
Abstract
We recently evaluated the capacity of Delta-24-RGD oncolytic adenovirus to trigger an antitumor immune response in a syngeneic mouse glioma model. This virotherapy elicited immunity against both tumor-associated antigens and viral antigens. An immunogenic cell death accompanied by pathogen- or damage- associated patterns (PAMPs and DAMPs) induced by the virus may be responsible for the adenoviral-mediated antitumor effect.
Replication-competent adenoviruses are among viruses being engineered for use as therapeutic agents for cancer.Citation1 In the current paradigm of oncolytic virotherapy, the development of an antitumor immune response is responsible for clinical success. In this model, the virus overcomes or reverses the characteristic immune evasion of solid tumorsCitation2 and facilitates specific recognition of tumor-associated antigens (TAAs) by the host immune system. Thus, the intratumoral necrosis (local effect) induced by the oncolysis serves as a mechanism for activation of the innate and adaptive immune responses that will eventually be responsible for the elimination of distant invasive cells, including metastases (systemic effect).Citation1
However, the immunodominance hypothesis seems to argue against the possibility of an adenovirus-mediated triggering of antitumor immunity.Citation3 The immunodominance model proposes that an immune response is directed against a few epitopes (dominant antigens) of all the many epitopes produced during an infection. Therefore, despite the fact that the virus-induced necrosis facilitates the flooding of the tumor milieu with TAAs, the adenoviral antigens expressed by the infected cancer cells are the dominant ones preventing the development of any efficacious immune response directed against tumor antigens (subdominant antigens).Citation3 In summary, according to the immunodominance model, if viral antigens and pathogen-associated molecular patterns (PAMPs) are responsible for triggering the immune response, the response will be directed exclusively against virus-derived antigens and will be clinically ineffective.
In our work, we specifically tested the capacity of an oncolytic adenovirus (Delta-24-RGD)Citation4 to trigger an antitumor immune response in a syngeneic mouse model.Citation5. The experiment consisted in the intracranial injection of mouse glioma cells into the brain of mice, followed by treatment of the tumors with intratumoral injections of Delta-24-RGD. In studies of co-cultures of splenocytes derived from virus-treated mice mixed with viral infected versus non-infected glioma cells, we were able to detect a strong immune response directed against virus-derived antigens.Citation5 However, these experiments also revealed that the splenocytes of the primed mice also reacted against uninfected glioma cells, strongly indicating that treated mice developed an adaptive immune response specifically against TAAs. Furthermore, using the OVA modeling system, we demonstrated that OVA-expressing glioma cells infected with oncolytic adenoviruses were recognized by OVA-specific CD8+ cells.Citation5 Thus, adenovirus infection induces the recognition of nonviral cellular antigens by the host immune system.
Our report seems to indicate that adenovirus-infected cancer cells expressed not only PAMPs but also danger- (damage-) associated molecular patterns (DAMPs).Citation6 Although not completely expected, our observations are in agreement with previous reports demonstrating that expression of DAMPs by cancer cells is observed in the course of infection by different types of viruses, including measles and Coxsackie B viruses.Citation1
We and others have also reported that adenoviruses induce potent autophagy in host cells.Citation4,7 Although the roles of autophagy during adenoviral infection have yet to be completely defined, it is clear that the autophagic process is one of the main pathways by which viral antigens are processed. In support, the Epstein-Barr nuclear antigen of the Epstein-Barr virus has been previously shown to be processed predominantly by autophago-lysosomes.Citation8 Interestingly, while autophagy can be key for the processing of virus-derived antigens, our experiments showed that the immune proteasome participates in processing tumor-associated cellular antigens in adenovirus-infected cancer cells.
Collectively, these observations suggest that viral antigens are processed mainly through autophagy and that TAAs are processed by the immunoproteasome. If proven true, this separation of functions between the autophagy and immunoproteasome epitope-producing pathways would be of clinical significance, particularly if we aim to enhance the antitumor immunity vs. antivirus immunity in cancer patients treated with oncolytic viruses.
Although PAMPs mediate the immune response against pathogens and DAMPs mediate the recognition of self and tissue antigens,Citation6 PAMPs and DAMPs generated in the context of viral infection may not be rivals for triggering the immune response. In fact, there is evidence of the opposite, and it has been hypothesized that both signaling pathways cooperate to regulate the ultimate immune response of the host. This intriguing hypothesis is based on several facts. Firstly, PAMPs induced during infection may trigger the expression of DAMPs. Accordingly, infection by viruses or bacteria has been linked with the production of DAMPs.Citation6 Furthermore, if PAMPs and DAMPs are produced during the infection, components of both groups may physically interact, and these heterodimer complexes of proteins may influence the immune response.Citation6 In this regard, it has been proposed that the relative amount of PAMPS and DAMPs, as well as their interactions, may modulate the direction of the immune response.Citation6 Consistent with this concept, DAMPs and PAMPs can share the same Toll-like receptors.Citation6 Collectively, these observations buttress a model in which adenovirus infection of cancer cells triggers an antitumor immune response by the combined effect of PAMPs and DAMPs.
