
Abstract
Aberrant STAT signaling is associated with the development and progression of many cancers and immune related diseases. Recent findings demonstrate that proteostasis modulators under clinical investigation for cancer therapy have a significant impact on STAT signaling, which may be critical for mediating their anti-cancer effects. Chaperones are critical for protein folding, stability and function and, thus, play an essential role in the maintenance of proteostasis. In this review we discuss the role of chaperones in STAT and tyrosine kinase (TK) protein folding, modulation of STAT and TK activity, and degradation of TKs. We highlight the important role of chaperones in STAT signaling, and how this knowledge has provided a framework for the development of new therapeutic avenues of targeting STAT signaling related pathologies.
Abbreviations
| Seventeen-AAG | = | 17-N-Allylamino-17-demethoxygeldanamycin |
| AD HIES | = | Autosomal Dominant Hyper-IgE Syndrome |
| DMAG | = | 17-allylamino-de-methoxy geldanamycin |
| EFGR | = | Epidermal Growth Factor Receptor |
| ERp57 | = | Endoplasmic reticulum protein 57 |
| ES cells | = | Embryonic stem cells |
| GA | = | Geldanamycin |
| H2O2 | = | Hydrogen peroxide |
| Hip | = | HSP70-interacting protein |
| Hop | = | HSP70-HSP90 organizing complex |
| HSC | = | Heat shock cognate |
| HSE | = | Heat shock element |
| Hsf1 | = | Heat shock factor |
| HSP | = | Heat shock protein |
| IL | = | Interleukin |
| IFN | = | Interferon |
| JAK | = | Janus Kinase |
| LPS | = | Lipopolysaccharide |
| mtHSP90 | = | Mitochondrial HSP90 |
| NAC | = | nascent polypeptide-associated complex |
| pY-Stat | = | phosphotyrosylated signal transducer and activator of transcription |
| RTK | = | receptor tyrosine kinases |
| SH2 domain | = | Src Homology 2 domain |
| SOCS | = | suppressor of cytokine signaling |
| STATs | = | signal transducers and activators of transcription |
| TK | = | tyrosine kinases |
| TOM/TIM | = | Translocase of the outer/ inner mitochondrial membrane |
| TRiC | = | tailless-complex polypeptide-1 ring complex |
| VSMC | = | Vascular smooth muscle cells |
| WT | = | Wild type |
Introduction
Chaperone proteins play a prominent role in the maintenance of proteostasis, or cellular protein homeostasis,Citation1-4 by modulating myriad cellular processes including protein synthesis, complex assembly, protein degradation, and protein trafficking.Citation5,6 Chaperones facilitate cellular adaptation in response to stress,Citation7 and also are actively involved in the modulation of major biological processes under normal conditions, including cell proliferation, differentiation and apoptosis.Citation8-10 Extensive data now reveal the foremost role chaperones play in modulating the cell signaling machinery,Citation11 including the de novo synthesis and folding of key cell signaling players, stabilization of the active conformations of these proteins, targeting and translocation of protein complexes into the nucleus, and disposing of used or misfolded proteins via proteasomal degradation.Citation10,11 In this review we highlight recent findings that contribute to our understanding of how chaperones impact cell-signaling pathways involving signal transducers and activators of transcription (STATs). This review is motivated by the flurry of recent investigations in this area and by studies demonstrating that the anticancer effects of drugs targeting chaperones, such as HSP90, are mediated, at least in part, through their impact on the STAT signaling pathway.
The STAT family of latent cytosolic transcription factors consists of 7 members (Stat1, Stat2, Stat3, Stat4, Stat5A, Stat5B, and Stat6).Citation12 Additional key components of the STAT signaling apparatus include: cytokines and growth factors, cell surface receptors, tyrosine kinases (TKs), receptor tyrosine kinases (RTKs), tyrosine phosphatases, and suppressor of cytokine signaling (SOCS) proteins. STAT proteins are located predominantly within the cytoplasm dimerized N-terminal-to-N-terminal (head-to-head) The canonical STAT signaling pathway is initiated when cytokines or growth factors bind the extracellular portion of specific cell surface receptors inducing receptor dimerization or oligomerization, resulting in juxtapositioning and activation of receptor-associated (JAK and Src) or receptor-intrinsic tyrosine kinases through kinase-mediated transphosphorylation. Dimerized STAT proteins are then recruited to activated receptor complexes via their Src homology 2 (SH2) domain and are phosphorylated at specific tyrosine residues by the activated kinase. The phosphotyrosylated (pY-) STATs undergo a conformational change, dimerizing C-terminal to C-terminal (tail-to-tail). Activated STAT dimers accumulate in the nucleus where they bind to promoters and modulate expression of genes that influence a wide variety of cellular processes.Citation13
As components of the STAT signaling apparatus are often large, multi-domain proteins with complex topologies, and as STAT signal transduction depends on sophisticated protein structural rearrangements,Citation12 it is not surprising that chaperones play a major role in STAT signaling. It is clear from the findings reviewed here that chaperones greatly influence the maturation and activation of STATs, TKs, and RTKs, and that further explorations of this area will yield new information useful for modulating chaperone activity in a variety of disease states.
STAT Proteins and Chaperones
Overview
STAT proteins are highly dependent on elaborate cellular machinery composed of chaperones for biogenesis and optimal activity, as depicted in . Here we review findings that reveal that STATs require interaction with the eukaryotic chaperonin protein, tailless-complex polypeptide-1 (TCP-1) ring complex (TRiC; also know as chaperonin containing TCP-1, CCT) for de novo folding. Multiple additional chaperones play a role in optimizing STAT protein activity, especially heat shock protein (HSP) 90. There are many exciting potential clinical applications of manipulating STAT-HSP90 interaction in the development of improved treatment approaches for human diseases such as cancer and atherosclerosis. Of all the STAT proteins, Stat3 interactions with chaperone proteins are most well characterized, and these interactions are summarized in .
Figure 1. STAT proteins interact with multiple chaperones to achieve nascent folding, activation, nuclear (and possibly mitochondrial) translocation, DNA binding, and possibly degradation.
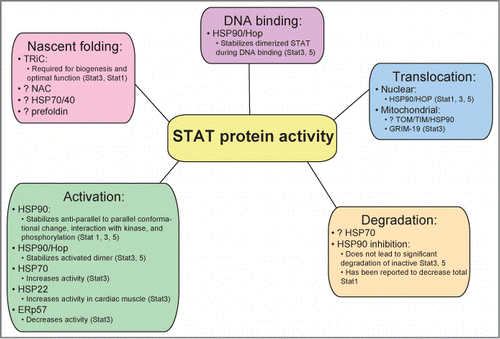
Figure 2. Interactions of chaperone proteins with Stat3 panel (A) and Jak2 panel (B). Stat3 requires interaction with TRiC for optimal biogenesis (portion 1). As most TRiC clients also interact with nascent polypeptide-associated complex (NAC), HSP70/HSP40, and prefoldin, we hypothesize that these chaperones are also involved in de novo Stat3 folding. Inactive Stat3 is found in large complexes in the cytoplasm containing chaperone proteins HSP90 and ERp57 (portion 2). Stat3 is recruited to activated cytokine receptors, and HSP90 is required for Stat3 interaction with JAK kinases, Stat3 phosphorylation, and dimerization of activated Stat3 (portion 3). It is thought that HSP90 stabilizes activated Stat3, making Stat3 phosphorylation and tail-to-tail dimerization more favorable. HSP90/Hop is required for Stat3 nuclear translocation (portion 4). HSP90/Hop co-localizes with Stat3, stabilizing Stat3 while it is bound to DNA (portion 5). Although data are limited, others have hypothesized that mtHSP90 is involved, in conjunction with TOM/TIM, with Stat3 mitochondrial translocation (portion 6). ERp57 activity within the ER decreases Stat3 activity in the cytoplasm (portion 7). The activity of other chaperone proteins, such as HSP70 and HSP22, is associated with increased Stat3 activity (not pictured). In panel B, tyrosine kinases are classic HSP90 clients that require interaction with HSP90 for de novo folding and maturation as well as activation. HSP70 plays an important role in the degradation of tyrosine kinases, which can occur at any stage of tyrosine kinase maturation or after activation. In the presence of HSP90 inhibitors tyrosine kinases are preferentially degraded.
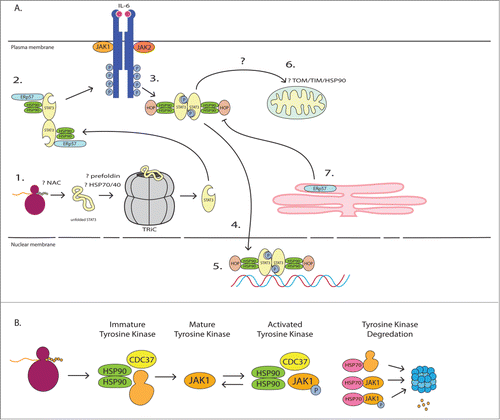
STAT Proteins and TRiC
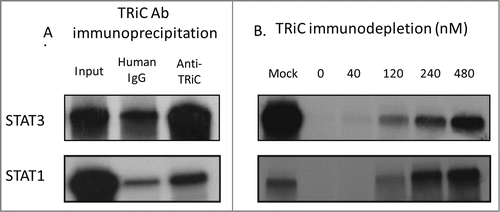
Chaperonins are a structurally conserved class of oligomeric, double-ring, high molecular weight, ATP-dependent chaperones with the unique ability to fold proteins that cannot be folded by simpler chaperone systems.Citation5 TRiC/CCT is a 1 MDa complex composed of 8 homologous subunits arranged in 2 octameric rings, forming a cage that facilitates protein folding.Citation14,15 TRiC is not induced by stress but instead participates in the de novo folding of > 10% of cytosolic proteins.Citation14 Our lab has recently shown that Stat3 is a TRiC substrate, and that Stat3 requires interaction with TRiC for biogenesis and optimal function within human cell lines.Citation16 Furthermore, manipulating the interaction between Stat3 and TRiC affects Stat3 activity: knock down of TRiC using shRNA reduces Stat3 sensitivity to IL-6-mediated activation, while increasing Stat3 affinity for TRiC through the addition of an extra TRiC binding domain improves in vitro Stat3 activity. In addition to Stat3, we have demonstrated that Stat1 requires interaction with TRiC for biogenesis ().
Figure 3. TRiC binds Stat1/Stat3 co-translationally and is required for Stat1/Stat3 synthesis in RRLs. In panel (A), TRiC was immunoprecipitated from rabbit reticulocyte lysate with a combination of antibodies to CCT2 and CCT5 (Anti-TRiC) or with a nonspecific control antibody (Human IgG) following translation of the indicated proteins in the presence ofCitation35S-methionine. Immunoprecipitates were separated by SDS-PAGE and autoradiographed. Half of each IP reaction prior to precipitation was run separately on SDS-PAGE and autoradiographed (Input). In panel (B), Stat1/Stat3 was translated in TRiC-depleted RRLs or following the addition of purified bovine TRiC in increasing amounts in the presence ofCitation35S-methionine followed by SDS-PAGE and autoradiography. The results shown are representative of 3 experiments.
TRiC client proteins usually require interaction with ribosome-associated chaperones for stabilization during translation and for facilitation of TRiC binding. The nascent polypeptide-associated complex (NAC) is assumed to aid in the co-translational folding of most peptides.Citation17,18 The HSP70/40 complex and prefoldin are cytosolic chaperones that act on newly synthesized proteins, aiding de novo protein folding and targeting newly synthesized substrates to the TRiC complex.Citation17,19 As we have shown that Stat1/3 are TRiC substrates, it is possible that Stat1/3 also interact with NAC, HSP70/40 and prefoldin during de novo folding.
