
Abstract
We have previously shown that the development of a major histocompatibility complex class I (MHC-I)-deficient tumor was favored in protein kinase C-θ knockout (PKC-θ−/−) mice compared to that occurring in wild-type mice. This phenomenon was associated with scarce recruitment of natural killer (NK) cells to the tumor site, as well as impaired NK cell activation and reduced cytotoxicity ex vivo. Poly-inosinic:cytidylic acid (poly I:C) treatment activated PKC-θ in NK cells depending on the presence of a soluble factor produced by a different splenocyte subset. In the present work, we sought to analyze whether interleukin-15 (IL-15) and/or interferon-α (IFNα) mediate PKC-θ-dependent antitumor NK cell function. We found that IL-15 improves NK cell viability, granzyme B expression, degranulation capacity and interferon-γ (IFNγ) secretion independently of PKC-θ. In contrast, we found that IFNα improves the degranulation capability of NK cells against target cancer cells in a PKC-θ-dependent fashion both ex vivo and in vivo. Furthermore, IFNα induces PKC-θ auto-phosphorylation in NK cells, in a signal transduction pathway involving both phosphatidylinositol-3-kinase (PI3K) and phospholipase-C (PLC) activation. PKC-θ dependence was further implicated in IFNα-induced transcriptional upregulation of chemokine (C-X-C motif) ligand 10 (CXCL10), a signal transducer and activator of transcription-1 (STAT-1)-dependent target of IFNα. The absence of PKC-θ did not affect IFNα-induced STAT-1 Tyr701 phosphorylation but affected the increase in STAT-1 phosphorylation on Ser727, attenuating CXCL10 secretion. This connection between IFNα and PKC-θ in NK cells may be exploited in NK cell-based tumor immunotherapy.
Keywords:
Abbreviations:
- CDK8, cyclin-dependent kinase 8
- CXCL10, (C-X-C motif) ligand 10/CXCL10
- FCS, fetal calf serum
- IFNα, interferon-α
- IFNA1
- IFNγ, interferon-γ, IFNG
- IL-15, interleukin-15/IL15
- mAb, monoclonal antibody
- MACS, magnetic cell separation
- MEF, murine embryonic fibroblast
- MHC-I, major histocompability complex class I/MHC-I
- NK, natural killer
- PI3K, phosphatidylinositol-3-kinase
- PKC-θ, protein kinase C-θ, PRKCQ
- PLC, phospholipase-C
- Poly I:C, poly-inosinic:cytidilic acid
- RT-PCR, real-time polymerase chain reaction
- STAT-1, signal transducer and activator of transcription-1/STAT1.
Introduction
The serine threonine specific protein kinase C-θ (PKC-θ) was initially isolated and characterized as a PKC isoenzyme expressed in T cells.Citation1 In this context, PKC-θ signal transduction downstream of the T cell receptor (TCR) triggers key transcription factors essential for T-cell activation such as activating protein-1 (AP-1, also known as JUN ), nuclear factor of k-light chain polypeptide gene enhancer in B cells (NF-κB) and nuclear factor of activated T cells (NF-AT).Citation2-6 However, PKC-θ expression is not exclusive to T cells.Citation7
The cancer immunosurveillance hypothesis proposes that the immune system detects and eliminates neoplastic cells undergoing malignant transformation and that the adaptive immune system is able to maintain small cancer lesions in an equilibrium, i.e., in a growth constricted state.Citation8-12 PKC-θ is essential for this immune function at least in the control of leukemia as evinced by prior findings demonstrating that PKC-θ-deficient mice exhibit increased lymphoproliferative disease incidence and onset.Citation13 This response presumably involved cytotoxic T lymphocyte (CTL) function, considering that these leukemic cancer cells express major histocompatibility complex class I (MHC-I).
Cells of the innate immune system, including γδ T cells, natural killer (NK) cells and natural killer T (NKT) cells, can also mediate anticancer immune responses. Specifically, NK cells play an important role in tumor immunosurveillance,Citation14-16 as they have been observed to control the progression of MHC-I-deficient tumors via perforin/granzyme- and FasL-mediated cytotoxicity.Citation17-19 Interestingly, we have previously shown that PKC-θ is required for FasL-expression in T cells, although not for CTL degranulation.Citation20,21 Since most cancer cells down-regulate MHC-I expression to escape the CTL-mediated cytotoxicityCitation22 it is important to evaluate further the role of NK cells in mediating antitumor immunity.
We have previously shown that development of a MHC-I-deficient tumor (RMA-S) is favored in PKC-θ−/− mice as compared to that occurring in wild-type (wt) mice.Citation23,24 This pathophysiological condition was found to be associated with a deficient recruitment of NK cells to the tumor site in PKC-θ−/− mice, correlating with impaired activation of recruited NK cells. Furthermore, in vitro testing showed that NK cells isolated from PKC-θ−/− mice exhibited reduced cytotoxicity against leukemic RMA-S cells after poly-inosinic:cytidiylic acid (poly I:C) treatment ex vivo.Citation23 We also observed that poly I:C treatment increased the relative expression levels and the activation status of PKC-θ in NK cells, both in vivo and in vitro. In the latter scenario, the presence of the complete splenocyte immune population was apparently needed, suggesting that the effect is presumably partially mediated by macrophages and/or dendritic cells.Citation23 Our finding is in accord with previous studies demonstrating a NK cell-dependent increase in anticancer activity of NK cells responding to poly I:C stimulation.Citation25 PKC-θ activation in this context is presumably mediated by soluble factor(s), which could be cytokine(s) secreted by macrophages and (or) dendritic cells (DCs) that are known to activate NK cells.Citation24 Thus, we have set out to define factors mediating NK activation and tumoricidal activity through PKC-θ. Since it has been previously reported that interleukin-12 (IL-12) signal transduction is affected in NK cells derived from PKC-θ−/− mice,Citation26 this cytokine was tested first. Interleukin-15 (IL-15) was also included in the mentioned studies, since it is important in regulating NK cell function and survival,Citation27,28 and for efficient antitumor NK cell activity.Citation29 Indeed, we reported that both, IL-12 and IL-15 activated PKC-θ in NK cells, with IL-15 being more potent at inducing PKC-θ phosphorylation. More importantly, in a mixed splenocyte culture stimulated ex vivo with poly I:C, neutralizing antibodies against IL-15 substantially reduced NK cell PKC-θ phosphorylation, whereas IL-12 antibody blockade was ineffective.Citation23 Therefore, IL-15 appeared to be the most feasible candidate to mediate PKC-θ-dependent antitumor NK cell immune function.24 In the present study, we initially set out to test this possibility, testing IL-15 in regards to PKC-θ activation status and NK cell immunophenotypes. Contrary to our expectations, our results implicate interferon-α (IFNα) as the principal cytokine that signals through PKC-θ in NK cells and, as a consequence of downstream trancriptional changes, is primarily responsible for PKC-θ-dependent NK cell anticancer immunity.Citation
Results
PKC-θ in IFNα and IL-15 effect on survival and immune function of NK cells
Our previous studies suggested that IL-15 could be the main cytokine responsible for the PKC-θ-dependent antitumor function of NK cells.23 In order to evaluate the necessity for PKC-θ-mediated signal transduction in a particular NK cell biological process, we comparatively analyzed IFNα and IL-15 responses in NK cells derived from PKC-θ−/− versus wt animals. As shown in , using an Annexin V externalization assay, we found that IL-15 is crucial for NK cell survival as although the majority (∼70%) of isolated murine NK cells were Annexin V positive within the first 24 h in culture, this programmed cell death was almost completely abolished by inclusion of IL-15 in the cultures. However, this effect was found to be independent of PKC-θ, since it was equally achieved in NK cells from wt or PKC−θ−/− mice. IFNα also seemed to improve survival, although less efficiently than IL-15 and also in a PKC-θ-independent manner. IL-15 also induced interferon-γ (IFNγ) production in purified NK cells in a PKC-θ independent fashion, whereas IFNα had no effect ().
