy lifestyle. While comorbidities of obesity such as type 2 diabetes and cardiovascular disorders have long been considered to be diseases, obesity itself was only recently recognized as a disease by the American Medical Association.Citation1 This delay has hampered progress toward a greater understanding of the underlying mechanisms of obesity that could lead to useful pharmaceutical interventions for prevention or treatment. There is a poor success rate for the long-term reduction of body weight through behavioral intervention alone, with the majority of obese patients achieving only a 5% decrease in body weight within a 2 year period.Citation2 More research is needed to investigate how increased adiposity is linked to other disease states and how changes in basal energy expenditure affect body weight.
Confounding our understanding of the mechanisms of obesity onset are conditions such as insulin resistance which often accompany increases in adiposity.Citation3 Insulin resistance is characterized by reductions in insulin-stimulated glucose uptake and metabolism in the skeletal muscle and adipose tissues and by reduced suppression of hepatic glucose output.Citation4 Obesity has long been assumed to be a causative factor in the progression from insulin resistance to type 2 diabetes. However, not all obese individuals are diabetic, indicating that obesity may not be directly responsible for increased insulin resistance.Citation5 In fact, insulin resistance is tightly linked to other pathologies, including hypertension, hyperlipidemia, and metabolic syndrome.Citation4 It is very likely that disruption of the insulin signaling pathway in adipose tissues contributes to at least some of these pathologies.
Phosphoinositide 3-Kinase (PI3K) mediates insulin signaling in adipocytes
PI3Ks are important transducers of downstream signals initiated by activation of the insulin receptor. This places PI3K at a pivotal junction of metabolic regulation in insulin-responsive tissues. PI3K activity is decreased in the muscle, liver and adipose tissue in almost all type 2 diabetic patients.Citation6 Therefore, many researchers have examined the actions of PI3K and the contributions of this enzyme to metabolic dysfunction. Class 1A PI3Ks are insulin-activated heterodimeric enzymes composed of a regulatory subunit tightly bound to a catalytic subunit. The catalytic subunit phosphorylates the 3′ hydroxyl position of the inositol ring in phosphatidylinositol 4,5-bisphosphate to generate the second messenger phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 binds to downstream targets to effect changes in signaling pathways that are relevant to a wide range of metabolic processes. The p110α catalytic subunit is thought to be the primary insulin-responsive PI3K in adipose tissues. Other catalytic subunits such as p110β may serve other functions but do not seem to be strong mediators of insulin signaling in the adipocyte.Citation7
Adipose tissue is a highly insulin-responsive organ, and PI3K plays a major role in regulating many insulin-dependent processes in adipocytes that provide systemic metabolic benefits. Insulin induces fatty acid and glucose uptake in adipocytes by promoting the translocation of transporters to the cell surface. Treatment of 3T3-L1 adipocytes with wortmannin, a PI3K inhibitor, decreased insulin-stimulated fatty acid and glucose uptake through reduced translocation of fatty acid transport protein 1 and glucose transporter 4, respectively.Citation8,9 The ability of insulin to suppress lipolysis is also PI3K dependent.Citation9 Attenuation of PI3K activity through the use of p110α-specific inhibitors blocked insulin signaling in adipocytes, demonstrating that the catalytic activity of PI3K is necessary to mediate the effects of insulin.Citation7 PI3K activity is also important in the brown adipose tissue (BAT), where it contributes to insulin-stimulated expression of thermogenic proteins.Citation10 However, the precise contribution of PI3K activity to BAT function is less well understood than its role in WAT.
Loss of p110α in the adipose tissues causes obesity
We recently reported that adipose-specific ablation of the p110α PI3K catalytic subunit in a mouse model resulted in an age-dependent increase in body weight beginning at 6 weeks of age.Citation11 By 4 months of age the p110α-null (α−/−) mice were obese and displayed the classic phenotypes of metabolic syndrome including glucose intolerance and increased liver lipid deposition. Mice with reduced PI3K activity in the adipose tissue consumed the same number of calories and exhibited the same ambulatory activity as the control animals, yet they had lower energy expenditure when compared to their lean controls.Citation11 This lack of balance between caloric intake and expenditure likely contributes to the α−/− obese phenotype. We provided evidence that the reduction in energy expenditure in the α−/− mouse is due at least in part to decreased BAT energy expenditure as a result of reduced expression of many key mitochondrial-specific and thermogenic genes including the classic BAT marker UCP-1 (uncoupling protein-1).Citation11
Many of the recent investigations into the function of BAT focused on the potential of BAT activation as a means of increasing energy expenditure and inducing weight loss for obesity therapy. While these investigations proved that BAT in obese as compared to lean subjects is less metabolically active, little progress has been made in elucidating the mechanisms involved in the reduction of BAT activity.Citation12 Mouse models with reduced UCP-1 expression show a variety of phenotypes, primarily heightened cold sensitivity and obesity induced only under thermoneutral conditions.Citation13-15 It is clear from this work that loss of UCP-1 alone is not sufficient to induce obesity under normal conditions.