STAT Proteins and HSP90
HSP90 is a highly conserved molecular chaperone that serves as a proteostasis buffer under environmentally stressful conditions and facilitates the maturation and activation of > 100 client proteins, many of which are critical for cell signaling and adaptive responses to stress. HSP90 exists as 2 isoforms in the cytosol; HSP90α is induced by stress and is commonly upregulated in cancer cells while HSP90β is expressed constitutively at high concentrations in most tissues.Citation20 HSP90 utilizes energy garnered from ATP hydrolysis and interacts with more than 20 co-chaperones—HSP70, HSP40, HSP70-interacting protein (Hip), HSP70-HSP90 organizing complex (Hop), CDC37, p23, and immunophilins—to help client proteins fold.Citation21-24
Physical association between HSP90 and Stat1/3 was initially reported in plasma membrane cholesterol-rich rafts [with caveolin-1, cytokine receptors (gp130), JAK1/JAK2, and Tyk2] and cytosolic complexes (with ERp57).Citation25 The DNA binding domain of Stat3 was shown, using domain deletion mutations and immunoprecipitation studies, to directly interact with the N-terminal ATPase domain of HSP90, and that HSP90 inhibitors, like geldanamycin (GA), inhibit this interaction by binding to the same HSP90 domain.Citation26,27 More recently, surface plasmon resonance spectroscopy showed that the high-affinity, specific interaction between the N-terminal domain of HSP90β and Stat3 requires a functional Stat3 DNA binding domain, and that this interaction has no preference for inactive vs. activated Stat3.Citation27 Domain deletion mutations and immunoprecipitation studies localized the Stat5 HSP90 binding domain to the C-terminal half of the coiled-coil domain.Citation28
Results using HSP90 inhibitors strongly suggest that STAT-HSP90 interactions contribute to optimal STAT activity, and that this contribution becomes essential when cells undergo increased stress. For example, treatment of Hep3B cells with GA results in reduced interleukin (IL)-6-induced-Stat3 signaling at 37°C and markedly decreased signaling when the cells are stressed at 39.5°C.Citation25 Overexpression of HSP90 in cells treated with GA restores IL-6-induced-Stat3 activity to normal levels.Citation26 Similarly, GA blocks the activation and phosphorylation of IL-2-induced Stat5 in an IL-2 dependent T cell line.Citation28
STAT proteins are not considered traditional HSP90 clients in that they do not require HSP90 for de novo folding; multiple studies show that HSP90 inhibitors decrease STAT activity through reduction of pY-STAT levels with minimal to no effect on total STAT protein levels.Citation29-32 Moulick et al. recently proposed that HSP90 promotes optimal STAT activity by serving as a scaffolding molecule, stabilizing STAT proteins in their active, phosphotyrosylated configuration.Citation33 In human chronic myelogenous leukemia cells treated with novel HSP90 inhibitor PU-H71, pY-Stat5 activity was reduced shortly after HSP90 inhibition, and this reduction could not be attributed to a change in kinase or phosphatase activity. Furthermore, binding to HSP90 appeared to change the conformation of Stat5, as Stat5 was more susceptible to trypsin cleavage when bound to HSP90. To investigate whether HSP90 binding results in a more favorable Stat5 configuration for phosphorylation and activation, the authors used a pulse-chase experiment after cells were treated with the phosphatase inhibitor, orthovanadate. Cells exposed to the HSP90 inhibitor demonstrated rapid decrease in pY-Stat5 concentration compared with unexposed cells, suggesting that Stat5 is more likely to remain phosphorylated when bound to HSP90.Citation33
Evidence also suggests that HSP90, along with the co-chaperone Hop, remains bound to dimerized pY-STATs, which targets and facilitates their translocation into the nucleus and stabilizes their interaction with DNA. Stat3-HSP90 interaction was shown to be essential for leukemia inhibitory factor (LIF)-induced, Stat3-mediated self-renewal of mouse embryonic stem (ES) cells.Citation34,35 Hop co-precipitates with HSP90 and Stat3 in ES cells, and Hop knockdown decreases pY-Stat3 nuclear translocation.Citation36 Using these observations, Setati et al. initially suggested that HSP90/Hop interacts with Stat3 at activated cytokine receptors, facilitating pY-Stat3 dimerization, nuclear targeting, nuclear translocation, and DNA binding.Citation34 Howard et al. also reported that Stat1 and HSP90 are both recruited to the plasma membrane in alveolar macrophage cells after stimulation with interferon (IFN)-γ, and that inhibition of HSP90 with 17-N-Allylamino-17-demethoxygeldanamycin (17-AAG) resulted in inhibition of Stat1 phosphorylation and DNA binding.Citation37 Finally, HSP90 has been shown to support Stat5-mediated transcription in that HSP90 inhibition led to decreased Stat5 DNA binding; quantitative chromatin immunoprecipitation assays demonstrated that HSP90 and Stat5 co-localize at critical Stat5 target promoters.Citation33
To summarize available data thus far, HSP90 clearly influences STAT protein activity through direct interaction; through this interaction, HSP90 stabilizes STAT dimers into a configuration that is more favorable for phosphorylation and activation. HSP90 (with co-chaperone Hop) then translocates with activated STAT dimers into the nucleus where it continues to stabilize STAT dimers bound to DNA.
Stat3/5 are constitutively upregulated in many cancers, and multiple studies in human cancer cell lines and animal models have demonstrated that HSP90 inhibition leads to decreased Stat3/5 signaling, thus highlighting the potential promise of this treatment strategy. HSP90 inhibitor STA-1474 potently decreased constitutive Stat3 phosphorylation in osteosarcoma cell lines with minimal effect on total and pY-Stat3 in nonmalignant canine osteoblasts.Citation29 Multiple myeloma cells treated with HSP90 inhibitors had decreased pY-Stat3 and Stat3 activityCitation30 and chronic lymphocytic leukemia cells treated with NVP-AUY922 and fludarabine had decreased Stat3 activity and increased apoptosis even when cultured on CD40L-stromal layer mimicking the supportive environment of the bone marrow.Citation38 Leukemia cells (with mutated FLT3 kinase) treated with 17-AAG had decreased Stat5 activity with increased cell cycle arrest and apoptosis.Citation39 Human pancreatic cancer cells treated with 17-AAG were found to have decreased Stat3/5 activation and signaling, as well as disruption of a novel IL-6/Stat3/HIF-α-mediated autocrine activation loop required to facilitate transcription of targets such as VEGF.Citation40 A subcutaneous xenograft tumor model and an orthotopic tumor model of pancreatic cancer cells both demonstrated potent reduction of tumor growth and tumor vascularization after treatment with HSP90 inhibitor 17-allylamino-de-methoxy geldanamycin (DMAG), and these effects were mediated by a reduction of IGF-IR and Stat3 signaling.Citation40 A recent review by Kim et al. summarizes the development of HSP90 inhibitors for clinical use in selectively inhibiting tumor growth.Citation41
HSP90 inhibitors are also being studied to block STAT signaling in other disease processes such as atherosclerosis. The use of 17-DMAG in hyperlipidemic ApoE−/− mice decreased the size of atherosclerotic lesions and decreased lipid and macrophage content of lesions, correlating with decreased pY-Stat3 and NF-κB signaling. The authors concluded that HSP90 inhibitors reduce inflammation in atherosclerosis.Citation42
As discussed above, Stat1/3/5 have been shown to require HSP90/Hop for nuclear translocation. These STAT proteins also are found in the mitochondria. Although this finding remains controversial, Stat3 has been shown to reduce oxidative stress and influence the electron transport chain,Citation43 leading to the question of whether Stat3 and/or other STAT proteins require chaperoning for mitochondrial translocation.Citation44 Mitochondrial HSP90 (mtHSP90), mtHSP60, and mtHSP70 are involved in the transport of multiple mitochondrial transcription factors such as p53, IRF3, CREB, NF-κB, and MEF2D, and some authors have hypothesized that mitochondrial chaperone proteins TOM/TIM (translocase of the outer/inner mitochondrial membrane) work with mtHSP90 to aid Stat3 entry into the mitochondria.Citation45 Although data addressing this hypothesis is limited, Stat3 does co-immunoprecipitate with TOM20 in rat left ventricular protein extracts.Citation46 Other authors have reported that GRIM-19 (gene associated with retinoid-interferon-induced mortality-19) is involved in the transport of Stat3 into the mitochondria.Citation47,48
STAT Proteins and Other Chaperones
HSP70
The human HSP70 family includes at least 12 proteins, including stress-induced HSP70 (HSP70–1A and HSP70–1B), constitutively expressed heat shock cognate (HSC) protein 70, glucose-regulated protein (GRP) 78 (Bip, primarily located in the ER), and mitochondrial HSP75 (mtHSP70).Citation49 The HSP70 family contributes to multiple biochemical functions in the cell, including nascent protein folding, protein homeostasis, protein translocation into the nucleus, ER and mitochondria, and trafficking of misfolded proteins to the ubiquitin/ proteasome machinery for degradation. Both HSC70 and HSP70 activity require interaction with multiple co-chaperones, such as J-domain protein HSP40, BAG family, Hip, Hop, HSPBP1, and CHIP (carboxyl-terminus of HSC70 interacting protein).Citation49
HSP70 has been shown through immunoprecipitation to directly interact with multiple STAT proteins, including Stat1/3/5B in normal rat kidney interstitial fibroblast (NRK-49F) cells; this interaction was increased when cells were stressed by exposure to advanced glycation end-product.Citation50 Although data are limited, a few reports suggest that manipulating HSC70/HSP70 activity impacts STAT protein activity within cells. Knock down of HSP70 (HSP70–1A and HSP70–1B) using siRNA further decreased constitutive Stat3 activity in an acute myeloid leukemia cell line (HEL) treated with arsenic trioxide and 17-DMAG.Citation51 Also, increasing HSP70 activity with geranylgeranylacetone in a rat model of intracerebral hemorrhage resulted in increased Stat3 phosphorylation, decreased neuronal apoptosis, and improved functional recovery.Citation52
HSP22
HSP22 is only expressed in heart and skeletal muscle, and is upregulated during cardiac stress. In an HSP22 knockout mouse model, there was decreased Stat3 signaling in response to overload stress, as well as decreased Stat3 levels and activity within mitochondria.Citation53 These findings support the hypothesis that HSP22 is critical for nuclear and mitochondrial Stat3 activity in cardiomyocytes. Preemptive overexpression of HSP22 also protected swine myocardium from post-ischemic or H2O2-mediated apoptosis through increased STAT and NF-κB signaling.Citation54
ERp57
The endoplasmic reticulum (ER) is involved in the response to cell stress and contains multiple chaperone proteins that are generally thought to perform quality control of newly synthesized glycoproteins and regulation of the Unfolded Protein Response (UPR). ERp57 (or glucose-regulated protein 58, Grp58) is a thiol disulfide oxidoreductase and chaperone protein that is found in the ER and predominantly acts as part of a complex (with calnexin and calreticulin) involved in the quality control of the secretory pathway. ERp57 is also found in the cytoplasm, and has been shown to interact with Stat3 within plasma membrane rafts and within the cytoplasm.Citation55-60 Stat3 and ERp57 have also been found to be associated in the nucleus bound to DNA in M14 melanoma cells and IL-6-stimulated HepG2 hepatoma cells.Citation61 The effect of Stat3-ERp57 interaction on Stat3 activity is controversial. When recombinant ERp57 was added in excess there was decreased pY-Stat3 in S100 cytosol, and when ERp57 was overexpressed in hepatoma Hep3B cell lines, both constitutive and IL-6-induced Stat3 activity was reduced.Citation60 The authors hypothesized that ERp57 may function to sequester activated pY-Stat3 in cytokine-stimulated cells leading to decreased Stat3 activity. However, anti-ERp57 antibodies decreased activated Stat3 DNA binding in melanoma and HepG2 cell lysates stimulated with IL-6, suggesting that ERp57 may be a critical component of the DNA-bound Stat3 complex.Citation61 Finally, ERp57-deficient cells have significantly increased Stat3 activity. This increased activity was restored to normal by expression of the ERp57 within the ER but not within the cytoplasm ERp57; ERp57-dependent modulation of Stat3 activity was further enhanced by interactions in the lumen of the ER between ERp57 and calreticulin.Citation62
Tyrosine Kinases and Chaperones
Overview
Unlike STAT proteins, kinases are considered classic HSP90 clients, requiring interaction with HSP90 and co-chaperones for de novo folding to achieve maturation i.e. their native folded state. Additionally HSP90 also has been shown to modulate kinase activity post-maturation i.e., to facilitate mature kinase conformational transitions between active and inactive forms, which are coupled to the HSP90 ATPase cycle.Citation63,64 In addition, others have suggested that HSP90 regulates mature kinase activity by sequestration, a process by which mature kinases in active or metastable conformation are rendered inactive when bound to the chaperone and released from the chaperone complex only when the kinase activity is needed.Citation65,66 Since HSP90 has a diverse group of client proteins, it depends on multiple co-chaperones to recruit specific substrates. One such polypeptide is the kinase specific adaptor CDC37, which is known to play a major role in modulating kinase function. Tyrosine kinase interactions with chaperone proteins are summarized in . Here, we will focus our discussion primarily on kinases involved in STAT signaling.
