
Abstract
NF-κB is a master transcriptional regulator of inflammation that plays an important role in oncogenesis, particularly in tumors that arise in the context of inflammation. Copper metabolism MURR1 domain-containing 1 (COMMD1) is a negative regulator of NF-κB. Recent genetic-based studies in both mice and human patients indicate that COMMD1 has an important role in controlling intestinal inflammation and constraining progression to colitis-associated cancer.
Abbreviations:
- BMDM, bone marrow derived myeloid cells
- CAC, colitis-associated cancer
- COMMD1, copper metabolism MURR1 domain containing 1
- GWAS, genome wide association studies
- IBD, inflammatory bowel disease
- IκB, inhibitor of κB
- IKK, IκB kinase
- K/O, knockout
- LPS, lipopolysaccharide
- Mye-K/O, myeloid-specific Commd1 knockout
- NF-κB, nuclear factor-κB
- SNP, single nucleotide polymorphism
- WT, wild-type
Introduction
Colon cancer is the second most common malignancy in men and women in North America. Inflammatory bowel disease (IBD), a chronic condition characterized by persistent inflammation in the gastrointestinal tract, leads to increased risk of colon cancer, an entity that has been termed colitis-associated cancer (CAC). It is thought that genetic and environmental factors in affected individuals drive an alteration in the dynamic balance between inflammatory responses to microbial colonization and tissue protective mechanisms of the intestine, ultimately resulting in disease.Citation1 Increased expression of a variety of pro-inflammatory factors is central to tissue inflammation in IBD patients, and animal models indicate that several of these factors can also promote tumor growth. A common denominator to many of these cytokines is that their expression is dependent on nuclear factor-κB (NF-κB), a master transcriptional regulator of the inflammatory response that is also implicated in the progression to cancer.Citation2
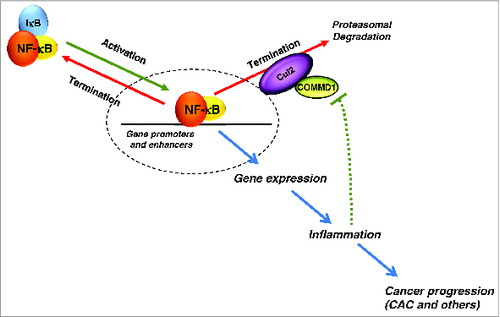
NF-κB activity is regulated by IκB, a constitutive inhibitor that is phosphorylated and degraded in response to a variety of pro-inflammatory and stress signals. Subsequently, NF-κB activity is terminated through a variety of mechanisms including the re-expression of IκB as well the induction of genes that limit IκB kinase activity, such as A20 and CYLD.Citation3 Another important regulatory process involves the ubiquitination and proteasomal degradation of the NF-κB subunit RelA (). Cul2 is the main ubiquitin ligase that targets RelA, and its activity is, in turn, dependent on copper metabolism MURR1 domain containing 1 (COMMD1).Citation4,5
Figure 1. The role of COMMD1 in NF-κB control and inflammation. Upon activation, NF-κB is released from NF-κB/IκB complex and translocates into nucleus to promote gene expression. Resynthesized IκB facilitates nuclear export of NF-κB to terminate the activation process. Unlike IκB, copper metabolism MURR1 domain containing 1 (COMMD1), along with the E3 ligase Cul2, targets chromatin-bound NF-κB for ubiquitination and proteasomal degradation. Inflammation can repress COMMD1 expression, establishing a positive feedback loop. Persistent NF-κB activation leads to inflammation and progression to colitis-associated cancer (CAC).
COMMD1 is a prototype of the COMM domain containing family, a group of 10 proteins that share a highly conserved C-terminal motif.Citation6 This domain serves as an interacting region that mediates COMMD protein dimerization and several other protein-protein interactions. Apart from its role in NF-κB regulation, COMMD1 is important for hypoxia responses, as well as copper and sodium transport. In view of these pleiotropic effects, it had remained unclear whether this gene contributed significantly to the pathophysiology of inflammation and cancer progression.
Commd1 deficiency leads to more robust pro-inflammatory responses
In order to address whether COMMD1 participates in the control of inflammation in vivo, we generated a mouse model of Commd1 deficiency, deleting this gene specifically in cells of myeloid lineage.Citation7 Bone marrow-derived myeloid cells (BMDM) from these Commd1−/− mice (termed (Mye-K/O) displayed altered expression of a substantial proportion of lipopolysaccharide (LPS)-regulated genes. Surprisingly, only about one third of Commd1 regulated genes were known NF-κB targets, suggesting that other pathways are also deregulated. In line with the effect of Commd1 on LPS-inducible genes, Mye-K/O mice were more sensitive to sepsis, exhibiting excess mortality and more robust pro-inflammatory cytokine release.