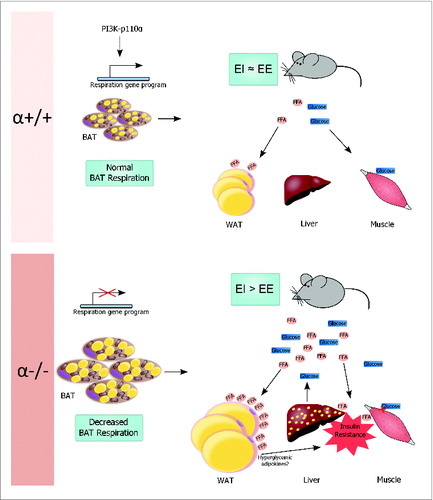
The insulin signaling pathway is a powerful regulator of gene expression, and we showed that p110α regulates the mitochondria at the transcriptional level in BAT.Citation11 The broad spectrum of genes affected by reduced p110α expression in BAT adds to the known importance of transcriptional regulation by the insulin signaling pathway.Citation11 The reduced respiration of α−/− BAT is likely a result of a global reduction in respiratory gene expression rather than loss of one target such as UCP-1. Reduced energy expenditure in the α−/− mouse without any alterations in food intake or activity results in an energy surplus exemplified by elevated serum lipids including free fatty acids (FFAs) which cause lipotoxicity in peripheral tissues (). The WAT and the liver cope with this lipid overload by increasing their lipid uptake, resulting in increased adiposity and hepatic steatosis, respectively. The adipose-specific α−/− mouse model is a strong example of how changes in BAT function are capable of driving systemic metabolic changes.
Figure 1. Model of metabolic dysfunction in the α−/− mouse. Top: Control mice (α+/+) express physiological levels of the p110α PI3K catalytic subunit in the adipose tissues and are lean. PI3K promotes transcription of the respiratory gene program to maintain normal respiration in the brown adipose tissue (BAT). BAT plays an important role in regulating systemic energy homeostasis so that energy intake (EI) approximately equals energy expenditure (EE). Excess free fatty acids (FFA) are taken up and stored by the white adipose tissue (WAT). The skeletal muscle relies heavily on glucose for energy and aids in glucose clearance from the circulation. Bottom: Mice with adipose-specific ablation of p110α (α−/−) have increased adiposity that affects BAT and WAT. Loss of p110α reduces transcription of genes critical for normal BAT respiration, so EE is lower than EI. It is likely that WAT initially copes with excess circulating FFAs, but chronic exposure finally overwhelms WAT storage capacity so that FFAs increase in the circulation or are taken up by the liver (spots indicate lipid). Insulin resistance in the liver and skeletal muscle, possibly induced by elevated FFAs or hyperglycemic adipokines secreted from WAT, increases hepatic glucose output and reduces glucose uptake by the muscle, further driving the α−/− mouse into metabolic disarray.
Reduced expression of adipose-specific p110α causes systemic insulin resistance
In addition to elevated fasting serum FFAs and increased lipid deposition in the α−/− liver, we also reported diminished insulin signaling in the liver and skeletal muscle of α−/− mice.Citation11 Impaired glucose tolerance and high fasting serum insulin levels were further evidence of systemic metabolic disorders caused by adipose-specific loss of p110α. These results strongly indicate that p110α signaling in the adipose tissues regulates systemic insulin sensitivity. As previously described, PI3K is important for glucose transport into the cell. It is likely that, in addition to decreased energetics in the brown adipocytes, loss of PI3K activity diminishes glucose uptake in the α−/− adipocytes and contributes to the observed systemic glucose intolerance. Adipose tissues also secrete several adipokines which can affect insulin sensitivity of peripheral tissues. It may be that loss of p110α in the adipose tissues increases secretion of hyperglycemic adipokines to promote insulin resistance ().Citation16 In addition, exposure of the peripheral tissues to FFAs can induce insulin resistance, and it is possible that inability of the adipose tissue to adequately take up FFAs from the circulation accentuates this effect.Citation17 Insulin resistance in the liver of α−/− mice would lead to increased hepatic glucose output, which would contribute to the systemic defect in glucose homeostasis ().