Figure 1. Dependence on PKC-θ for IL-15 and IFNα-induced NK cell survival and immune function. (A–D) Natural killer (NK) cells derived from C57BL6 mice null for protein kinase C-θ (PKC-θ−/−) vs. wild-type animals (PKC-θ+/+) were functionally characterized for responses to interleukin 15 (IL-15) and interferon-α (IFNα). NK cells were isolated by magnetic cell separation (MACS) technology and cultured in complete medium at 2 × 106 cells/mL in the absence (−) or in the presence of the indicated concentrations of IL-15 or IFNα for 24 h. Afterwards, cells were labeled with an anti-NK1.1 mAb labeled with phycoerythrin (PE) to restrict the cytofluorimetric analysis to NK cells. (A) Cell death was determined by surface staining with Annexin-V labeled with fluorescein isothiocyanate (FITC) and flow cytometry. (B and C) After 24 h cytokine stimulation, NK cells were incubated with YAC-1 cells at a 1:1 ratio during 6h in the presence of monensin and the production of interferon-γ (IFNγ) (B) and degranulation (C) was tested by flow cytometry. IFNγ was detected by intracellular staining with a specific anti-IFNγ antibody labeled with FITC and degranulation was determined by analysis of CD107a surface expression using an anti-CD107a antibody labeled with FITC. (D) The content of granzyme B was analyzed by permeabilization and intracellular staining with a specific anti-granzyme B antibody detected using an FITC-conjugated secondary and flow cytometry. Data are the mean ± SD of 2 independent experiments performed in triplicate; statistical analyses were performed by Student's t test: *P < 0.05; **P < 0.02.
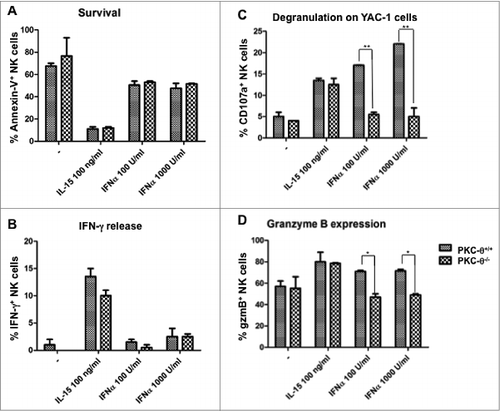
As shown in , IL-15 improved NK cell degranulation when co-cultured with YAC-1 target cells as measured by an increase in the percentage of NK cells expressing CD107a, but this effect was again PKC-θ-independent. In sharp contrast, IFNα increased degranulation against YAC-1 cells to a higher magnitude, and this was entirely dependent upon PKC-θ expression, since this immunity-related biological process was abolished in NK cells from PKC-θ−/− mice (). Furthermore, although both IL-15 and IFNα modestly increased granzyme B expression in NK cells from wt mice over the already high basal expression level characteristic of spleen NK cells,23 this increase was dependent on PKC-θ only in the case of IFNα (). In sum, these experiments show that although IL-15 is important to maintain NK cell viability and in the induction of IFNγ secretion, these immune functions were independent of PKC-θ. On the other hand, our findings are the first to provide evidence that the increase in NK cell cytotoxic potential induced by IFNα is dependent on PKC-θ, with implications in the antitumor function of these molecules.
IFNα-mediated NK cell activation in vivo depends on PKC-θ
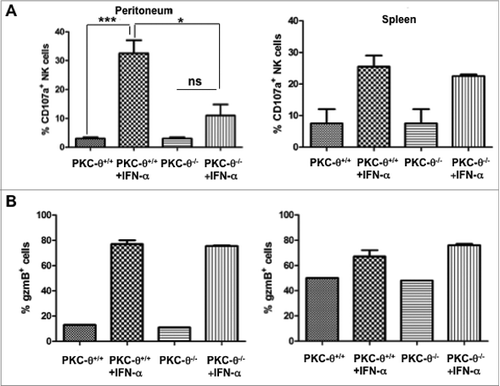
We next set out to determine the physiological dependence of IFNα-induced increase of NK cell cytotoxic potential by stimulating NK cells with IFNα in vivo. To this end, we injected 10,000 IU of IFNα intraperitoneally into wt or PKC-θ−/− mice and, 24 h later, obtained purified peritoneal or splenic NK cells, and assayed NK cell degranulation (as measured by expression of 107a) against YAC-1 cells and the percentage of NK cells expressing granzyme B. We found that injection of IFNα increased the cytotoxic potential of peritoneal or splenic NK cells against YAC-1 cells (). This effect was significantly (p-value <0.05) reduced in peritoneal NK cells obtained from PKC-θ−/− mice, confirming the in vitro result and implicating PKC-θ−/− as a key mediator of NK cell immune responses to IFNα. However, despite our findings using NK cells from the peritoneum, no significant difference was observed using splenic NK cells. In vivo injection of IFNα also resulted in a net increase in the expression of granzyme B, especially in peritoneal NK cells (), but this effect was independent of PKC-θ expression, since granzyme B was observed in a similar fraction of NK cells derived from PKC-θ−/− mice.
Figure 2. Dependence on PKC-θ for IFNα-induced NK cell immune function in vivo. (A and B) Protein kinase C-θ knockout (PKC-θ−/−) or wild-type (PKC-θ+/+) C57BL/6 mice were injected intraperitoneally with 10,000 IU/mL of interferon-α (IFNα) or the same volume of PBS. After 24 h, mice were sacrificed, and natural killer (NK) cells were magnetically isolated by MACS technology either from total spleen cells or cells derived from flushing the peritoneum. NK cells were functionally characterized following a 6 h stimulation with YAC-1 target cells at a 1:1 ratio, immunofluorescence staining and cytofluorimetric analysis. (A) The degranulation potential of isolated NK cells against YAC-1 cells was estimated using a fluorophore-conjugated anti-CD107a antibody and flow cytometry. (B) The granzyme B content of YAC-1 stimulated NK cells was estimated via immunofluorescence staining and flow cytometry. Data are the mean ± SD of assaying NK cells derived from 3 different mice in each experimental condition; statistical analyses were performed by Student's t test; *P < 0.05; ***P < 0.01; ns: difference not statistically significant.
IFNα activates PKC-θ in NK cells through a PI3K- and PLC-dependent pathway
As shown in , IFNα treatment of splenic NK cells from wt mice induced a rapid, efficient and sustained auto-phosphorylation of PKC-θ on Ser676, thus indicating enzymatic activation of the kinase. In contrast, the protein expression level of PKC-θ was not affected ().