Src Kinases and Chaperones
Src kinases constitute a family of 8 members including Src, Yes, Fyn, and Fgr (that form the SrcA subfamily), Lck, Hck, Blk, and Lyn (that form the SrcB subfamily) and Frk. Src-family protein kinases are involved in a wide variety of cellular activities including cell differentiation, motility, proliferation, and survival.Citation67 Although some of the Src kinases also play a prominent role in non-STAT signaling networks, there is an abundance of biochemical evidence showing that most Src kinases are capable of specifically activating STATs, especially Stat3 and to some extent Stat5.Citation68-72
The first oncogene discovered, v-Src, is one of the earliest HSP90 kinase substrates described. In an attempt to pinpoint the inhibitory mechanism behind benzoquinone ansamycins toward tyrosine kinase induced oncogenic transformations, Whitesell et. al. discovered that v-Src was a client of HSP90. These studies demonstrated that the antibiotic geldanamycin (GA) was a pharmacological inhibitor of HSP90 chaperone, thereby identifying the basis for GA's anti-oncogenic activity.Citation73,74 It is now well established that the oncogenic transformation ability of v-Src is due to its unrestrained tyrosine kinase catalytic activity toward multiple substrates, including Stat3/5, which is highly dependent on HSP90.Citation75-77 The proto-oncogene counterpart of v-Src, c-Src, also was shown to depend on HSP90 for activity. Interestingly, when compared to its oncogenic mutant counterpart, c-Src kinase showed markedly weaker binding and less sensitivity to HSP90 inhibition,Citation78 suggesting that each has a different requirement for HSP90 chaperone activity. In addition, Koga et al. showed that HSP90 binds to c-Src, and sequesters it in an intermediate state between active and inactive conformations; in contrast to v-Src, HSP90 inhibition was accompanied by activation of c-Src and its downstream targets after dissociation from HSP90. Because of the potential toxicity that might arise from HSP90 inhibition in normal tissues, the benefit of targeting HSP90 activity for therapeutic purposes may not be straightforward. Thus, this approach warrants further studies.Citation65,79
HSP90 and CDC37 also modulate the activity of other Src kinase family members such as Hck, Lck, Fyn, and Lyn. GA treatment of macrophages abrogates lipopolysaccharide (LPS)-induced macrophage cell adhesion and results in a concomitant decrease in the concentration of Hck, and to lesser extent Fyn and Lyn, all well established effector molecules downstream of LPS.Citation80,81 Similar observations have been made with Lck.Citation82 Additionally, the specific catalytic activity of the kinases is markedly diminished in the presence of HSP90 inhibitors, implying that in the absence of chaperone activity, the kinases do not achieve their native functional configuration. To further examine the role of HSP90 pre- and post-maturation, pulse chase experiments were performed on wild type (WT) kinases, demonstrating that mainly nascent kinases are prone to degradation (short half-life) in the presence of HSP90 inhibitors, while the mature kinases exhibit a longer half-life.Citation80 These results suggest that WT kinases rely on HSP90 primarily for maturation.
Interestingly, however, the impact of HSP90 activity on kinase function after maturation appears to be conditional. This has been demonstrated through stability assays. Constitutively active mutants of Hck and Fyn remain dependent on HSP90 for stability post-maturation, whereas WT kinases do not.Citation80,83 This observation suggests that either the mutations are destabilizing and require increased HSP90 activity, or that the constitutively active (open) kinase conformation may be inherently unstable, thus more dependent on chaperone activity for stability than the inactive kinase (closed).Citation80 If the latter is true, it implies that WT kinases may require HSP90 transiently during activation (while in the open conformation). Therefore, it is possible that the role of chaperones does not stop at nascent kinase folding, as under certain circumstances HSP90 remains actively engaged to maintain stability and modulate kinase activity post-maturation.
HPS90 is intimately associated with its co-chaperones, they represent the major way in which HSP90 activity is modulated within the cell and have been shown to target specific substrates to HSP90. Although there are over 20 HSP90 co-chaperones known to date, CDC37 stands out as the predominant co-chaperone in kinase biology. Indeed, disruption of CDC37/HSP90 association by siRNA depletion or inhibition of its activator, CK2 kinase, in human multiple myeloma cells, results in the degradation of client kinases, including c-Src, thereby decreasing levels of constitutively active Stat3 and other downstream effectors.Citation84 Interestingly, some HSP90 client kinases have been reported to phosphorylate key sites on the chaperone, implying a link between HSP90 regulatory mechanism and the kinase activity of its substrates.Citation85,86 These studies showed that kinases such as pp60 v-Src and c-Src phosphorylate HSP90 at specific sites affecting its activity and substrate specificity.Citation87,88 How this impacts downstream STAT signaling is not clear, however, a recent study indicates that phosphorylation of the C-terminal end of HSP70 and HSP90 results in the preferential binding to co-chaperone HOP; this then leads to increased protein folding activity of HSP90. The same study showed an association between phosphorylation of HSP90 and increased proliferation rates in neoplastic cells.Citation89
JAK Kinases and Chaperones
JAK kinases make up the predominant non-receptor kinases involved in STAT activation. There are 4 known JAKs—Jak1, Jak2, Jak3 and Tyk2.Citation90 The available sequence and structural information, show that JAK kinases bear a structural organization similar to other kinases.Citation91 Thus, it is not surprising that HSP90 is also essential for JAK kinase folding and maturation. Indeed, the cellular protein levels of all 4 JAK kinases are markedly reduced when cells are exposed to HSP90 inhibitors, suggesting that JAK kinases are dependent on HSP90 for de novo folding and maturation. This, as expected, is accompanied by the loss of STAT phosphorylation, especially Stat1/3/5/6.Citation92
The involvement of HSP90 in the regulation of normal JAK kinase activity was suggested by findings that HSP90 chaperone is necessary for IFN I and II signaling. IFN-γ, is a key player in both innate and adaptive immunity. It has been established that the canonical IFN-γ- signaling occurs through JAK/STAT pathway. Recent studies indicate that HSP90/CDC37 protein complex is a molecular chaperone for Jak1/2 and is important to the normal immune response. Co-immunoprecipitation experiments reveal a physical association between JAK kinases and the CDC37/HSP90 complex that is disrupted when cells are treated with HSP90 inhibitors. Additionally, studies examining IFN-γ-induced T-cell activation, showed that chemical inhibition of HSP90 or siRNA knockdown of HSP90 and CDC37 results in depletion of Jak1/2; which accordingly led to diminished pY-Stat1, decreased expression of IFN-γ-inducible genes and consequently, impaired T cell activation, suggesting that JAK kinases are bona fide HSP90 clients.Citation93
As mentioned above, it is clear that HSP90 is essential for maintaining the proper functioning of JAK signaling, thereby enabling appropriate responses to cytokine stimuli. However, when overexpressed in certain cell lines, HSP90 has been shown to result in JAK kinase dysregulation, thereby leading to inappropriate JAK/STAT signaling. For instance, in classical Hodgkin's lymphoma, HSP90 is overexpressed and thought to support an oncogenic signaling network in which Stat3, Stat5 and Stat6 are constitutively activated. Inhibition of HSP90 leads to JAK kinase depletion, reduced phosphorylation of Stat3/5/6 and proliferation of cHL cells.Citation94
Another way in which HSP90 contributes to aberrant JAK/STAT signaling, is by stabilizing constitutively active oncogenic mutations. It has recently been demonstrated that mutation V617F in Jak2 kinase is the genetic cause for several myeloproliferative disorders.Citation95-97 This mutation has been shown to abolish JAK kinase autoinhibition resulting in a constitutively active kinase that, in turn, constitutively actives Stat3/5, leading to transformation and proliferation of these cells. As has been observed in other kinases, mutant and aberrantly activated JAK kinases found in association with malignant phenotypes, also appear to be highly dependent on HSP90 for stability. Pharmacological inhibition of HSP90 in cells containing Jak2 oncogenic mutations results in Jak2 degradation, diminished constitutive Stat5 activation, and reduced proliferation.Citation98,99 Although both wild type and mutant JAK kinases to some extend depend on HSP90 chaperone for stability, data on Jak2 V617F seems to mirror observations made on other kinases showing that constitutively active mutant kinases are more dependent on HSP90 than their WT counterparts, a difference that could be potentially exploited for therapeutic benefit.Citation100 In addition to V617F, other interesting Jak2 mutations such as E864K, Y913C and G935R, confer resistance to JAK kinase inhibitors; however, when cells harboring the resistance mutations are exposed to both HSP90 and kinase inhibitors, the resistance is overcome. Indeed, in the same studies, cytotoxicity assays show that the cells having the resistant mutations were more sensitive than WT cells to HSP90 inhibition using AUY22.Citation101 Similarly, inhibition of HSP90 activity has also been shown to be effective in cases where cells are able to overcome Jak2 inhibition and sustain aberrant JAK/STAT signaling by co-opting other kinases to trans-activate Jak2.Citation102 These examples, while not exhaustive, highlight the importance of HSP90 in the development and maintenance of many pathological states mediated by aberrant JAK/STAT signaling, including both solid and hematologic malignancies,Citation99 along with the therapeutic potential of pharmacological inhibition HSP90 as potential treatment for such malignancies.
Receptor Tyrosine Kinase and Chaperones
As might be expected, receptor tyrosine kinases (RTK) pose protein-folding challenges for cells, because, in addition to the proper folding and maturation of the chaperone-dependent kinase domain, they also have to be targeted to the membrane. There is abundant evidence showing that molecular chaperones play an important role in the folding and maturation of many membrane proteins, particularly RTK, in addition to facilitating degradation and modulating the activities of mature receptors.Citation103 Biochemical and genetic studies in mammalian cells have provided great insight into the role of HSP90 and HSP70 in the biogenesis and maturation of RTK. The use of HSP90-specific inhibitors as chemical probes has contributed immensely to the identification of the majority of RTK that are HSP90 clients. Several RTK that are involved in the STAT signaling network,Citation103-106 including EGFR, MET, PDGF, IGF-IR and FGFR,Citation40,107,108 have been identified as HSP90 clients.