Our data and several other reports suggest that viruses, in addition to subverting immunogenic cell death,Citation9 may, under certain circumstances, induce this key type of cell death. Immunogenic cell death is characterized by the pre-apoptotic exposure of calreticulin and is thought to regulate the immune response to tumor cells observed during anticancer therapeutic strategies,Citation9 including treatment with oncolytic adenoviruses.Citation1 Thus, measles and Coxsackie B viruses have been shown to induce immune-stimulatory cell death in melanoma and lung cancer.Citation1 Importantly, combining immune modulators with adenoviruses may enhance virus-mediated immunogenic cell death. In this regard, viruses have been successfully combined with pharmacological treatments that trigger immunogenic cell death, such as mitoxantrone, temozolomide/cyclophosphamide, and bortezomib.Citation1 This combined therapy strategy may be more efficient than treatment with virus alone in terms of overcoming the deeply immunosuppressive environment of solid tumors.
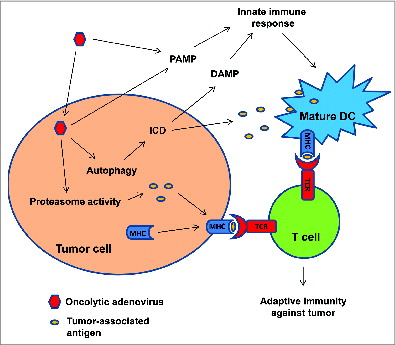
In summary, we have reported that infection of glioma cells with oncolytic adenoviruses triggers an immune response against cancer cells. We propose that this immune response is mediated by the cooperation of presentation of TAAs to T cells by infected tumor cells and dendritic cells stimulated by PAMP and DAMP signals, resulting in the prevention of the development of immune dominance and the development of an immune response exclusively directed against viral antigens (). We also speculate that the positive therapeutic effects of virotherapy can be enhanced by combining the oncolytic virus with treatments capable of inducing immunogenic cell death, including radiotherapy.Citation10
Figure 1. Delta-24-RGD elicits antitumor immunity during adenoviral-mediated cancer therapy. Adenoviral infection of the tumor cells increases the activity of proteasome and the presentation of tumor-associate antigens (TAAs) to T cells. Adenovirus also induces autophagic cell death which is immunogenic (immunogenic cell death, ICD), resulting in the release of DAMPs and TAAs. DAMPs, together with PAMPs from the viral infection, stimulate innate immune response and the presentation of TAAs to T cells by activated immune cells. Thus, adaptive immunity against tumor is instigated.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther 2014; 22:251-6; PMID:24048442; http://dx.doi.org/10.1038/mt.2013.220
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Yewdell JW. Confronting complexity: real-world immunodominance in antiviral CD8+ T cell responses. Immunity 2006; 25:533-43; PMID:17046682; http://dx.doi.org/10.1016/j.immuni.2006.09.005
- Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, Xu J, Kondo Y, Bekele BN, Colman H, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst 2007; 99:1410-14; PMID:17848677; http://dx.doi.org/10.1093/jnci/djm102
- Jiang H, Clise-Dwyer K, Ruisaard KE, Fan X, Tian W, Gumin J, Lamfers ML, Kleijn A, Lang FF, Yung WK, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One 2014; 9:e97407; PMID:24827739; http://dx.doi.org/10.1371/journal.pone.0097407
- Escamilla-Tilch M, Filio-Rodriguez G, Garcia-Rocha R, Mancilla-Herrera I, Mitchison NA, et al. The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol Cell Biol 2013; 91:601-10.
- Jiang H, White EJ, Rios-Vicil CI, Xu J, Gomez-Manzano C, Fueyo J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J Virol 2011; 85:4720-9; PMID:21367888; http://dx.doi.org/10.1128/JVI.02032-10
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Münz C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005; 307:593-6; PMID:15591165; http://dx.doi.org/10.1126/science.1104904
- Kepp O, Senovilla L, Galluzzi L, Panaretakis T, Tesniere A, Schlemmer F, Madeo F, Zitvogel L, Kroemer G. Viral subversion of immunogenic cell death. Cell Cycle 2009; 8:860-9; PMID:19221507; http://dx.doi.org/10.4161/cc.8.6.7939
- Galluzzi L, Kepp O, Kroemer G. Immunogenic cell death in radiation therapy. Oncoimmunology 2013; 2:e26536.