Figure 3. IFNα-induced PKC-θ activation in NK cells. (A) Natural killer (NK) cells were magnetically isolated by MACS technology from spleens of wild-type (wt) C57BL/6 mice and cultured in complete medium at 2 × 106 cells/mL in the absence (−) or presence (+) of the indicated concentrations of interferon-α (IFNα) for 30 min (left panel), or in the presence of 100 IU/mL of IFNα for the indicated periods of time (right panel). (B and C) Isolated NK cells from wt C57BL/6 mouse spleens were pre-incubated for 30 min in the absence (control) or in the presence of either 10 μM U73122 or 100 nM of wortmannin or rapamycin, as indicated. Afterwards, 100 IU/mL of IFNα were added (+) or not (−) and cells were incubated in the presence of the indicated inhibitors for an additional 30 min. (B) PKC-θ phosphorylation in Ser676 determined by immunoblot. Total PKC-θ and/or β-actin expression was determined in parallel by immunoblot as loading controls. (C) The phospho-PKC-θ/β-actin ratios were determined by densitometry and results expressed as the increase in this ratio. Data shown are the mean ± SD of at least 2 different experiments for each condition.
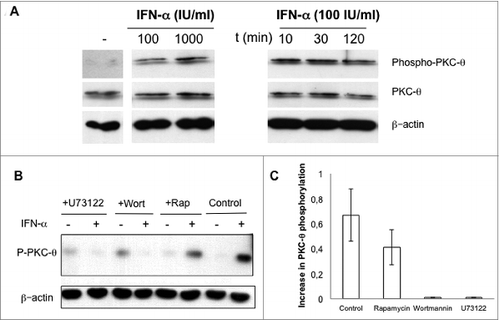
We next sought to identify the signal transduction pathway that connects IFNα to PKC-θ. Interestingly, PKC-θ is the only T-cell expressed PKC isoform that is activated through a PI3K-dependent pathway triggered by TCR ligation.30 Although the main signaling pathway elicited by cytokine receptors is mediated by Janus kinases (JAKs) and signal transducer and activator of transcription (STATs), cytokines such as IFNα, IL-2 and IL-15 also activate the phosphatidylinositol-3-kinase (PI3K) pathway in B and T cells.31-34 IFNα is also known to activate phospholipase-C (PLC) signaling cascades.35 Hence, we tested the ability of wortmannin, a PI3K inhibitor, and U73122, a PLC inhibitor, or rapamycin, a mammalian target of rapamycin (mTOR) inhibitor, to attenuate PKC-θ activation induced by IFNα in NK cells. As shown in , both wortmannin and U73122 abrogated the increase in PKC-θ auto phosphorylation induced by IFNα in NK cells, while rapamycin only partially inhibited this process.
The induction of some IFNα−target genes in NK cells is PKC-θ-dependent
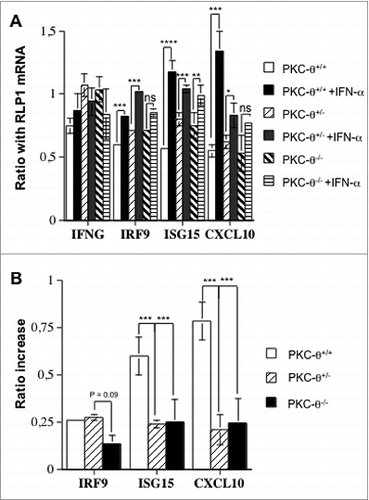
We next set out to analyze if transcription of IFNα-responsive genes was affected in NK cells from PKC-θ−/− mice. We used quantitative reverse transcription polymerase chain reaction (qRT-PCR) to assay the transcriptional levels of interferon-regulatory factor 9 (IRF-9) and interferon-stimulated 15 kDa protein (ISG15), 2 IFNα-responsive genes that have been previously reported to be dependent upon PKC-θ signaling in T cells,36 as well as the mRNA levels of (C-X-C motif) ligand 10 (CXCL10), a candidate IFNα-responsive gene dependent on STAT1.37 We used interferon-γ (IFNγ) as a control gene that is not affected by IFNα in NK cells. As shown in , IFNα treatment significantly increased the transcription of IRF9 in NK cells obtained from either wt or heterozygotic PKC-θ+/− mice, but not in those purified from PKC-θ−/− mice. ISG15 transcription was significantly increased in NK cells from all 3 genetic backgrounds, although to a different extent (). Finally, CXCL10 transcript levels were substantially increased in NK cells from wt mice, reduced in heterozygotic PKC-θ+/− mice, and abolished in NK cells from PKC-θ−/− mice (). On the contrary, we did not detect a significant increase in the transcription of IFNγ in response to treatment with IFNα, demonstrating specificity in transcriptional responses. shows the increase in the ratio of gene expression of each experimental transcript relative to the transcript levels of Rab interacting lysosomal protein-like 1 (RILPL1, also known as RLP1), a stable and abundant mRNA, upon IFNα treatment in the same experiments. The IFNα−dependent increase in the ratio was moderate for IRF9 and equal in NK cells obtained from either wt or heterozygotic PKC-θ+/− mice, but decreased by more than 50% in NK cells from PKC-θ−/− mice, although this difference was not statistically significant (P-value>0.05). In comparison, the relative increase in the transcript levels of ISG15 was higher than that of IRF9, an increase significantly inhibited in NK cells derived from either PKC-θ+/− or PKC-θ−/− mice. Finally, the highest magnitude of IFNα-induced transcriptional increase corresponded to CXCL10, a molecular phenotype significantly inhibited in NK cells from either PKC-θ+/− and PKC-θ−/− mice. The fact that IRF9 gene expression was not affected in NK cells from PKC-θ+/−, whereas ISG15 and CXCL10 mRNA levels were significantly affected by PKC-θ deficiency, suggests a dosage effect at promoters, some of which require full activation of the transcription factors involved. Our data confirmed that IFNα-mediated IRF9 and ISG15 expression is dependent upon PKC-θ in NK cells, as previously reported in T cells.36 However, our data showing a dependency on PKC-θ for the IFNα-mediated induction of CXCL10 gene expression are new and intriguing.
Figure 4. PKC-θ-dependent transcriptional changes in NK cells induced by IFNα. (A and B) Spleens of wild-type (wt), protein kinase C-θ null heterozygotes (PKC-θ+/−) or knockout (PKC-θ−/−) C57BL/6 mice were used to magnetically (MACS) isolate 10 × 106 natural killer (NK) cells. NK cells from each group were cultured in complete medium for 90 min with or without 100 IU/mL of IFNα, as indicated. RNA was extracted, reverse transcribed and cDNAs amplified via real time qPCR. (A) mRNA expression level of interferon-γ (IFNG) interferon regulatory factor 9 (IRF-9), ISG15 ubiquitin-like modifier (ISG15) or chemokine (C-X-C motif) ligand 10 (CXCL10) as a ratio of the reference RLP1 mRNA level used for standardization. (B) qPCR results from (A) shown as the increase in the ratio of the indicated mRNA level expressed in control cells (untreated) versus cells treated with IFNα. Results are the mean ± SD of duplicate determinations in 2 independent experiments; statistical analyses were performed by Student's t test; *P < 0.05; **P< 0.02; ***P < 0.01; ns: difference not statistically significant.