Studies of the ErbB family of RTK especially highlight the critical role of chaperones in the modulation of STAT signaling. The epidermal growth factor receptor (EGFR) is the most studied of these perhaps because of its prominent role in many human cancers. EGFR and other ErbB family members have been found in association with a handful of molecular chaperones and co-chaperones, most prominently HSP90 and CDC37. Like the non-receptor kinases discussed above, it appears that all studied RTK, including the ErbB family, are dependent on HSP90 at the nascent stage.Citation109 However, ErbB2 and the majority of the active mutants in the ErbB family appear to remain engaged and require chaperones throughout their lifespan, including nascent protein folding, trafficking, modulating activity of the mature receptor, and assisting in the degradation process.Citation110,111The contribution of chaperones to EGFR/ErbB biology has been excellently reviewed elsewhere.Citation112 Consequently, we will only highlight a few key studies demonstrating the direct effects of inhibiting HSP90 on STAT signaling. For example, when HSP90 activity in cells is inhibited with the HSP90 inhibitor, SNX-2112, both WT and mutant EGFR fail to fold properly and are in turn degraded, thereby preventing EGF activation of pY-Stat3.Citation113 Certain mutations on exon 20 of ErbB2 and EGFR are of great clinical interest because they result in constitutive activation of the receptors and confer resistance to EGFR tyrosine kinase inhibitors. Analysis of these mutants shows a strong dependence on HSP90, and, as expected, the mutants were not sensitive to the kinase inhibitor gefitinib. However treatment with 17-AAG alone, and in combination with gefitinib, abolished the unrestrained EGFR activity and persistent phosphorylation of its downstream signal transducers Stat3 and Akt.Citation114 These studies again emphasize the therapeutic potential of HSP90 inhibitors in the treatment of aggressive malignancies.
Recent studies reveal that molecular chaperones also exert their activity extracellularly. HSP90/CDC37 and HSP70 and its co-chaperones have been found in extracellular complexes with cell surface receptors including EGFR, HER2 and matrix metalloproteinase 2. Extracellular HSP90 and HSP70 are thought to be involved in polypeptide complex assembly and in facilitating cell invasion signaling pathways. HSP90 has been shown to be essential for HRG-induced HER-2 activation and its downstream signaling events that lead to cytoskeletal rearrangements necessary for cell invasion. Thus, inhibition of extracellular chaperone activity results in diminished cancer cell migration and invasion.Citation115,116,117 In addition, recent work looking at the mechanism of heat stress-induced transactivation of EGFR suggests a role for secreted HSP70 in the initial stages of heat shock-mediated signaling. The ligand independent activation of EGFR and subsequent downstream activation of Stat3 observed during the initial stages of heat stress response in a squamous carcinoma cell line was shown to be mediated by extracellular HSP70.Citation118
Contribution of Chaperones to Degradation of STAT Pathway Proteins
Part of the therapeutic rationale behind HSP90 inhibition is based on observations that molecular chaperones collaborate with the ubiquitin/proteosome system during polypeptide triageCitation119 such that when cells are treated with HSP90 inhibitors, traditional HSP90 client proteins, including kinases, are shuttled to the proteasome for degradation. Several studies have implicated HSP70 as having a major role in this degradation process,Citation120,121 likely serving as a sensor for stressed proteins and determining the fate of the proteins.Citation122 The emerging view is that, depending on the circumstances, HSP90 and HSP70 are involved in either the stabilization or degradation of client polypeptides. Exactly how the decision to stabilize versus degrade proteins is reached is not clear, but evidence from several labs has begun to provide insight into a possible mechanism. For example, it has been observed that GA treatment of cells induces a change in the composition of cofactors in the HSP90/HSP70 complex to include polypeptides with E3 ligase activity. This, in turn, results in the ubiquitination of client proteins within the chaperon complex.Citation119,123 Indeed, with regards to v-Src, An et al. elegantly demonstrated that the composition of the complex of HSP90 and cofactors changes from Hsp90/p23/p50 to Hsp90/Hsp70/Hop in the presence of GA, which results in increased proteosomal degradation of v-Src.Citation124 Others have reported similar dynamics involving other chaperone co-factors in conjunction with HSP70.Citation125 It is also clear that although HSP90 is the predominant chaperone involved in the modulation of kinase activity, other chaperones such as HSP70 and co-chaperones/co-factors p23, p50, Hop, CHIP and HSP40 also play an important supporting role.
Results are not as straightforward in defining the role of chaperones in STAT protein degradation. As stated above, STAT proteins are not classic HSP90 clients, in that they do not require HSP90 for de novo folding or maturation. Multiple studies with HSP90 inhibitors demonstrate a reduction of pY-Stat3/5 with minimal to no effect on total Stat3.Citation29-32 Howard et al. did show that inhibition of HSP90 with 17-AAG resulted in inhibition of Stat1 phosphorylation which lead to Stat1 insolubility and ultimately degradation via the proteasome, but the role of HSP70 or other chaperones in this degradation process was not explored.Citation37 As discussed above regarding the kinases, although HSP90 is the predominant chaperone involved in the modulation of STAT protein activity, HSP70 activity has been associated with increased STAT protein activity – especially when cells are undergoing increased stress. Further studies are needed to clarify the potential role that chaperones play in the degradation of STAT proteins.
STAT Proteins and Regulation of Chaperones
While the focus of this review is to highlight the dependence of STAT signaling pathways on chaperone proteins, we should acknowledge that it is also well recognized that STAT signaling pathways play an important role in the induction of HSP expression and activity as well. It is clear from the reports summarized below that STAT signaling can upregulate the HSPs that are responsible for optimizing STAT protein activity, thereby serving as a positive feedback mechanism for STAT protein activity.
Stephanou, et. al., initially reported that Stat1/3 are involved in the regulation of HSP90 and HSP70 by showing that IL-6-induced Stat3 signaling increased expression of HSP90β and that IFN-γ-activated Stat1 signaling induced expression of HSP70 and HSP90β.Citation126-130 Additional evidence for this relationship includes the following: Hydrogen peroxide (H2O2) activates Stat1/3 in vascular smooth muscle cells (VSMC) leading to HSP70 expression.Citation131 Thrombin-induced HSP70/HSP90 expression in VSMC also appears to be mediated through the JAK/STAT pathway (Stat1/2/3).Citation132 HSP105β, a member of the HSP70 family that is expressed during stress and functions in suppressing protein aggregation, has been shown to induce HSP70 expression through Stat3 in mammalian cells.Citation133 Stat3 activation via IL-6 administration as a resuscitation adjuvant in a rat model of trauma-hemorrhagic shock leads to upregulation of HSP70 expression in the liver, which contributes to prevention of hepatocyte apoptosis.Citation134 Finally, sodium 4-phenylbutyrae (4PBA), a drug that corrects trafficking and function of the mutant ΔF508 cystic fibrosis transmembrane conductance regulator (CFTR) in epithelial cells, appears to increase HSP70 expression, at least in part, through Stat3 activation.Citation135
Additional HSPs also appear to be induced by Stat1/3/5 signaling. HSP27 is induced by stress and participates in stabilizing proteins, inhibiting apoptosis, mediating thermotolerance, and regulating cell differentiation. Constitutively activated Stat3 in breast cancer cells upregulates HSP27, which may play a part in the mechanism by which Stat3 contributes to oncogenesis.Citation136 Stat3/5B both appear to be mediators of thrombin-induced HSP27 expression in VSMC.Citation137 Stat1 also appears to induce HSP27 in cells after exposure to heat or chemical stress; cells infected with viral pathogens have dramatically reduced levels of Stat1 leading to decreased HSP27, and therefore more readily undergo apoptosis after exposure to heat.Citation138 HSP60/HSP10 comprise the mitochondrial chaperonin complex that stabilizes mitochondrial proteins. Upregulation of HSP60/10 in a rat model of the post-ischemic brain appeared to be mediated through Stat3 signaling,Citation139 as was IFN-γ-induced expression of HSP60/HSP10 in C6 astroglioma cells.Citation140
Finally, Stat1/3 have also been shown to directly interact with heat shock factor (Hsf) 1, the transcription factor most notably responsible for inducing HSPs in response to stress. After stimulation with IFN-γ, Stat1 and Hsf1 form a DNA binding complex that binds to the heat shock element, leading to enhanced activation of transcription.Citation128 The effect of Stat3-Hsf1 interaction on Stat3 or Hsf1 signaling remains controversial. It has been reported that Stat3 and Hsf1 are antagonistic with regard to HSP90β gene expression.Citation126,127 Stat3-Hsf1 complexes in gastric carcinoma cells infected with Helicobacter pylori appeared to be transcriptionally inactive; infected cells became depleted of HSP70 and eventually underwent apoptosis.Citation141 In contrast, interaction of Stat3 with Hsf1 also has been shown to result in increased Stat3 activity: G-CSF-stimulated Stat3 signaling mediated cardio-protection by decreasing cardiomyocyte apoptosis in a mouse heart ischemia/reperfusion model; decrease in cardiomyocyte apoptosis was shown to be critically dependent on the association of Stat3 and Hsf1.Citation142
Conclusions
Chaperone proteins play a substantial role in STAT signaling; they are clearly involved in the de novo folding and maturation of STAT proteins and kinases, in the optimization of STAT protein and kinase activity, and in the degradation of kinases. STAT proteins require interaction with TRiC for biogenesis and optimal function, and while STAT proteins are not traditional HSP90 clients, in that they do not require HSP90 for de novo folding or maturation, they do depend on HSP90 for activation, nuclear translocation, and DNA binding. STAT protein activity is also positively regulated by a number of additional chaperones, especially in cells experiencing increased stress. Kinases, on the other hand, are true HSP90 clients as they depend on HSP90 and co-chaperones for de novo folding, maturation, and activation.
The potential therapeutic implications of the relationship between chaperones and STAT signaling pathways are substantial (). Stat3, for example, plays a major role in many disease states involving dysregulated or dysfunctional apoptotic signaling pathways. Stat3 activity is decreased when cells undergo oxidative stress during ischemia/reperfusion (from trauma-hemorrhagic shock, myocardial infarction, or intracerebral hemorrhage) leading to increased pathologic apoptosis. Up-regulating chaperone activity in these conditions has been associated with increased Stat3 activity, decreased apoptosis, and improved functional recovery.
Table 1. Current and future strategies for modulating Stat3 activity for therapeutic benefit through targeting of chaperones.
Stat3 activity is aberrantly increased in a number of human pathologies as well, including > 50% of all cancers and atherosclerosis. HSP90 inhibitors, and to a lesser extent HSP70 inhibitors, have been used to decrease Stat3 activity and increase apoptosis of cancer cells in multiple cancer systems and models. Because HSP90 inhibitors affect other components of the Stat3 pathway in cancer cells besides Stat3, as summarized above, increased cancer cell apoptosis following HSP90 inhibitor treatment likely is due to the effects of reduced HSP90 activity on more than just Stat3 alone. HSP90 inhibitors also have been reported to decrease Stat3 activity and inflammation contributing to atherosclerosis in a mouse model of coronary artery disease. HSP90 inhibitors may be a therapeutic option for additional diseases that result from increased inflammation secondary to Stat3 activity, such as inflammatory bowel disease, cachexia, asthma and fibrosis. Decreasing Stat3-TRiC interaction could also represent a novel therapeutic approach to these disease states. We are excited about the therapeutic potential of manipulating the interaction between chaperones and STAT signaling pathways, while also recognizing that more research clearly is needed to understand the full nature of these interactions and to best exploit them for the benefit of patients.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Jane Bocchini for her help in Adobe Illustrator, especially with Figures 1 and 2.
Funding
Supported, in part, by grants RP110291 (to DJT) from the Cancer Prevention and Research Institute of Texas, PN1EY016525 (to DJT) from the NIH/NIE, T32 AI055413 (to DJT and CEB) from the NIH/NIAID, and a Stanley A. Plotkin Sanofi Pasteur Pediatric Infectious Diseases Society Fellowship Award (to CEB).