Age-dependent effect of PI3K on BAT function
We reported that the α−/− mice had increased body weight when compared to controls beginning at 6 weeks of age, and this difference gradually increased with age.Citation11 To determine if impaired BAT was a direct cause of obesity in the α−/− mice, we measured UCP-1 as a marker for BAT respiration. We did not see a difference in UCP-1 expression in BAT from α−/− vs. control mice at 3–4 weeks of age, before the divergence in body weight. By 8 weeks of age there was an obvious decrease in UCP-1 protein expression accompanied by an increase in adiposity of the α−/− mice (unpublished data).Citation11 These results support the notion that the primary driver of obesity in the α−/− mouse is impairment of BAT respiration.
Adipose p110α controls leptin levels
The adipose tissue is known to play an important role in modulating the hypothalamic-pituitary-gonadal (HPG) endocrine axis that controls puberty.Citation18 The normal function of the hypothalamus is dependent on normal energy balance. Obesity is clearly a state of disrupted energy homeostasis and is strongly associated with changes in sex hormone production and impaired reproductive health.Citation18 Obesity during childhood strongly affects male and female pubertal development and can also compromise normal glucose and lipid metabolism later in life.Citation19-21
Input by the adipose tissue to the reproductive axis is thought to be mediated largely by adipose-derived hormones such as leptin.Citation18 Adipose tissue also expresses the enzyme aromatase, which converts testosterone to estrogen. Leptin is thought to be a ‘permissive’ factor in puberty onset, and loss of leptin signaling inhibits pubertal development and promotes obesity.Citation22 The leptin-deficient ob/ob mouse develops severe reproductive defects including delayed puberty onset, attenuated testosterone, hypertrophied testes and infertility, all of which are corrected with exogenous leptin administration.Citation23 Our adult α−/− mice exhibited elevated serum leptin levels but no decrease in food intake, suggesting that they are leptin-resistant.Citation11 Furthermore, we found that puberty was delayed in male α−/− mice (unpublished data). Leptin is generally thought to be produced in direct proportion to fat mass. Surprisingly, we found that pubescent α−/− mice have elevated leptin levels as compared to their wildtype littermates and are not yet obese (unpublished data). This raises the possibility that high leptin in the α−/− mouse is not a result of increased adiposity, but rather is an independent phenotype due directly to the loss of p110α in the WAT. The roles of adipose p110α in regulating leptin production and reproductive development should be further studied in the α−/− mouse model.
Summary
Obesity is increasing across the world in epidemic proportions, yet a unifying mechanism for how people become and remain obese despite behavioral interventions remains elusive. Increased adiposity alone cannot account for the development of diabetes and metabolic syndrome, as obese but otherwise healthy individuals certainly exist.Citation5 There is a strong link between childhood insulin resistance and obesity which persists throughout adulthood, and the α−/− mouse provides a relevant model to examine the driving mechanisms behind these age-dependent metabolic changes.Citation24 Our research suggests that obesity and glucose intolerance result, at least in part, from insulin resistance (i.e., reduced PI3K activity) in adipose tissue. The α−/− mouse is a powerful tool for examining the effects of adipose-specific insulin resistance on systemic metabolic regulation and may provide information that will lead to the development of new therapies for the treatment and prevention of obesity.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by a NIH grant DK62722 and a VA Merit Award BX002263.