Reduced IFNα-induced STAT-1 phosphorylation in Ser727 and CXCL10 secretion in NK cells derived from PKC-θ−/− mice
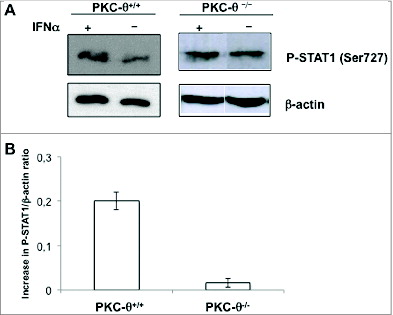
Our observation that IFNα stimulated CXCL10 transcription is impaired in NK cells from PKC-θ−/− mice suggested that PKC-θ is involved in STAT1 signaling. IFNα readily induced STAT1 phosphorylation at Tyr701 in NK cells obtained from the spleens of wild-type mice. However, the phospho-STAT1 level was only slightly reduced in splenic NK cells derived from PKC-θ−/− mice. Considering that STAT1 phosphorylation depends on the direct action of JAK1,38 PKC-θ presumably affects the STAT1 pathway downstream of JAK1-mediated tyrosine phosphorylation. Although tyrosine phosphorylation is indispensable for the transcriptional activity of STAT1, complete transcriptional capacity has been ascribed to additional phosphorylation events, including serine phosphorylation. For example, in macrophages, upon IFNγ treatment, PKC-α has been shown to phosphorylate STAT1 on Ser727, generating a fully functional STAT1 at the transcription level.39 Furthermore, PKC-δ, a close homolog of PKC-θ, has been shown to be able to associate with STAT1 and mediate IFNγ-induced STAT1 phosphorylation in a promyelocytic cell line.40 Interestingly, rotlerin, the PKC-δ inhibitor used in that study, also inhibits PKC-θ.20,21 Hence, we next tested whether IFNα treatment increased STAT-1 phosphorylation on Ser727 in NK cells. As shown in , 30 min of exposure to IFNα increased STAT1 phosphorylation on Ser727 in NK cells from wt mice. In contrast, NK cells from PKC-θ−/− mice showed basal Ser727 phosphorylation that did not increase upon IFNα treatment ().
Figure 5. IFNα-induced STAT1 phosphorylation at Tyr701 in NK cells is independent of PKC-θ. (A and B) Natural killer (NK) cells were isolated by MACS technology from spleens of wild type (PKC-θ+/+) or protein kinase C-θ (PKC-θ−/−) knockout C57BL/6 mice and cultured in complete medium at 2 × 106 cells/mL in the absence or presence of the indicated concentrations of interferon-α (IFNα) for 30 min. Reactions were stopped in the cold, cells lysed, protein extracts prepared and signal transducer and activator of transcription-1 (STAT1) phosphorylation at residue Tyr701 determined by phospho-STAT1 (P-STAT1) specific immunoblot. β-actin expression was determined in parallel as loading control. (B) The phospho-STAT1/β-actin ratios of the experiment shown in (A) were determined by densitometry and represented as a bar diagram. Data shown are representative of 2 independent experiments.
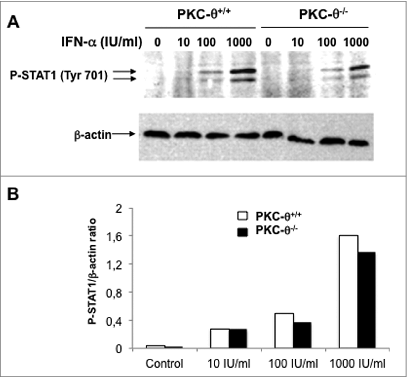
Figure 6. IFNα-mediated STAT-1 phosphorylation on Ser727 is partially dependent upon PKC-θ in NK cells. (A and B) Natural killer (NK) cells were isolated by MACS technology from spleens of wt (PKC-θ+/+) or protein kinase C-θ knockout (PKC-θ−/−) C57BL/6 mice and cultured in complete medium at 2 × 106 cells/mL in the absence (−) or in the presence (+) of 100 IU/mL of IFNα for 30 min. (A) Reactions were stopped in the cold, cells lysed, protein extracts prepared and STAT1 phosphorylation in Ser727 determined by immunoblot. β-actin expression was determined in parallel by immunoblot as loading control. (B) The phospho-STAT1/β-actin ratios were determined by densitometry and results expressed as the increase in this ratio between control cells and cells treated with IFNα in each case. Data shown are the mean ± SD of at least 2 different experiments for each condition.
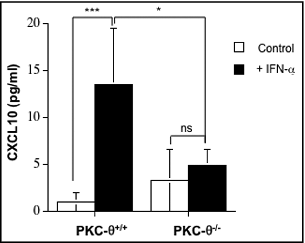
Finally, considering that IFNα induced transcription of CXCL10 through PKC-θ in NK cells, we next sought to ascertain the PKC-θ−dependent effects of IFNα on CXCL10 protein levels. We found that IFNα induced a significant increase in CXCL10 secretion by NK cells, which was statistically decreased in NK cells derived from PKC-θ−/− mice (). Taken together, these data elucidate a new signal transduction pathway in NK cells, connecting IFNα signaling with STAT-1-regulated genes through PKC-θ enzymatic activity.
Figure 7. IFNα-induced CXCL10 secretion from NK cells is dependent on PKC-θ. Natural killer (NK) cells were isolated by MACS technology from spleens of wild type (PKC-θ+/+) or protein kinase C-θ knockout (PKC-θ−/−) C57BL/6 mice and cultured in complete medium at 5 × 106 cells/mL in the absence (control) or in the presence (+IFNα) of 100 IU/mL of IFNα for 8 h. After this time, cell culture supernatants were collected and the presence of CXCL10 detected using an ELISA kit. Data are the mean ± SD of 3 different determinations made in duplicate; statistical analyses were performed by Student's t test; *P <0 .05; ***P <0 .01; ns: difference not statistically significant.
Discussion
Our previous studies have shown that PKC-θ is essential for the antitumor function of NK cells.23 We also previously showed that PKC-θ activation in this context is mediated by soluble factor(s), probably cytokines secreted by macrophages and (or) DCs that are known to activate NK cells.23,24 Thus, we prospectively evaluated whether IL-12 or IL-15 could be among the key cytokines activating PKC-θ signaling cascades. In this prior study, we found that although both IL-12 and IL-15 efficiently activated PKC-θ in NK cells, only neutralizing antibodies to IL-15 reduced PKC-θ auto-phosphorylation induced in vitro by poly I:C in the context of a mixed splenocyte population.23 In the present study, we initially tested the possibility that IL-15 could be the main cytokine controlling these responses. Although we confirmed our prior data indicating that IL-15 is essential for NK cell survival and for the secretion of IFNγ, we report here that these functions are actually PKC-θ independent.
Unexpectedly, we have found that the main DC-derived cytokine whose signaling is dependent upon the presence of PKC-θ in NK cells, and further is most likely responsible for PKC-θ-dependent NK-cell mediated antitumor activity is rather IFNα. This interpretation is consistent with the increased sensitivity of IFNAR−/− or PKC-θ−/− mice to the development of NK cell-sensitive tumors.23,41 We have shown that IFNα achieves this immune function by fostering at least 3 different responses in NK cells in a PKC-θ-dependent manner, including: i) augmenting the degranulation capacity of NK cells; ii) contributing to the increase in granzyme B expression in activated NK cells; and iii) inducing CXCL10 secretion from activated NK cells.