References
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. 2009; 78:959-91. Available from: www.annualreviews.orgdoiabs10.1146annurev.biochem.052308.114844">http:www.annualreviews.orgdoiabs10.1146annurev.biochem.052308.114844; PMID:19298183
- Hartl FU. Molecular chaperones in cellular protein folding. 1996; 381:571-9. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubm-ed&id=8637592&retmode=ref&cmd=prlinks; PMID:8637592
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. 2002; 295:1852-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11884745&retmode=ref&cmd=prlinks; PMID:11884745
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem [Internet] 2001; 70:603-47. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11395418&retmode=ref&cmd=prlinks; PMID:11395418; http://dx.doi.org/10.1146/annurev.biochem.70.1.603
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell 1998; 92:351-66; PMID:9476895; http://dx.doi.org/10.1016/S0092-8674(00)80928-9
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. 2011; 475:324-32. Available from: www.nature.comdoifinder10.1038nature10317"http:www.nature.comdoifinder10.1038nature10317; PMID:21776078
- Morimoto RI, Santoro MG. Stress-inducible responses and heat shock proteins: new pharmacologic targets for cytoprotection. 1998; 16:833-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9743115&retmode=ref&cmd=prlinks; PMID:9743115
- Dun MD, Aitken RJ, Nixon B. The role of molecular chaperones in spermatogenesis and the post-testicular maturation of mammalian spermatozoa. 2012; 18:420-35. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22523110&retmode=ref&cmd=prlinks; PMID:22523110
- Joly A-L, Wettstein G, Mignot G, Ghiringhelli F, Garrido C. Dual role of heat shock proteins as regulators of apoptosis and innate immunity. 2010; 2:238-47. Available from: http:eutils.ncbi.nlm.nih.govent-rezeutilselink.fcgi?dbfrom=pubmed&id=20375559&retmode=ref&cmd=prlinks; PMID:20375559
- Caplan AJ, Mandal AK, Theodoraki MA. Molecular chaperones and protein kinase quality control. 2007; 17:87-92. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17184992&retmode=ref&cmd=prlinks; PMID:17184992
- Nollen EAA, Morimoto RI. Chaperoning signaling pathways: molecular chaperones as stress-sensing “heat shock” proteins. 2002; 115:2809-16. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12082142&retmode=ref&cmd=prlinks; PMID:12082142
- Levy DE, Darnell JEJ. Stats: transcriptional control and biological impact. 2002; 3:651-62. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12209125&retmode=ref&cmd=prlinks; PMID:12209125
- Yu H, Jove R. The STATs of cancer–new molecular targets come of age. 2004; 4:97-105. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=14964307&retmode=ref&cmd=prlinks; PMID:14964307
- Dunn AY, Melville MW, Frydman J. Review: cellular substrates of the eukaryotic chaperonin TRiCCCT. J Struct Biol [Internet] 2001; 135:176-84. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11580267&retmode=ref&cmd=prlinks; PMID:11580267; http://dx.doi.org/10.1006/jsbi.2001.4380
- Gutsche I, Essen LO, Baumeister W. Group II chaperonins: new TRiC(k)s and turns of a protein folding machine. J Mol Biol [Internet] 1999; 293:295-312. Available from: http:eutils.ncbi.nlm.nih.goventrezeu-tilselink.fcgi?dbfrom=pubmed&id=10550210&retmode=ref&cmd=prlinks; PMID:10550210; http://dx.doi.org/10.1006/jmbi.1999.3008
- Kasembeli M, Lau WCY, Roh S-H, Eckols TK, Frydman J, Chiu W, Tweardy DJ. Modulation of STAT3 folding and function by TRiCCCT chaperonin. PLoS Biol 2014; 12:e1001844; PMID:24756126; http://dx.doi.org/10.1371/journal.pbio.1001844
- Preissler S, Deuerling E. Ribosome-associated chaperones as key players in proteostasis. 2012; 37:274-83. Available from: http:dx.doi.org10.1016j.tibs.2012.03.002; PMID:22503700
- del Alamo M, Hogan DJ, Pechmann S, Albanese V, Brown PO, Frydman J. Defining the specificity of cotranslationally acting chaperones by systematic analysis of mRNAs associated with ribosome-nascent chain complexes. PLoS Biol [Internet] 2011; 9:e1001100. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=21765803&retmode=ref&cmd=prlinks; PMID:21765803; http://dx.doi.org/10.1371/journal.pbio.1001100
- Vainberg IE, Lewis SA, Rommelaere H, Ampe C, Vandekerckhove J, Klein HL, Cowan NJ. Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell [Internet] 1998; 93:863-73. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilse-link.fcgi?dbfrom=pubmed&id=9630229&retmode=ref&cmd=prlinks; PMID:9630229; http://dx.doi.org/10.1016/S0092-8674(00)81446-4
- Millson SH, Truman AW, Racz A, Hu B, Panaretou B, Nuttall J, Mollapour M, Soti C, Piper PW. Expressed as the sole Hsp90 of yeast, the alpha and beta isoforms of human Hsp90 differ with regard to their capacities for activation of certain client proteins, whereas only Hsp90beta generates sensitivity to the Hsp90 inhibitor radicicol. FEBSJ [Internet] 2007; 274:4453-63. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17681020&retmode=ref&cmd=prlinks; PMID:17681020; http://dx.doi.org/10.1111/j.1742-4658.2007.05974.x
- Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis:emerging mechanistic insights. Nat Rev Mol Cell Biol 2010; 11:515-28. Available from: http:dx.doi.org10.1038nrm2918
- Wandinger SK, Richter K, Buchner J. The Hsp90 chaperone machinery. J Biol Chem [Internet] 2008; 283:18473-7. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=18442971&retmode=ref&cmd=prlinks; PMID:18442971; http://dx.doi.org/10.1074/jbc.R800007200
- Richter K, Soroka J, Skalniak L, Leskovar A, Hessling M, Reinstein J, Buchner J. Conserved conformational changes in the ATPase cycle of human Hsp90. J Biol Chem [Internet] 2008; 283:17757-65. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=18400751&retmode=ref&cmd=prlinks; PMID:18400751; http://dx.doi.org/10.1074/jbc.M800540200
- Zhang H, Burrows F. Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med (Berlin, Germany) [Internet] 2004; 82:488-99. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15168026&retmode=ref&cmd=prlinks; PMID:15168026
- Shah M, Patel K, Fried VA, Sehgal PB. Interactions of STAT3 with caveolin-1 and heat shock protein 90 in plasma membrane raft and cytosolic complexes. Preservation of cytokine signaling during fever. J Biol Chem [Internet] 2002; 277:45662-9. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12235142&retmode=ref&cmd=prlinks; PMID:12235142; http://dx.doi.org/10.1074/jbc.M205935200
- Sato N, Yamamoto T, Sekine Y, Yumioka T, Junicho A, Fuse H, Matsuda T. Involvement of heat-shock protein 90 in the interleukin-6-mediated signaling pathway through STAT3. Biochem Biophys Res Commun [Internet] 2003; 300:847-52. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12559950&retmode=ref&cmd=prlinks; PMID:12559950; http://dx.doi.org/10.1016/S0006-291X(02)02941-8
- Prinsloo E, Kramer AH, Edkins AL, Blatch GL. STAT3 interacts directly with Hsp90. IUBMB Life [Internet] 2012; 64:266-73. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22271514&retmode=ref&cmd=prlinks; PMID:22271514; http://dx.doi.org/10.1002/iub.607
- Xu W, Yu F, Yan M, Lu L, Zou W, Sun L, Zheng Z, Liu X. Geldanamycin, a heat shock protein 90-binding agent, disrupts Stat5 activation in IL-2-stimulated cells. J Cell Physiol [Internet] 2004; 198:188-96. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=14603521&retmmode=ref&cmd=prlinks; PMID:14603521; http://dx.doi.org/10.1002/jcp.10403
- McCleese JK, Bear MD, Fossey SL, Mihalek RM, Foley KP, Ying W, Barsoum J, London CA. The novel HSP90 inhibitor STA-1474 exhibits biologic activity against osteosarcoma cell lines. Int J Cancer J [Internet] 2009; 125:2792-801. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=19544563&retmode=ref&cmd=; PMID:19544563; http://dx.doi.org/10.1002/ijc.24660
- Stuhmer T, Zollinger A, Siegmund D, Chatterjee M, Grella E, Knop S, Kortum M, Unzicker C, Jensen MR, Quadt C, et al. Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia [Internet] 2008; 22:1604-12. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=18480838&retmode=ref&cmd=prlinks; http://dx.doi.org/10.1038/leu.2008.111
- Rahmani M, Reese E, Dai Y, Bauer C, Kramer LB, Huang M, Jove R, Dent P, Grant S. Cotreatment with suberanoylanilide hydroxamic acid and 17-allylamino 17-demethoxygeldanamycin synergistically induces apoptosis in Bcr-Abl+ Cells sensitive and resistant to STI571 (imatinib mesylate) in association with down-regulation of Bcr-Abl, abrogation of signal transducer and activator of transcription 5 activity, and Bax conformational change. Mol Pharmacol [Internet] 2005; 67:1166-76. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15625278&retmode=ref&cmd=prlinks; PMID:15625278; http://dx.doi.org/10.1124/mol.104.007831
- Chatterjee M, Jain S, Stuhmer T, Andrulis M, Ungethum U, Kuban R-J, Lorentz H, Bommert K, Topp M, Kramer D, et al. STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood [Internet] 2007; 109:720-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17003370&retmode=ref&cmd=prlinks; PMID:17003370; http://dx.doi.org/10.1182/blood-2006-05-024372
- Moulick K, Ahn JH, Zong H, Rodina A, Cerchietti L, Gomes DaGama EM, Caldas-Lopes E, Beebe K, Perna F, Hatzi K, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol [Internet] 2011; 7:818-26. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=21946277&retmode=ref&cmd=prlinks; PMID:21946277; http://dx.doi.org/10.1038/nchembio.670
- Setati MM, Prinsloo E, Longshaw VM, Murray PA, Edgar DH, Blatch GL. Leukemia inhibitory factor promotes Hsp90 association with STAT3 in mouse embryonic stem cells. IUBMB Life [Internet] 2010; 62:61-6. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20014282&retmode=ref&cmd=prlinks; PMID:20014282
- Bradley E, Bieberich E, Mivechi NF, Tangpisuthipongsa D, Wang G. Regulation of embryonic stem cell pluripotency by heat shock protein 90. Stem Cells (Dayton, Ohio) [Internet] 2012; 30:1624-33. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22696450&retmode=ref&cmd=prlinks; PMID:22696450; http://dx.doi.org/10.1002/stem.1143
- Longshaw VM, Baxter M, Prewitz M, Blatch GL. Knockdown of the co-chaperone Hop promotes extranuclear accumulation of Stat3 in mouse embryonic stem cells. EurJ Cell Biol [Internet] 2009; 88:153-66. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=18996616&retmode=ref&cmd=prlinks; PMID:18996616; http://dx.doi.org/10.1016/j.ejcb.2008.09.003
- Howard M, Roux J, Lee H, Miyazawa B, Lee J-W, Gartland B, Howard AJ, Matthay MA, Carles M, Pittet J-F. Activation of the stress protein response inhibits the STAT1 signalling pathway and iNOS function in alveolar macrophages: role of Hsp90 and Hsp70. Thorax [Internet] 2010; 65:346-53. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20388761&retmode=ref&cmd=prlinks; PMID:20388761; http://dx.doi.org/10.1136/thx.2008.101139