References
- Breymaier S. AMA adopts new policies on second day of voting at annual meeting. Chicago, IL: American Medical Association; 2013. Available from: http://www.ama-assn.org/ama/pub/news/news/2013/2013-06-18-new-ama-policies-annual-meeting.page
- Wadden TA, Volger S, Sarwer DB, Vetter ML, Tsai AG, Berkowitz RI, Kumanyika S, Schmitz KH, Diewald LK, Barg R, et al. A two-year randomized trial of obesity treatment in primary care practice. N Engl J Med 2011; 365:1969-79; PMID:22082239; http://dx.doi.org/
- Haslam DW, James WPT. Obesity. Lancet 2005; 366:1197-209; PMID:16198769; http://dx.doi.org/10.1056/NEJMoa1109220
- Reaven GM. Pathophysiology of insulin resistance in human disease. Physiol Rev 1995; 75:473-86; PMID:7624391; http://dx.doi.org/10.1016/S0140-6736(05)67483-1
- Vistisen D, Witte DR, Tabák AG, Herder C, Brunner EJ, Kivimäki M, Færch K. Patterns of obesity development before the diagnosis of type 2 diabetes: the whitehall II cohort study. PLoS Med 2014; 11:e1001602; PMID:24523667; http://dx.doi.org/10.1371/journal.pmed.1001602
- Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med 2006; 119:S10-16; PMID:16563942; http://dx.doi.org/10.1016/j.amjmed.2006.01.009
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 2006; 125:733-47; PMID:16647110; http://dx.doi.org/10.1016/j.cell.2006.03.035
- Stahl A, Evans JG, Pattel S, Hirsch D, Lodish HF. Insulin causes fatty acid transport protein translocation and enhanced fatty acid uptake in adipocytes. Dev Cell 2002; 2:477-88; PMID:11970897; http://dx.doi.org/10.1016/S1534-5807(02)00143-0
- Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M. Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem 1994; 269:3568-73; PMID:8106400
- Valverde AM. Insulin-induced up-regulated uncoupling protein-1 expression is mediated by insulin receptor substrate 1 through the phosphatidylinositol 3-kinase/akt signaling pathway in fetal brown adipocytes. J Biol Chem 2003; 278:10221-31; PMID:12525499; http://dx.doi.org/10.1074/jbc.M209363200
- Nelson VLB, Jiang Y-P, Dickman KG, Ballou LM, Lin RZ. Adipose tissue insulin resistance due to loss of PI3K p110α leads to decreased energy expenditure and obesity. Am J Physiol Endocrinol Metab 2014; 306:E1205-16; PMID:24691033; http://dx.doi.org/10.1152/ajpendo.00625.2013
- Vijgen GHEJ, Bouvy ND, Teule GJJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD. Brown adipose tissue in morbidly obese subjects. PloS One 2011; 6:e17247; PMID:21390318; http://dx.doi.org/10.1371/journal.pone.0017247
- Lowell BB, S-Susulic V, Hamann A, Lawitts JA, Himms-Hagen J, Boyer BB, Kozak LP, Flier JS. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 1993; 366:740-2; PMID:8264795; http://dx.doi.org/10.1038/366740a0
- Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab 2009; 9:203-9; PMID:19187776; http://dx.doi.org/10.1016/j.cmet.2008.12.014
- Enerbäck S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 1997; 387:90-4; http://dx.doi.org/10.1038/387090a0
- Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006; 444:847-53; PMID:17167472; http://dx.doi.org/10.1038/nature05483
- Delarue J, Magnan C. Free fatty acids and insulin resistance. Curr Opin Clin Nutr Metab Care 2007; 10:142-8; PMID:17285001; http://dx.doi.org/10.1097/MCO.0b013e328042ba90
- Michalakis K, Mintziori G, Kaprara A, Tarlatzis BC, Goulis DG. The complex interaction between obesity, metabolic syndrome and reproductive axis: a narrative review. Metabolism 2013; 62:457-78; PMID:22999785; http://dx.doi.org/10.1016/j.metabol.2012.08.012
- Lee M-J, Fried SK. Integration of hormonal and nutrient signals that regulate leptin synthesis and secretion. AJP Endocrinol Metab 2009; 296:E1230-8; http://dx.doi.org/10.1152/ajpendo.90927.2008
- Frisch RE. Fatness, menarche, and female fertility. Perspect Biol Med 1985; 28:611-33; PMID:4034365
- Kelsey MM, Zaepfel A, Bjornstad P, Nadeau KJ. Age-related consequences of childhood obesity. Gerontology 2014; 60:222-8; PMID:24434909; http://dx.doi.org/10.1159/000356023
- Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 2002; 110:1093-103; PMID:12393845; http://dx.doi.org/10.1172/JCI0215693
- Donato J, Cravo RM, Frazão R, Elias CF. Hypothalamic sites of leptin action linking metabolism and reproduction. Neuroendocrinology 2011; 93:9-18; PMID:21099209; http://dx.doi.org/10.1159/000322472
- Steinberger J, Moorehead C, Katch V, Rocchini AP. Relationship between insulin resistance and abnormal lipid profile in obese adolescents. J Pediatr 1995; 126:690-5; PMID:7751990; http://dx.doi.org/10.1016/S0022-3476(95)70394-2