The augmentation of the degranulation capacity of NK cells was demonstrated by treating isolated splenic NK cells in vitro with IFNα, or alternatively, by activating NK cells in vivo by injecting IFNα, followed by assaying their degranulation responses to YAC-1 cells. In either scenario, IFNα effects were strictly dependent upon PKC-θ, since NK cells from PKC-θ−/− mice did not increase their basal degranulation potential. In the case of the in vivo activation, this was only observed in peritoneal NK cells, probably reflecting the basal activated phenotype of splenic NK cells, especially against YAC-1 cells, target cells that are extremely sensitive to NK cell cytolysis.17,23
It has been previously reported that PKC-θ clusters at the NK cell immunological synapse, potentially amplifying effector responses, and possibly regulating NK cell degranulation.42 However, in this study the inhibition of degranulation was only partial in NK cells from PKC-θ−/− mice as compared with wild type mice (41.8% vs. 64.5%), and there were no effects observed on the cytotoxicity against YAC-1 or BaF3 cells transfected with KAR ligands. However, IFNγ secretion was affected,42 as has been previously described.43
Our group,21 and others,44,45 have demonstrated that PKC-θ is dispensable for degranulation induced by the TCR in mature cytotoxic T lymphocyte (CTL) clones and primary T cells, and further, that degranulation is not affected in CTLs from PKC-θ−/− mice. However, the induction of FasL expression in these cells is strictly dependent on PKC-θ.20,21,30
We have shown previously that basal NK cell degranulation or cytotoxicity against YAC-1 cells was not affected using freshly isolated splenic NK cells from PKC-θ−/− mice,23 in agreement with previous reports.43 However, YAC-1 cells are extremely sensitive to NK cells and unable to generate tumors in either syngeneic wt or PKC-θ−/− mice. On the other hand, degranulation or cytotoxicity against RMA-S cells has been shown to be reduced in NK cells obtained from PKC-θ−/− mice, in agreement with the increased tumorigenic potential of RMA-S cells in these knockout mice.23 Moreover, in this system, splenic NK cells must be previously activated by poly I:C to demonstrate cytotoxicity or degranulation potential against RMA-S cells, in contrast to unprimed NK cell activity against YAC-1 cells. Altogether, these data suggest that PKC-θ is a necessary constituent of in vivo activation of the degranulation potential of NK cells responding to poly I:C, or alternatively, endogenous tumor danger signals. Further, PKC-θ does not appear to be essential to the degranulation process itself, although it could amplify this response, as previously suggested.42 The present data also suggest that one of the PKC-θ dependent mechanisms by which poly I:C or danger signals activate NK cells in vivo is via the secretion of IFNα by DC or macrophages.24
Granzyme B is the archetypical effector molecule of CTL and NK cells.46 Granzyme B mRNA is abundant in NK cells and cytokine stimulation such as that meted by IL-15 and IFNα affects a net increase in protein content,47 a response independently confirmed here. Furthermore, we also found that the intracellular content of granzyme B was lower in IFNα−activated NK cells derived from PKC-θ−/− mice than from those of wt mice. In vitro, IFNα action is restricted to NK cells, and in this case the increase in granzyme B is PKC-θ-dependent.
These differences are not observed in vivo, in which IFNα injection readily induced an increase in granzyme B expression, especially in peritoneal NK cells, irrespective of PKC-θ status. This indicates that in vivo, other cellular and molecular mechanisms independent of PKC-θ contribute to the observed increase in granzyme B expression. IFNα could induce the secretion of several other cytokines from other cell populations that induce granzyme B expression in NK cells in a PKC-θ-independent fashion. Indeed, it has been demonstrated that Type I IFN induces IL-15 secretion from activated DCs, and that DCs, through cross-presentation and IL-15 signaling, can thus induce NK cell priming.48
CXCL10 is a chemokine involved in NK cell recruitment to inflamed tissues and sites of tumor development.49-51 In fact, it has recently been demonstrated that the efficacy of adenovirus-engineered dendritic cells as a cancer vaccine correlates with their ability to secrete CXCL10 and CXCL8 and to efficiently recruit antitumor NK cells.51 CXCL10 secretion has been, so far, primarily attributed to DCs and monocytes/macrophages.50,51 Our data indicate that NK cells also secrete CXCL10 upon IFNα treatment, albeit at a lower concentrations than DCs and macrophages. Furthermore, in a recent transcriptomic analysis we found that NK cell activation by target cells in the presence of whole peripheral blood mononuclear cells (PBMCs) induces the expression of CXCL10, as well as its receptor CXCR3, suggesting the presence of an autocrine migration loop occurring during NK cell activation.52 The results obtained here suggest that IFNα produced by DC and macrophages/monocytes present in the PBMC population is contributing at least to the upregulation of this chemokine in NK cells. Previously, we demonstrated that recruitment of NK cells to the tumor site was impaired in PKC-θ−/− mice, thus contributing to their increased sensitivity to tumor development. Hence, this defective response could be partly explained by the defective upregulation of CXCL10 by NK cells that we have uncovered here.
This observation has shed light on a new NK cell signal-transduction pathway connecting the IFNα receptor and PKC-θ activation, and further encompassed both PI3K and PLC activities, similar to that observed in T cells and reported by Srivastava et al. 36 PKC-θ is the only T cell expressed PKC isoform known to be activated through a PI3K-dependent pathway after TCR ligation.30 However, in T cells PLC does not seem to play a primary role in PKC-θ activation by TCR ligation.30 Therefore, unlike the route in T cells, it appears as though in IFNα-activated NK cells, both pathways should be activated to guarantee an efficient PKC-θ activation. The partial effect of the mTOR inhibitor rapamycin could be explained since this pathway is downstream of PI3K. These data are also in accord with PLC and PI3K activation in response to IFNα in other cellular contexts as well.34,35
Although the impaired transcription of ISG15 and IRF9 in PKC-θ deficient NK cells responding to IFNα is concordant with prior observations in the context of T cells.36 However, the dependence on PKC-θ of IFN-α-mediated CXCL10 transcription and secretion is novel. CXCL10 is a archetypical STAT1-dependent IFN-responsive gene.37 In agreement with observations in T cells,36 the main post-translational modification responsible for the activation of the transcriptional activity of STAT1, specifically JAK1-mediated phosphorylation at Tyr701, is intact in NK cells arising in PKC-θ−/− mice. Nevertheless, our data show a partially defective IFNα-mediated phosphorylation of STAT1 at Ser727, an alternative regulatory site in these cells. This post-translational modification may be a required signal transduction event for efficient CXCL10 transcription and secretion.
Recently, it has been reported that the main kinase mediating STAT1 Ser727 phosphorylation in macrophages and murine embryonic fibroblasts (MEFs) is cyclin-dependent kinase 8 (CDK8).53 CDK8 has been implicated as a STAT1 Ser727 phosphorylating enzyme in NK cells as well,54 although in this case the enzymatic activity seems rather related with the presence of a basal level of Ser727 STAT1 phosphorylation. With these observations in mind, the basal phosphorylation status of STAT1 in freshly isolated NK cells from both wt and PKC-θ−/− mice reported here is likely attributable to CDK8.