- Best OG, Mulligan SP. Heat shock protein-90 inhibitor, NVP-AUY922, is effective in combination with fludarabine against chronic lymphocytic leukemia cells cultured on CD40L-stromal layer and inhibits their activatedproliferative phenotype. Leuk Lymphoma [Internet] 2012; 53:2314-20. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22646928&retmode=ref&cmd=prlinks; PMID:22646928; http://dx.doi.org/10.3109/10428194.2012.698278
- Yao Q, Nishiuchi R, Kitamura T, Kersey JH. Human leukemias with mutated FLT3 kinase are synergistically sensitive to FLT3 and Hsp90 inhibitors: the key role of the STAT5 signal transduction pathway. Leukemia [Internet] 2005; 19:1605-12. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=16034464&retmode=ref&cmd=prlinks; http://dx.doi.org/10.1038/sj.leu.2403881
- Lang SA, Moser C, Gaumann A, Klein D, Glockzin G, Popp FC, Dahlke MH, Piso P, Schlitt HJ, Geissler EK, et al. Targeting heat shock protein 90 in pancreatic cancer impairs insulin-like growth factor-I receptor signaling, disrupts an interleukin-6signal-transducer and activator of transcription 3hypoxia-inducible factor-1alpha autocrine loop, and reduces orthotopic tumor growth. Clin Cancer Res [Internet] 2007; 13:6459-68. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17975158&retmode=ref&cmd=prlinks; PMID:17975158; http://dx.doi.org/10.1158/1078-0432.CCR-07-1104
- Kim HL, Cassone M, Otvos LJ, Vogiatzi P. HIF-1alpha and STAT3 client proteins interacting with the cancer chaperone Hsp90: therapeutic considerations. Cancer Biol Ther [Internet] 2008; 7:10-4. Available from: http:eutils.ncbi.nlm.nih.goventrezlselink.fcgi?dbfrom=pubmedeuti&id=18347418&retmode=ref&cmd=prlinks; PMID:18347418; http://dx.doi.org/10.4161/cbt.7.1.5458
- Madrigal-Matute J, Lopez-Franco O, Blanco-Colio LM, Munoz-Garcia B, Ramos-Mozo P, Ortega L, Egido J, Martin-Ventura JL. Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc Res [Internet] 2010; 86:330-7. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20154064&retmode=ref&cmd=prlinks; PMID:20154064; http://dx.doi.org/10.1093/cvr/cvq046
- Szczepanek K, Chen Q, Larner AC, Lesnefsky EJ. Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion [Internet] 2012; 12:180-9. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=21930250&retmode=ref&cmd=prlinks; PMID:21930250; http://dx.doi.org/10.1016/j.mito.2011.08.011
- Cimica V, Chen H-C, Iyer JK, Reich NC. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PLoS One 2011; 6:e20188; PMID:21625522; http://dx.doi.org/10.1371/journal.pone.0020188
- Szczepanek K, Lesnefsky EJ, Larner AC. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol [Internet] 2012; 22:429-37. Available from: http:dx.doi.org10.1016j.tcb.2012.05.001; http://dx.doi.org/10.1016/j.tcb.2012.05.001
- Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemiareperfusion. Basic Res Cardiol [Internet] 2010; 105:771-85. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20960209&retmode=ref&cmd=prlinks; http://dx.doi.org/10.1007/s00395-010-0124-1
- Shulga N, Pastorino JG. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J Cell Sci [Internet] 2012; 125:2995-3003. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22393233&retmode=ref&cmd=prlinks; http://dx.doi.org/10.1242/jcs.103093
- Tammineni P, Anugula C, Mohammed F, Anjaneyulu M, Larner AC, Sepuri NBV. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J Biol Chem 2013; 288:4723-32; PMID:23271731; http://dx.doi.org/10.1074/jbc.M112.378984
- Liu T, Daniels CK, Cao S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol Thers [Internet] 2012; 136:354-74. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22960394&retmode=ref&cmd=prlinks; PMID:22960394; http://dx.doi.org/10.1016/j.pharmthera.2012.08.014
- Chen S-C, Guh J-Y, Chen H-C, Yang Y-L, Huang J-S, Chuang L-Y. Advanced glycation end-product-induced mitogenesis is dependent on Janus kinase 2-induced heat shock protein 70 in normal rat kidney interstitial fibroblast cells. Transl Res [Internet] 2007; 149:274-81. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17466927&retmode=ref&cmd=prlinks; PMID:17466927; http://dx.doi.org/10.1016/j.trsl.2006.08.005
- Ghoshal S, Rao I, Earp JC, Jusko WJ, Wetzler M. Down-regulation of heat shock protein 70 improves arsenic trioxide and 17-DMAG effects on constitutive signal transducer and activator of transcription 3 activity. Cancer Chemother Pharmacol [Internet] 2010; 66:681-9. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20035426&retmode=ref&cmd=prlinks; PMID:20035426; http://dx.doi.org/10.1007/s00280-009-1210-7
- Sinn D-I, Chu K, Lee S-T, Song E-C, Jung K-H, Kim E-H, Park D-K, Kang K-M, Kim M, Roh J-K. Pharmacological induction of heat shock protein exerts neuroprotective effects in experimental intracerebral hemorrhage. Brain Res [Internet] 2007; 1135:167-76. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17208204&retmode=ref&cmd=prlinks
- Qiu H, Lizano P, Laure L, Sui X, Rashed E, Park JY, Hong C, Gao S, Holle E, Morin D, et al. H11 kinase heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation [Internet] 2011; 124:406-15. Available from: http:circ.ahajournals.orgcgidoi10.1161CIRCULATIONAHA.110.013847; PMID:21747053; http://dx.doi.org/10.1161/CIRCULATIONAHA.110.013847
- Chen L, Lizano P, Zhao X, Sui X, Dhar SK, Shen Y-T, Vatner DE, Vatner SF, Depre C. Preemptive conditioning of the swine heart by H11 kinaseHsp22 provides cardiac protection through inducible nitric oxide synthase. Am J PhysHeart Circ Physiol [Internet] 2011; 300:H1303-10. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=21317305&retmode=ref&cmd=prlinks; PMID:21317305; http://dx.doi.org/10.1152/ajpheart.00979.2010
- Ndubuisi MI, Guo GG, Fried VA, Etlinger JD, Sehgal PB. Cellular physiology of STAT3: where's the cytoplasmic monomer? J Biol Chem [Internet] 1999; 274:25499-509. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=10464281&retmode=ref&cmd=prlinks; PMID:10464281; http://dx.doi.org/10.1074/jbc.274.36.25499
- Sehgal PB. STAT-signalling through the cytoplasmic compartment: consideration of a new paradigm. Cell Signal [Internet] 2000; 12:525-35. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11027945&retmode=ref&cmd=prlinks; PMID:11027945; http://dx.doi.org/10.1016/S0898-6568(00)00098-X
- Sehgal PB. Cytokine-induced STAT signalling through the cytoplasmic compartment. Adv Exp Med Biol [Internet] 2001; 495:161-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11774561&retmode=ref&cmd=prlinks; PMID:11774561; http://dx.doi.org/10.1007/978-1-4615-0685-0_21
- Sehgal PB, Guo GG, Shah M, Kumar V, Patel K. Cytokine signaling: STATS in plasma membrane rafts. J Biol Chem [Internet] 2002; 277:12067-74. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11815625&retmode=ref&cmd=prlinks; PMID:11815625; http://dx.doi.org/10.1074/jbc.M200018200
- Sehgal PB. Plasma membrane rafts and chaperones in cytokineSTAT signaling. Acta Biochimica Polonica [Internet] 2003; 50:583-94. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=14515141&retmode=ref&cmd=prlinks; PMID:14515141
- Guo GG, Patel K, Kumar V, Shah M, Fried VA, Etlinger JD, Sehgal PB. Association of the chaperone glucose-regulated protein 58 (GRP58ER-60ERp57) with Stat3 in cytosol and plasma membrane complexes. J Interferon Cytokine Resh [Internet] 2002; 22:555-63. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12060494&retmode=ref&cmd=prlinks; http://dx.doi.org/10.1089/10799900252982034
- Eufemi M, Coppari S, Altieri F, Grillo C, Ferraro A, Turano C. ERp57 is present in STAT3-DNA complexes. Biochem Biophys Res Commun [Internet] 2004; 323:1306-12. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15451439&retmode=ref&cmd=prlinks; PMID: 15451439; http://dx.doi.org/10.1016/j.bbrc.2004.09.009
- Coe H, Jung J, Groenendyk J, Prins D, Michalak M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J Biol Chem [Internet] 2010; 285:6725-38. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20022947&retmode=ref&cmd=prlinks; http://dx.doi.org/10.1074/jbc.M109.054015
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell [Internet] 2002; 109:275-82. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12015977&retmode=ref&cmd=prlinks; PMID:12015977; http://dx.doi.org/10.1016/S0092-8674(02)00741-9
- Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MMU, Prodromou C, Robinson CV, Saibil HR, Pearl LH. Structure of an Hsp90-Cdc37-Cdk4 Complex. Mol Cell [Internet] 2006; 23:697-707. Available from: http:linkinghub.elsevier.comretrievepiiS1097276506004977; PMID:16949366; http://dx.doi.org/10.1016/j.molcel.2006.07.016
- Koga F, Xu W, Karpova TS, McNally JG, Baron R, Neckers L. Hsp90 inhibition transiently activates Src kinase and promotes Src-dependent Akt and Erk activation. Proc Natl Acad Sci U S A [Internet] 2006; 103:11318-22. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=16844778&retmode=ref&cmd=prlinks; PMID:16844778; http://dx.doi.org/10.1073/pnas.0604705103
- Citri A, Gan J, Mosesson Y, Vereb G, Szollosi J, Yarden Y. Hsp90 restrains ErbB-2HER2 signalling by limiting heterodimer formation. EMBO Rep 2004; 5:1165-70; PMID:15568014; http://dx.doi.org/10.1038/sj.embor.7400300
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol [Internet] 1997; 13:513-609. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9442882&retmode=ref&cmd=prlinks; PMID:9442882; http://dx.doi.org/10.1146/annurev.cellbio.13.1.513
- Schreiner SJ, Schiavone AP, Smithgall TE. Activation of STAT3 by the Src family kinase Hck requires a functional SH3 domain. J Biol Chem [Internet] 2002; 277:45680-7. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12244095&retmode=ref&cmd=prlinks; PMID:12244095; http://dx.doi.org/10.1074/jbc.M204255200
- Zhao L, Li W, Marshall C, Griffin T, Hanson M, Hick R, Dentchev T, Williams E, Werth A, Miller C, et al. Srcasm inhibits fyn-induced cutaneous carcinogenesis with modulation of notch1 and p53. Cancer Res [Internet] 2009; 69:9439-47. Available from: http:cancerres.aacrjournals.orgcgidoi10.11580008-5472.CAN-09-2976; PMID:19934324; http://dx.doi.org/10.1158/0008-5472.CAN-09-2976
- Shi M. A constitutively active lck kinase promotes cell proliferation and resistance to apoptosis through signal transducer and activator of transcription 5b activation. Mol Cancer Res [Internet] 2006; 4:39-45. Available from: http:mcr.aacrjournals.orgcgidoi10.11581541-7786.MCR-05-0202; PMID:16446405; http://dx.doi.org/10.1158/1541-7786.MCR-05-0202
- Welte T, Leitenberg D, Dittel BN, al-Ramadi BK, Xie B, Chin YE, Janeway CAJ, Bothwell AL, Bottomly K, Fu XY. STAT5 interaction with the T cell receptor complex and stimulation of T cell proliferation. Science [Internet] 1999; 283:222-5. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9880255&retmode=ref&cmd=prlinks; PMID:9880255; http://dx.doi.org/10.1126/science.283.5399.222
- Xi S, Zhang Q, Dyer KF, Lerner EC, Smithgall TE, Gooding WE, Kamens J, Grandis JR. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem [Internet] 2003; 278:31574-83. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12771142&retmode=ref&cmd=prlinks; PMID:12771142; http://dx.doi.org/10.1074/jbc.M303499200
- Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A [Internet] 1980; 77:1311-5. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=6246487&retmode=ref&cmd=prlinks; PMID:6246487; http://dx.doi.org/10.1073/pnas.77.3.1311
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A [Internet] 1994; 91:8324-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=8078881&retmode=ref&cmd=prlinks; PMID:8078881; http://dx.doi.org/10.1073/pnas.91.18.8324
- Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, Jove R. Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science [Internet] 1995; 269:81-3. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=7541555&retmode=ref&cmd=prlinks; PMID:7541555; http://dx.doi.org/10.1126/science.7541555
- Kazansky AV, Rosen JM. Signal transducers and activators of transcription 5B potentiates v-Src-mediated transformation of NIH-3T3 cells. Cell Growth Differ [Internet] 2001; 12:1-7. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11205741&retmode=ref&cmd=prlinks; PMID:11205741
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JEJ. Stat3 as an oncogene. Cell [Internet] 1999; 98:295-303. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=10458605&retmode=ref&cmd=prlinks; PMID:10458605; http://dx.doi.org/10.1016/S0092-8674(00)81959-5
- Xu Y, Singer MA, Lindquist S. Maturation of the tyrosine kinase c-src as a kinase and as a substrate depends on the molecular chaperone Hsp90. Proc Natl Acad Sci U S A [Internet] 1999; 96:109-14. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9874780&retmode=ref&cmd=prlinks; PMID:9874780; http://dx.doi.org/10.1073/pnas.96.1.109
- Yano A, Tsutsumi S, Soga S, Lee M-J, Trepel J, Osada H, Neckers L. Inhibition of Hsp90 activates osteoclast c-Src signaling and promotes growth of prostate carcinoma cells in bone. Proc Natl Acad Sci U S A [Internet] 2008; 105:15541-6. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=18840695&retmode=ref&cmd=prlinks; PMID:18840695; http://dx.doi.org/10.1073/pnas.0805354105
- Scholz GM, Hartson SD, Cartledge K, Volk L, Matts RL, Dunn AR. The molecular chaperone Hsp90 is required for signal transduction by wild-type Hck and maintenance of its constitutively active counterpart. Cell Growth Differ 2001; 12:409-17; PMID:11504706
- Stefanova I, Corcoran ML, Horak EM, Wahl LM, Bolen JB, Horak ID. Lipopolysaccharide induces activation of CD14-associated protein tyrosine kinase p5356lyn. J Biol Chem [Internet] 1993; 268:20725-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=7691802&retm-ode=ref&cmd=prlinks; PMID:7691802
- Bijlmakers MJ, Marsh M. Hsp90 is essential for the synthesis and subsequent membrane association, but not the maintenance, of the Src-kinase p56(lck). Mol Biol Cell [Internet] 2000; 11:1585-95. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=10793137&retmode=ref&cmd=prlinks; PMID:10793137; http://dx.doi.org/10.1091/mbc.11.5.1585
- Yun B-G, Matts RL. Differential effects of Hsp90 inhibition on protein kinases regulating signal transduction pathways required for myoblast differentiation. Exp Cell Res [Internet] 2005; 307:212-23. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15922741&ret-mode=ref&cmd=prlinks; PMID:15922741; http://dx.doi.org/10.1016/j.yexcr.2005.03.003
- Zhao M, Ma J, Zhu H-Y, Zhang X-H, Du Z-Y, Xu Y-J, Yu X-D. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol Cancer [Internet] 2011; 10:104. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=21871133&retmode=ref&cmd=prlinks; PMID:21871133; http://dx.doi.org/10.1186/1476-4598-10-104
- Scroggins BT, Neckers L. Post-translational modification of heat-shock protein 90: impact on chaperone function. Expert Opin Drug Discov 2007; 2:1403-14; PMID:23484535; http://dx.doi.org/10.1517/17460441.2.10.1403
- Li J, Soroka J, Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta 2012; 1823:624-35; PMID:21951723; http://dx.doi.org/10.1016/j.bbamcr.2011.09.003
- Mimnaugh EG, Worland PJ, Whitesell L, Neckers LM. Possible role for serinethreonine phosphorylation in the regulation of the heteroprotein complex between the hsp90 stress protein and the pp60v-src tyrosine kinase. J Biol Chem 1995; 270:28654-9; PMID:7499384; http://dx.doi.org/10.1074/jbc.270.48.28654
- Duval M, Le Boeuf F, Huot J, Gratton J-P. Src-mediated phosphorylation of Hsp90 in response to vascular endothelial growth factor (VEGF) is required for VEGF receptor-2 signaling to endothelial NO synthase. Mol Biol Cell 2007; 18:4659-68; PMID:17855507; http://dx.doi.org/10.1091/mbc.E07-05-0467
- Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, Vojtesek B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein foldingdegradation balances. Oncogene 32:3101-10; PMID:22824801; http://dx.doi.org/10.1038/onc.2012.314
- Liu KD, Gaffen SL, Goldsmith MA. JAKSTAT signaling by cytokine receptors. Curr Opin Immunol [Internet] 1998; 10:271-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9638363&retmode=ref&cmd=prlinks; PMID:9638363; http://dx.doi.org/10.1016/S0952-7915(98)80165-9
- Yamaoka K, Saharinen P, Pesu M, Holt VET3, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol [Internet] 2004; 5:253. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15575979&retmode=ref&cmd=prlinks; PMID:15575979; http://dx.doi.org/10.1186/gb-2004-5-12-253
- Schoof N, Bonin von F, Trümper L, Kube D. HSP90 is essential for Jak-STAT signaling in classical Hodgkin lymphoma cells. Cell Commun Signal [Internet] 2009; 7:17. Available from: www.biosignaling.comcontent7117">http:www.biosignaling.comcontent7117; PMID:19607667; http://dx.doi.org/10.1186/1478-811X-7-17
- Shang L, Tomasi TB. The heat shock protein 90-CDC37 chaperone complex is required for signaling by types I and II interferons. J Biol Chem [Internet] 2006; 281:1876-84. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=16280321&retmode=ref&cmd=prlinks; PMID:16280321; http://dx.doi.org/10.1074/jbc.M509901200
- Schoof N, Bonin von F, Trümper L, Kube D. HSP90 is essential for Jak-STAT signaling in classical Hodgkin lymphoma cells. Cell Commun Signal 2009; 7:17; PMID:19607667; http://dx.doi.org/10.1186/1478-811X-7-17
- Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJP, Boggon TJ, Wlodarska I, Clark JJ, Moore S, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell [Internet] 2005; 7:387-97. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15837627&retmode=ref&cmd=prlinks; PMID:15837627; http://dx.doi.org/10.1016/j.ccr.2005.03.023
- James C, Ugo V, Le Couedic J-P, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature [Internet] 2005; 434:1144-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15793561&retmode=ref&cmd=prlinks; PMID:15793561; http://dx.doi.org/10.1038/nature03546
- Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, Score J, Seear R, Chase AJ, Grand FH, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood [Internet] 2005; 106:2162-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=15920007&retmode=ref&cmd=prlinks; PMID:15920007; http://dx.doi.org/10.1182/blood-2005-03-1320
- Bareng J, Jilani I, Gorre M, Kantarjian H, Giles FJ, Hannah A, Albitar M. A potential role for HSP90 inhibitors in the treatment of JAK2 mutant-positive diseases as demonstrated using quantitative flow cytometry. Leuk Lymphoma [Internet] 2007; 48:2189-95. Available from: http:informahealthcare.comdoiabs10.108010428190701607576; PMID:17926180; http://dx.doi.org/10.1080/10428190701607576
- Proia DA, Foley KP, Korbut T, Sang J, Smith D, Bates RC, Liu Y, Rosenberg AF, Zhou D, Koya K, et al. Multifaceted intervention by the Hsp90 inhibitor ganetespib (STA-9090) in cancer cells with activated JAKSTAT signaling. PLoS One [Internet] 2011; 6:e18552. Available from: http:dx.plos.org10.1371journal.pone.0018552.t001; PMID:21533169; http://dx.doi.org/10.1371/journal.pone.0018552
- Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R, Joshi A, Balusu R, Koul S, Chen J, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood [Internet] 2009; 114:5024-33. Available from: www.bloodjournal.orgcgidoi10.1182blood-2009-05-222133">http:www.bloodjournal.orgcgidoi710.1182blood-2009-05-222133; PMID:19828702; http://dx.doi.org/10.1182/blood-2009-05-222133
- Weigert O, Lane AA, Bird L, Kopp N, Chapuy B, van Bodegom D, Toms AV, Marubayashi S, Christie AL, McKeown M, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med [Internet] 2012; 209:259-73. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22271575&retmode=ref&cmd=prlinks; PMID:22271575; http://dx.doi.org/10.1084/jem.20111694
- Koppikar P, Bhagwat N, Kilpivaara O, Manshouri T, Adli M, Hricik T, Liu F, Saunders LM, Mullally A, Abdel-Wahab O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012; 489:155-9; PMID:22820254; http://dx.doi.org/10.1038/nature11303
- Bohonowych JE, Gopal U, Isaacs JS. Hsp90 as a gatekeeper of tumor angiogenesis: clinical promise and potential pitfalls. J Oncol [Internet] 2010; 2010:412985. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20628489&retmode=ref&cmd=prlinks; PMID:20628489
- Nichane M, Ren X, Bellefroid EJ. Self-regulation of Stat3 activity coordinates cell-cycle progression and neural crest specification. EMBO J [Internet] 2010; 29:55-67. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=19851287&retmode=ref&cmd=prlinks; PMID:19851287; http://dx.doi.org/10.1038/emboj.2009.313
- Zong CS, Chan J, Levy DE, Horvath C, Sadowski HB, Wang L-H. Mechanism of STAT3 Activation by Insulin-like Growth Factor I Receptor. J Biol Chem [Internet] 2000; 275:15099-105. Available from: www.jbc.orgcontent2752015099.abstract">http:www.jbc.orgcontent2752015099.abstract; PMID:10747872; http://dx.doi.org/10.1074/jbc.M000089200
- Dudka AA, Sweet SMM, Heath JK. Signal transducers and activators of transcription-3 binding to the fibroblast growth factor receptor is activated by receptor amplification. Cancer Res [Internet] 2010; 70:3391-401. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20388777&retmode=ref&cmd=prlinks; PMID:20388777; http://dx.doi.org/10.1158/0008-5472.CAN-09-3033
- Matei D, Satpathy M, Cao L, Lai Y-C, Nakshatri H, Donner DB. The platelet-derived growth factor receptor alpha is destabilized by geldanamycins in cancer cells. J Biol Chem [Internet] 2007; 282:445-53. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17079230&retmode=ref&cmd=prlinks; PMID:17079230; http://dx.doi.org/10.1074/jbc.M607012200
- Ou W-B, Hubert C, Corson JM, Bueno R, Flynn DL, Sugarbaker DJ, Fletcher JA. Targeted inhibition of multiple receptor tyrosine kinases in mesothelioma. Neoplasia 2011; 13:12-22; PMID:21245936
- Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L. Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J Biol Chem [Internet] 2001; 276:3702-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11071886&retmode=ref&cmd=prlinks; PMID:11071886; http://dx.doi.org/10.1074/jbc.M006864200