Role of COMMD1 in colitis and CAC
Next, we assessed the possible involvement of this gene in human diseases. In IBD patients, we found that COMMD1 expression was suppressed both in inflamed tissue and circulating white cells. This reduction in Commd1 expression was recapitulated in a murine model of colitis, indicating that this is an inducible phenomenon in response to tissue inflammation that likely represents a positive feedback loop that favors further inflammation (). In line with these observations, decreased COMMD1 expression has also been found in a variety of solid tumors, although a linkage to tumor inflammation has not been made.Citation8
To assess if COMMD1 could be further implicated in IBD pathogenesis we also examined genetic data from genome-wide associated studies (GWAS) in IBD and found a suggestive association between single nucleotide polymorphisms (SNPs) in the 3′ region of COMMD1 and ulcerative colitis risk. Interestingly, GWAS have already uncovered that Cul2, the ubiquitin ligase that along with COMMD1 promotes RelA ubiquitination, is linked to IBD risk.Citation9 The risk polymorphism near the COMMD1 gene is located over a putative enhancer and interestingly, is associated with reduced expression of this gene in the general population.
The physiologic significance of COMMD1 expression in the pathogenesis of colitis was subsequently assessed using mouse models. Acute colitis induced by dextran sodium sulfate (DSS) was more severe in Mye-K/O mice and was accompanied by increased pro-inflammatory gene expression. Similar findings were made using a chronic colitis model, with Mye-K/O mice again exhibiting worse disease and exaggerated pro-inflammatory cytokine expression. Moreover, in the context of chronic inflammation, myeloid cell deficiency of Commd1 led to increased progression to dysplasia, characterized by more frequent and more advanced dysplastic foci. In line with these findings, inflamed tissue in Mye-K/O mice that progressed to dysplasia also demonstrated increased activation of NF-κB. In contrast with the effects of myeloid cell deficiency, deletion of Commd1 in the intestinal epithelium did not affect disease course in any of these models. This latter observation is in line with a recent report examining models of liver inflammation where deletion in immune cells but not in hepatocytes affected inflammation severity.Citation10
Altogether, our findings indicate that COMMD1 plays an important role in limiting NF-κB activity in myeloid cells, and through these effects, modulates intestinal inflammation and the development of CAC (). The mechanism by which inflammation leads to suppressed expression of COMMD1 remains unclear but could have important implications to cancer progression in IBD and other inflammatory conditions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010; 28:573-621; PMID:20192811; http://dx.doi.org/10.1146/annurev-immunol-030409-101225
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKb links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004; 118:285-96; PMID:15294155; http://dx.doi.org/10.1016/j.cell.2004.07.013
- Hayden MS, Ghosh S. NF-kB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012; 26:203-34; PMID:22302935; http://dx.doi.org/10.1101/gad.183434.111
- Maine GN, Mao X, Komarck CM, Burstein E. COMMD1 promotes the ubiquitination of NF-kB subunits through a Cullin-containing ubiquitin ligase. EMBO J 2007; 26:436-47; PMID:17183367; http://dx.doi.org/10.1038/sj.emboj.7601489
- Mao X, Gluck N, Li D, Maine GN, Li H, Zaidi IW, Repaka A, Mayo MW, Burstein E. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kB/RelA. Genes Dev 2009; 23:849-61; PMID:19339690; http://dx.doi.org/10.1101/gad.1748409
- Burstein E, Hoberg JE, Wilkinson AS, Rumble JM, Csomos RA, Komarck CM, Maine GN, Wilkinson JC, Mayo MW, Duckett CS. COMMD proteins: A novel family of structural and functional homologs of MURR1. J Biol Chem 2005; 280:22222-32; PMID:15799966; http://dx.doi.org/10.1074/jbc.M501928200
- Li H, Chan L, Bartuzi P, Melton SD, Weber A, Ben-Shlomo S, Varol C, Raetz M, Mao X, Starokadomskyy P, et al. Copper Metabolism Domain-containing 1 Represses Genes that Promote Inflammation and Protects Mice From Colitis and Colitis-associated Cancer. Gastroenterology 2014; 147:184-195; PMID:24727021; http://dx.doi.org/10.1053/j.gastro.2014.04.007
- van de Sluis B, Mao X, Zhai Y, Groot AJ, Vermeulen JF, van der Wall E, van Diest PJ, Hofker MH, Wijmenga C, Klomp LW, et al. COMMD1 disrupts HIF-1a/b dimerization and inhibits human tumor cell invasion. J Clin Invest 2010; 120:2119-30; PMID:20458141; http://dx.doi.org/10.1172/JCI40583
- Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 2011; 43:1066-73; PMID:21983784; http://dx.doi.org/10.1038/ng.952
- Bartuzi PWT, Dekker D, Fedoseienko A, Kloosterhuis N, Youssef S, Li H, Shiri-Sverdlov R, Kuivenhoven J, de Bruin A, Burstein E, Hofker M, van de Sluis B. A cell-type-specific role for murine Commd1 in liver inflammation. Biochim Biophys Acta 2014; PMID:25072958; http://dx.doi.org/10.1016/j.bbadis.2014.06.035