We further found that this phosphorylation increases in NK cells from wt but not from PKC-θ−/− mice treated with IFNα. NK cells from mice expressing a mutant STAT1 recalcitrant to phosphorylation in Ser727 have been previously shown to exhibit higher cytotoxicity and granzyme B expression.54 Considering that granzyme B expression has been shown to be regulated by a post-transcriptional mechanism in NK cells,47 this effect of mutant STAT1 may relate to an unknown, non-transcriptional role of STAT1. In MEFs expressing mutant STAT1 and stimulated with IFNγ the transcription of CXCL10 is augmented rather than diminished.53 Our data have been obtained in NK cells stimulated with IFNα, and the situation is quite distinct, since CDK8-mediated basal STAT1 phosphorylation is present, and the role of PKC-θ seems rather to enhance Ser727 phosphorylation, resulting in an increase in CXCL10 gene transcription. It is likely that phosphorylation on STAT1 Ser727 induces different responses in specific cellular contexts.
In summary, the present study uncovers the connection between IFNα and PKC-θ in NK cells and point to IFNα and PKC-θ-dependent signaling pathways as crucial regulators of NK cell function that can be exploited in NK cell-based tumor immunotherapy.
Materials and Methods
Mice
PKC-θ−/− mice in the C57/BL6 background were a generous gift from Dr. D. Littman 6 and were housed under the same conditions as the C57/BL6 parental (wt) mice. All experiments involving animals were performed according to the guidelines and regulations of the Center National de la Recherche Scientifique, France. Authors have official degrees for performing animal experimentation delivered by corresponding authorities in Spain or France, respectively.
Tumor cell lines
YAC-1 is a Moloney murine leukemia virus-induced lymphoma of the H-2Kb haplotype that lacks MHC-I expression and is sensitive to lysis by NK cells.55 Cell lines were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS), Glutamax and antibiotics (complete medium).
In vitro analysis of the effect of IFN-α and IL-15 on functional parameters in isolated NK cells
NK cells were isolated from the spleens of C57/BL6 wt or PKC-θ−/− mice by magnetic cell separation (MACS) technology, using magnetic beads coated with anti-CD49b monoclonal antibody (mAb), as indicated previously.23 NK cells were cultured in complete medium. Briefly, NK cells were isolated by positive selection using LS columns and anti-CD49b magnetic microbeads (Myltenyi Biotec, Madrid) from spleens of wild-type and PKC-θ−/− C57BL/6 mice. Isolated NK cells were cultured for 24 h in this complete medium, supplemented with or without different concentrations of IL-15 or IFNα, both from Myltenyi Biotec, Madrid. Then, several functional assays were performed via marker staining and cytofluorimetric analyses using a FACScan (BD Biosciences, Madrid) flow cytometer as follows:
Viability was determined by trypan blue staining and labeling with fluorescein isothiocyanate (FITC) conjugated Annexin-V and flow cytometry.
IFNγ expression was determined by intracellular staining with an anti-IFNγ rat monoclonal antibody (mAb) conjugated to FITC (BD, Madrid), as previously indicated.56 In brief, NK cells were mixed at 1:1 ratio for 6 h with activating YAC-1 cells in the presence of monensin (GolgiStop reagent) to inhibit vesicular trafficking, thereby attenuating IFNγ secretion and favoring its intracellular accumulation. At the end of the incubation, cells were washed, fixed with 1% paraformaldehyde in PBS and permeabilized using a solution of 0.1% saponin in PBS with 5% FCS. The last solution contained the anti-IFNγ-FITC and also a mouse anti-NK1.1 mAb labeled with phycoerythrin (PE; BD, Madrid), both at a 1/1000 dilution and cells were incubated for 1 h at room temperature. IFNγ staining was analyzed by flow cytometry via gating within the NK1.1+ population.
Degranulation was analyzed following a similar protocol as indicated for IFN-γ secretion, using also YAC-1 cells as stimulus for NK cells, by analyzing CD107a externalization using a FITC-labeled anti-CD107a rat mAb, as indicated in.23 Briefly, purified NK cells were incubated with target cells at a 1:1 ratio during 5 hours in the presence of GolgiStop (BD Biosciences, Madrid) and FITC-conjugated anti-CD107a mAb (BD Biosciences, Madrid). Then, cells were labeled with PE-conjugated anti-NK1.1 and analyzed via flow cytometry.
The intracellular expression of the cytotoxic effector granzyme B was analyzed using staining with a specific non-commercial rabbit polyclonal antibody on permeabilized NK cells, as we previously described.23,57
Effect of IFNα on NK cell activation in vivo
For in vivo characterization of responses, 10,000 IU of IFNα per mouse were injected in wt or PKC-θ−/− mice and immune cells were obtained 24 h later from dissociated spleen cell preparations or flushed from their peritoneum, essentially as previously described.23 The NK cells were then purified by MACS technology, as indicated above. Next, their intracellular expression of granzyme B, and their degranulation potential stimulated by YAC-1 cells was analyzed as indicated above.
Analysis of PKC-θ activation in NK cells induced by IFNα
NK cells were isolated from the spleens of wt mice and treated during various times (10 to 120 min) with different doses of IFNα (100 to 1000 IU/mL). The auto phosphorylation of PKC-θ on Ser676 and its level of expression was analyzed by immunoblot using a rabbit pAb (Upstate/Millipore, Madrid) and a mouse mAb (BD, Madrid), respectively. Anti-β-actin antibody (Sigma, Madrid) was used in immunoblots as loading controls. Anti-β-actin reprobing on the same unstrapped membrane in which phospho-PKC-θ had been analyzed was used as a preferred loading control, since stripping of the anti-phospho-PKC−θ antibody resulted in protein loss, and anti-PKC-θ blotting in a separate membrane of a parallel set of samples gave sometimes inconsistent results. The effect of several enzymatic inhibitors on the auto-phosphorylation of PKC-θ at Ser676 following treatment with 100 IU/mL IFNα for 30 min was also analyzed by preincubating isolated NK cells with the indicated inhibitors for 30 min prior to adding IFNα. These included the PLC inhibitor U73122 (Calbiochem/Merck, Madrid) at 10 μM, the PI3K inhibitor wortmannin (Sigma, Madrid), and the mTOR inhibitor rapamycin (Sigma, Madrid), both at 100 nM. The phospho-PKC-θ/β-actin ratio in each sample was determined using the ImageJ software.
RNA isolation and qRT-PCR of IFNα-responsive genes
ISG15 and IRF9 mRNA levels were assayed as controls as these have previously been described to be PKC-θ-dependent genes, at least in T cells 36; CXCL10 was tested as a prototype of a STAT1-dependent, IFN-responsive gene 37; and IFNG was tested as a control gene, not affected by IFNα, at least in NK cells.