- Xu W, Mimnaugh EG, Kim J-S, Trepel JB, Neckers LM. Hsp90, not Grp94, regulates the intracellular trafficking and stability of nascent ErbB2. Cell Stress Chaperones [Internet] 2002; 7:91-6. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11892991&retmode=ref&cmd=prlinks; PMID:11892991; http://dx.doi.org/10.1379/1466-1268(2002)007%3c0091:HNGRTI%3e2.0.CO;2
- Shimamura T. Epidermal Growth Factor Receptors Harboring Kinase Domain Mutations Associate with the Heat Shock Protein 90 Chaperone and Are Destabilized following Exposure to Geldanamycins. Cancer Res [Internet] 2005; 65:6401-8. Available from: http:cancerres.aacrjournals.orgcgidoi10.11580008-5472.CAN-05-0933; PMID:16024644; http://dx.doi.org/10.1158/0008-5472.CAN-05-0933
- Citri A, Kochupurakkal BS, Yarden Y. The achilles heel of ErbB-2HER2: regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle (Georgetown, Tex) [Internet] 2004; 3:51-60. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=14657666&retmode=ref&cmd=prlinks; PMID:14657666; http://dx.doi.org/10.4161/cc.3.1.607
- Rice JW, Veal JM, Barabasz A, Foley B, Fadden P, Scott A, Huang K, Steed P, Hall S. Targeting of multiple signaling pathways by the Hsp90 inhibitor SNX-2112 in EGFR resistance models as a single agent or in combination with erlotinib. Oncol Res [Internet] 2009; 18:229-42. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20225761&retmode=ref&cmd=prlinks; PMID:20225761; http://dx.doi.org/10.3727/096504009X12596189659240
- Xu W, Soga S, Beebe K, Lee M-J, Kim YS, Trepel J, Neckers L. Sensitivity of epidermal growth factor receptor and ErbB2 exon 20 insertion mutants to Hsp90 inhibition. Br J Cancer [Internet] 2007; 97:741-4. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17712310&retmode=ref&cmd=prlinks; PMID:17712310; http://dx.doi.org/10.1038/sj.bjc.6603950
- Sidera K, Gaitanou M, Stellas D, Matsas R, Patsavoudi E. A critical role for HSP90 in cancer cell invasion involves interaction with the extracellular domain of HER-2. J Biol Chem [Internet] 2008; 283:2031-41. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=18056992&retmode=ref&cmd=prlinks; PMID:18056992;http://dx.doi.org/10.1074/jbc.M701803200
- Hamidieh El A, Grammatikakis N, Patsavoudi E. Cell surface Cdc37 participates in extracellular HSP90 mediated cancer cell invasion. PLoS One [Internet] 2012; 7:e42722. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22912728&retmode=ref&cmd=prlinks; PMID:22912728; http://dx.doi.org/10.1371/journal.pone.0042722
- Sims JD, McCready J, Jay DG. Extracellular Heat shock protein (Hsp)70 and Hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS On [Internet] 2011; 6:e18848. Available from: http:dx.plos.org10.1371journal.pone.0018848.g007; PMID:21533148; http://dx.doi.org/10.1371/journal.pone.0018848
- Evdonin AL, Guzhova IV, Margulis BA, Medvedeva ND. Extracellular heat shock protein 70 mediates heat stress-induced epidermal growth factor receptor transactivation in A431 carcinoma cells. FEBS Lett 2006; 580:6674-8; PMID:17126326; http://dx.doi.org/10.1016/j.febslet.2006.11.024
- Esser C, Alberti S, Hohfeld J. Cooperation of molecular chaperones with the ubiquitinproteasome system. Biochim Biophys Acta 2004; 1695:171-88; PMID:15571814; http://dx.doi.org/10.1016/j.bbamcr.2004.09.020
- Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz AL, Ciechanover A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J Biol Chem [Internet] 1997; 272:9002-10. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9083024&retmode=ref&cmd=prlinks; PMID:9083024; http://dx.doi.org/10.1074/jbc.272.14.9002
- Lee DH, Sherman MY, Goldberg AL. Involvement of the molecular chaperone Ydj1 in the ubiquitin-dependent degradation of short-lived and abnormal proteins in Saccharomyces cerevisiae. Mol Cell Biol [Internet] 1996; 16:4773-81. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=8756635&retmode=ref&cmd=prlinks; PMID:8756635
- Pratt WB, Morishima Y, Peng HM, Osawa Y. Proposal for a role of the Hsp90Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp Biol Med [Internet] 2010; 235:278-89. Available from: http:ebm.rsmjournals.comcgidoi10.1258ebm.2009.009250; PMID:20404045; http://dx.doi.org/10.1258/ebm.2009.009250
- Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2Neu. Proc Natl Acad Sci U S A 2002; 99:12847-52; PMID:12239347; http://dx.doi.org/10.1073/pnas.202365899
- An WG, Schulte TW, Neckers LM. The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome. Cell Growth Differ [Internet] 2000; 11:355-60. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=10939589&retmode=ref&cmd=prlinks; PMID:10939589
- Mandal AK, Gibney PA, Nillegoda NB, Theodoraki MA, Caplan AJ, Morano KA. Hsp110 chaperones control client fate determination in the hsp70-Hsp90 chaperone system. Mol Biol Cell [Internet] 2010; 21:1439-48. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=20237159&retmode=ref&cmd=prlinks; PMID:20237159; http://dx.doi.org/10.1091/mbc.E09-09-0779
- Stephanou A, Amin V, Isenberg DA, Akira S, Kishimoto T, Latchman DS. Interleukin 6 activates heat-shock protein 90 beta gene expression. Biochem J [Internet] 1997; 321 (Pt 1):103-6. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9003407&retmode=ref&cmd=prlinks; PMID:9003407
- Stephanou A, Isenberg DA, Akira S, Kishimoto T, Latchman DS. The nuclear factor interleukin-6 (NF-IL6) and signal transducer and activator of transcription-3 (STAT-3) signalling pathways co-operate to mediate the activation of the hsp90beta gene by interleukin-6 but have opposite effects on its inducibility by heat shock. Biochem J [Internet] 1998; 330(Pt 1):189-95. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9461509&retmode=ref&cmd=prlinks; PMID:9461509
- Stephanou A, Isenberg DA, Nakajima K, Latchman DS. Signal transducer and activator of transcription-1 and heat shock factor-1 interact and activate the transcription of the Hsp-70 and Hsp-90beta gene promoters. J Biol Chem [Internet] 1999; 274:1723-8. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=9880553&retmode=ref&cmd=prlinks; PMID:9880553; http://dx.doi.org/10.1074/jbc.274.3.1723
- Stephanou A, Latchman DS. Transcriptional regulation of the heat shock protein genes by STAT family transcription factors. Gene Exp [Internet] 1999; 7:311-9. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=10440232&retmode=ref&cmd=prlinks; PMID:10440232
- Fritchley SJ, Kirby JA, Ali S. The antagonism of interferon-gamma (IFN-gamma) by heparin: examination of the blockade of class II MHC antigen and heat shock protein-70 expression. Clin Exp Immunol [Internet] 2000; 120:247-52. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=10792372&retmode=ref&cmd=prlinks; PMID:10792372; http://dx.doi.org/10.1046/j.1365-2249.2000.01178.x
- Madamanchi NR, Li S, Patterson C, Runge MS. Reactive oxygen species regulate heat-shock protein 70 via the JAKSTAT pathway. Arterioscl Thromb Vasc Biol [Internet] 2001; 21:321-6. Available from: http:atvb.ahajournals.orgcgidoi10.116101.ATV.21.3.321; PMID:11231909; http://dx.doi.org/10.1161/01.ATV.21.3.321
- Madamanchi NR, Li S, Patterson C, Runge MS. Thrombin regulates vascular smooth muscle cell growth and heat shock proteins via the JAK-STAT pathway. J Biol Chem [Internet] 2001; 276:18915-24. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=11278437&retmode=ref&cmd=prlinks; PMID:11278437; http://dx.doi.org/10.1074/jbc.M008802200
- Yamagishi N, Fujii H, Saito Y, Hatayama T. Hsp105β upregulates hsp70 gene expression through signal transducer and activator of transcription-3. FEBS J [Internet] 2009; 276:5870-80. Available from: http:doi.wiley.com10.1111j.1742-4658.2009.07311.x; PMID:19754877; http://dx.doi.org/10.1111/j.1742-4658.2009.07311.x
- Thacker SA, Robinson P, Abel A, Tweardy DJ. Modulation of the unfolded protein response during hepatocyte and cardiomyocyte apoptosis in traumahemorrhagic shock. Sci Rep 2013; 3:1187; PMID:23378918; http://dx.doi.org/10.1038/srep01187
- Suaud L, Miller K, Panichelli AE, Randell RL, Marando CM, Rubenstein RC. 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J Biol Chem [Internet] 2011; 286:45083-92. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=22069317&retmode=ref&cmd=prlinks; PMID:22069317; http://dx.doi.org/10.1074/jbc.M111.293282
- Song H, Ethier SP, Dziubinski ML, Lin J. Stat3 modulates heat shock 27kDa protein expression in breast epithelial cells. Biochem Biophys Res Commun [Internet] 2004; 314:143-50. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=14715258&retmode=ref&cmd=prlinks; PMID:14715258; http://dx.doi.org/10.1016/j.bbrc.2003.12.048
- Cao H, Dronadula N, Rizvi F, Li Q, Srivastava K, Gerthoffer WT, Rao GN. Novel role for STAT-5B in the regulation of Hsp27-FGF-2 axis facilitating thrombin-induced vascular smooth muscle cell growth and motility. Circ Res [Internet] 2006; 98:913-22. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=16527988&retmode=ref&cmd=prlinks; PMID:16527988; http://dx.doi.org/10.1161/01.RES.0000216954.55724.a2
- Yokota S-I, Yokosawa N, Kubota T, Okabayashi T, Arata S, Fujii N. Suppression of thermotolerance in mumps virus-infected cells is caused by lack of HSP27 induction contributed by STAT-1. J Biol Chem [Internet] 2003; 278:41654-60. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=12917439&retmode=ref&cmd=prlinks; PMID:12917439; http://dx.doi.org/10.1074/jbc.M305701200
- Kim S-W, Lee J-K. NO-induced downregulation of HSP10 and HSP60 expression in the postischemic brain. J Neurosci Res [Internet] 2007; 85:1252-9. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17348040&retmode=ref&cmd=prlinks; PMID:17348040; http://dx.doi.org/10.1002/jnr.21236
- Kim S-W, Kim J-B, Kim J-H, Lee J-K. Interferon-gamma-induced expressions of heat shock protein 60 and heat shock protein 10 in C6 astroglioma cells: identification of the signal transducers and activators of transcription 3-binding site in bidirectional promoter. Neuroreport [Internet] 2007; 18:385-9. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=17435608&retmode=ref&cmd=prlinks; PMID:17435608; http://dx.doi.org/10.1097/WNR.0b013e32801299cc
- Pierzchalski P, Krawiec A, Ptak-Belowska A, Baranska A, Konturek SJ, Pawlik WW. The mechanism of heat-shock protein 70 gene expression abolition in gastric epithelium caused by Helicobacter pylori infection. Helicobacter [Internet] 2006; 11:96-104. Available from: http:eutils.ncbi.nlm.nih.goventrezeutilselink.fcgi?dbfrom=pubmed&id=16579839&retmode=ref&cmd=prlinks; PMID:16579839; http://dx.doi.org/10.1111/j.1523-5378.2006.00383.x
- Ma H, Gong H, Chen Z, Liang Y, Yuan J, Zhang G, Wu J, Ye Y, Yang C, Nakai A, et al. Association of Stat3 with HSF1 plays a critical role in G-CSF-induced cardio-protection against ischemiareperfusion injury. J Mol Cell Cardiol 2012; 52:1282-90; PMID:22426029; http://dx.doi.org/10.1016/j.yjmcc.2012.02.011