Ribonucleic acid (RNA) isolation was performed and reverse transcribed as previously described.58 cDNAs (cDNAs) were amplified using the SyberGreen polymerase chain reaction (PCR) Master Mix from Invitrogen. Amplification products were detected by Real Time qPCR using the light cycler 480 (Roche), according to manufacturer specifications. Reactions were carried out for 5 min at 95°C followed by 40 cycles of: 30 sec at 95°C, 30 sec at 64°C and 30 sec at 72°C. qRT-PCR data was analyzed using the Light Cycler 480 Software release 1.5 (Roche), according to the manufacturer specifications. The housekeeping gene β−actin was used to first normalize the mRNA expression levels. RLP13A (RLP1) was then used as a standard reference mRNA and relative mRNA levels were calculated according to this standard. Primers were designed and selected using the Primer3 (v. 0.4.0) program and were the following:
IFNγ: Forward, 5′-GCTCTGAGACAATGAACGCTAC -3′,
IFNγ: Reverse, 5′-TCTTCCACATCTATGCCACTTG -3′
CXCL10: Forward, 5′-CCCACGTGTTGAGATCATTG-3′,
CXCL10: Reverse, 5′-TCCATCACAGCACCGGG-3′,
IRF9: Forward, 5′- CGC TGC TGC TCA CCT TCA T -3′,
IRF9 : Reverse, 5′-GGC TGT GCA CCT GGG TCT TA -3′
ISG15: Forward, 5′-GATTTCCTGGTGTCCGTGAC-3′
ISG15: Reverse, 5′-CTAGCATCACTGTGCTGCTG-3′
β-actin: Forward, 5′-GAGGGAAATCGTGCGTGACA-3
β-actin: Reverse, 5′-AATAGTGATGACCTGGCCGT-3
RLP13A: Forward, 5′-GGTCCTCAAGACCAACGGACTC-3′
RPL13A: Reverse, 5′-GTGCGCTGTCAGCTCTCTAATG-3′
Analysis of STAT1 phosphorylation induced by IFNα in NK cells
STAT1 phosphorylation in Tyr701 38 or Ser727 39 was analyzed in control NK cells or in cells treated with IFNα from wt or PKC-θ−/− mice by immunoblot using anti-STAT1 phospho-specific rabbit pAbs (Cell Signaling, Barcelona). Anti-β-actin reprobing on the same membrane in which phospho-STAT1 was analyzed has been used as a preferred loading control, since stripping of the anti-phospho-STAT1 antibodies resulted in protein loss, and anti-STAT1 blotting in a separate membrane of a parallel set of samples sometimes yielded inconsistent results. In addition, IFNα treatment for short times did not affect to the total STAT1 content (data not shown). The phospho-STAT1/β-actin ratio in each sample was determined using the ImageJ software.
Analysis of CXCL10 secretion stimulated by IFNα in NK cells
CXCL10 secretion was determined by analyzing the supernatants of purified NK cells incubated for 8 h in complete medium in the presence or absence of 100 IU/mL IFNα. CXCL10 concentrations in the supernatants were measured using a specific quantitative ELISA kit (R&D systems, Abingdon, UK).
Statistical analysis
Results are shown as the average ± SD and statistical significance was evaluated using the Student's t test. Differences were not considered significant if P-values were ≥0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the collaborative projects CliNK SOE2/P1/E341 from Sudoe/Interreg IV B (EU) and Grant CTPP5/12 from “Communauté de Travail des Pyrénées”; by grant SAF2010–15341 to AA and grant SAF2011–25390 to JP, both from Ministerio de Economía y Competitividad (Spain); by Fondo Social Europeo and by the association CIEL, the association L'Un pour l'Autre and the federation Ensangble to MV; and by one AOI from the CHU Montpellier and fellowships from the ARC and Higher Education Commission, Pakistan to MGR.
References
- Baier G, Telford D, Giampa KM, Coggeshall KM, Baier-Bitterlich G, Isakov N, Altman A. Molecular cloning and characterization of PKC-q, a human novel member of the protein kinase C (PKC) gene family expressed predominantly in hematopoietic cells. J Biol Chem 1993; 268:4997-5004; PMID:8444877
- Altman A, Kaminski S, Busuttil V, Droin N, Hu J, Tadevosyan Y, Hipskind RA, Villalba M. Positive feedback regulation of PLCg1/Ca2+ signaling by PKCq in restimulated T cells via a tec kinase-dependent pathway. Eur J Immunol 2004; 34:2001-11; PMID:15214048; http://dx.doi.org/10.1002/eji.200324625
- Baier-Bitterlich G, Uberall F, Bauer B, Fresser F, Wachter H, Grunicke H, Utermann G, Altman A, Baier G. Protein kinase C-q isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol Cell Biol 1996; 16:1842-50; PMID:8657160
- Coudronniere N, Villalba M, Englund N, Altman A. NF-kB activation induced by TCR/CD28 costimulation is mediated by PKC-q. Proc Natl Acad Sci USA 2000; 97:3394-9; PMID:10716728
- Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, Leitges M, Baier G. Protien kinase C q affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med 2003; 197:1525-35; PMID:12782715; http://dx.doi.org/10.1084/jem.20020234
- Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg PL, et al. PKC-q is required for TCR-induced NF-kB activation in mature but not in immature T lymphocytes. Nature 2000; 404:402-7; PMID:10746729; http://dx.doi.org/10.1038/35006090
- Isakov N, Altman A. Protein kinase C q in T cell activation. Annu Rev Immunol 2002; 20:761-94; PMID:11861617; http://dx.doi.org/10.1146/annurev.immunol.20.100301.064807
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting : from immunosurveillance to tumor escape. Nature Immunol 2002; 3:991-8; http://dx.doi.org/10.1038/ni1102-991
- Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nature Rev Immunol 2006; 6:836-48; http://dx.doi.org/10.1038/nri1961
- Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007; 450:903-7; PMID:18026089; http://dx.doi.org/10.1038/nature06309
- Villalba M, López-Royuela N, Krzywinska E, Rathore M, Hipskind R, Haouas H, Allende-Vega N. Chemical metabolic inhibitors for the treatment of blood-borne cancers. Anti-Cancer Ag Med Chem 2014; 14:223-32;
- Villalba M, Rathore MG, López-Royuela N, Krzywinska E, Garaude J, Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol 2013; 45:106-13; PMID:22568930; http://dx.doi.org/10.1016/j.biocel.2012.04.024
- Garaude J, Kaminski S, Charni S, Aguiló JI, Jacquet C, Plays M, Rodriguez F, Hernández J, Hipskind RA, Anel A, et al. Impaired anti-leukemic immune response in PKCq-deficient mice. Mol Immunol 2008; 45:3463-9; PMID:18462800; http://dx.doi.org/10.1016/j.molimm.2008.03.016
- Ljunggren HG, Karre K. Host resistance directed selectively against H-2-deficient lymphoma variants. Analysis of the mechanism. J Exp Med 1985; 162:1745-95; PMID:3877776; http://dx.doi.org/10.1084/jem.162.6.1745
- Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell–directed therapies: moving from unexpected results to successful strategies. Nature Immunol 2008; 9:486-94; http://dx.doi.org/10.1038/ni1580
- Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nature Immunol 2008; 9:503-10; http://dx.doi.org/10.1038/ni1582
- Pardo J, Balkow S, Anel A, Simon MM. Granzymes are critically involved in NK-mediated control of RMA-S tumor growth in vivo. Eur J Immunol 2002; 32:2881-6; PMID:12355441; http://dx.doi.org/10.1002/1521-4141(2002010)32:10<2881::AID-IMMU2881>3.0.CO;2-K
- Van den Broek MF, Kägi D, Zinkernagel RM, Hengartner H. Perforin dependence of natural killer cell-mediated tumor control in vivo. Eur J Immunol 1995; 25:3514-6; PMID:8566046; http://dx.doi.org/10.1002/eji.1830251246
- Screpanti V, Wallin RPA, Ljunggren HG, Grandien A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol 2001; 167:2068-73; PMID:11489989; http://dx.doi.org/10.4049/jimmunol.167.4.2068
- Villalba M, Kasibhatla S, Genestier L, Mahboubi A, Green DR, Altman A. Protein kinase C-q cooperates with calcineurin to induce Fas ligand expression during activation-induced T cell death. J Immunol 1999; 163:5813-9; PMID:10570264
- Pardo J, Buferne M, Martínez-Lorenzo MJ, Naval J, Schmitt-Verhulst AM, Boyer C, Anel A. Differential implication of PKC isoforms in TCR/CD3-induced Fas ligand expression and in CTL degranulation. Int Immunol 2003; 15:1441-50; PMID:14645153; http://dx.doi.org/10.1093/intimm/dxg141
- Garrido F, Algarra I, García-Lora AM. The escape of cancer from T lymphocytes: immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol Immunother 2010; 59:1601-6; PMID:20625726; http://dx.doi.org/10.1007/s00262-010-0893-2
- Aguiló JI, Garaude J, Pardo J, Villalba M, Anel A. Protein kinase C-q is required for NK cell activation and in vivo control of tumor progression. J Immunol 2009; 182:1972-81; http://dx.doi.org/10.4049/jimmunol.0801820
- Anel A, Aguiló JI, Catalán E, Garaude J, Rathore MG, Pardo J, Villalba M. Protein kinase C-q (PKC-q) in natural killer cell function and anti-tumor immunity. Front Immunol 2012; 3:187; PMID:22783260; http://dx.doi.org/10.3389/fimmu.2012.00187
- Akazawa T, Ebihara T, Okuno M, Okuda Y, Shingai M, Tsujimura K, Takahashi T, Ikawa M, Okabe M, Inoue N, et al. Antitumor NK activation induced by the Toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci USA 2007; 104:252-7; PMID:17190817; http://dx.doi.org/10.1073/pnas.0605978104
- Page KM, Chaudhary D, Goldman SJ, Kasaian MT. Natural killer cells from protein kinase C q–/– mice stimulated with interleukin-12 are deficient in production of interferon-g. J Leukoc Biol 2008; 83:1267-76; PMID:18263766; http://dx.doi.org/10.1189/jlb.1107745
- Carson WE, Giri JG, Lindemann MJ, Linett ML, Ahdieh M, Paxton R, Anderson D, Eisenmann J, Grabstein K, Caligiuri MA. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med 1994; 180:1395-403; PMID:7523571; http://dx.doi.org/10.1084/jem.180.4.1395
- Cooper MA, Fehniger TA, VanDeusen JB, Waite RE, Liu Y, Aguila HL, Caligiuri MA. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. Blood 2002; 100:3633-8; PMID:12393617; http://dx.doi.org/10.1182/blood-2001-12-0293
- Liu RB, Engels B, Arina A, Schreiber K, Hyjek E, Schietinger A, Binder DC, Butz E, Krausz T, Rowley DA, et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res 2012; 72:1964-74; PMID:22374983; http://dx.doi.org/10.1158/0008-5472.CAN-11-3208
- Villalba M, Bi K, Hu J, Altman Y, Bushway P, Reits E, Neefjes J, Baier G, Abraham RT, Altman A. Translocation of PKCq in T cells is mediated by a nonconventional, PI3-K- and Vav-dependent pathway, but does not absolutely require phospholipase C. J Cell Biol 2002; 157:253-63; PMID:11956228; http://dx.doi.org/10.1083/jcb.200201097
- Ben Ahmed M, Belhadj Hmida N, Moes N, Buyse S, Abledadhim M, Louzir H, Cerf-Bensussan N. IL-15 renders conventional lymphocytes resistant to suppressive functions of regulatory T cells through activation of the phosphatidylinositol 3-kinase pathway. J Immunol 2009; 182:6763-70; PMID:19454671; http://dx.doi.org/10.4049/jimmunol.0801792
- Brennan P, Babage JW, Burgering BMT, Gromer B, Reif K, D.A. C. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity 1997; 7:679-89; PMID:9390691; http://dx.doi.org/10.1016/S1074-7613(00)80388-X
- Truitt KE, Mills GB, Turck CW, Imboden JB. SH2-dependent association of phosphatidylinositol 3'-kinase 85-kDa regulatory subunit with the interleukin-2 receptor b chain. J Biol Chem 1994; 269:5937-43; PMID:7509794
- Uddin S, Fish E, Sher D, Gardziola C, White M, Platanias L. Activation of the phosphatidylinositol 3-kinase serine kinase by IFN-a. J Immunol 1997; 158:2390-7; PMID:9036989
- Premecz G, Markovits A, Bagi G, Farkas T, Földes I. Phospholipase C and phospholipase A2 are involved in the antiviral activity of human interferon-a. FEBS Lett 1989; 249:257-60; PMID:2544450; http://dx.doi.org/10.1016/0014-5793(89)80635-0
- Srivastava KK, Batra S, Sassano A, Li Y, Majchrzak B, Kiyokawa H, Altman A, Fish EN, Platanias LC. Engagement of protein kinase C-q in interferon signaling in T-cells. J Biol Chem 2004; 279:29911-20; PMID:15150272; http://dx.doi.org/10.1074/jbc.M401997200
- Wang J, Campbell IL. Innate STAT1-dependent genomic response of neurons to the antiviral cytokine alpha interferon. J Virol 2005; 79:8295-302; PMID:15956575; http://dx.doi.org/10.1128/JVI.79.13.8295-8302.2005
- Fu YY. A transcription factor with SH2 and SH3 domains is directly activated by an interferon alpha-induced cytoplasmic protein tyrosine kinase(s). Cell 1992; 70:323-35.
- Hardy PO, Diallo TO, Matte C, Descoteaux A. Roles of phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase in the regulation of protein kinase C-a activation in interferon-g-stimulated macrophages. Immunology 2009; 128:e652-e60; PMID:19740326; http://dx.doi.org/10.1111/j.1365-2567.2009.03055.x
- Deb D, Sassano A, Lekmine F, Majchrzak B, Verma A, Kambhampati S, Uddin S, Rahman A, Fish E, Platanias L. Activation of protein kinase Cd by IFN-g. J Immunol 2003; 171:267-73; PMID:12817007; http://dx.doi.org/10.4049/jimmunol.171.1.267
- Swann JB, Hayakawa Y, Zerafa N, Sheelman KCF, Scott B, Schreiber RD, Hertzog P, Smyth MJ. Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol 2007; 178:7540-9; PMID:17548588; http://dx.doi.org/10.4049/jimmunol.178.12.7540
- Merino E, Abeyweera TP, Firth MA, Zawislak CL, Basu R, Liu X, Sun JC, Huse M. Protein kinase C-q clustering at immunological synapses amplifies effector responses in NK cells. J Immunol 2012; 189:4859-69; PMID:23077238; http://dx.doi.org/10.4049/jimmunol.1200825
- Tassi I, Cella M, Presti R, Colucci A, Gilfillan S, Littman DR, Colonna M. NK cell activating receptors require PKCq for sustained signaling, transcriptional activation and IFN-g secretion. Blood 2008; 112:4109-16; PMID:18784374; http://dx.doi.org/10.1182/blood-2008-02-139527
- Grybko MJ, Pores-Fernando AT, Wurth GA, Zweifach A. Protein kinase C activity is required for cytotoxic T cell lytic granule exocytosis, but the q isoform does not play a preferential role. J Leukoc Biol 2007; 81:509-19; PMID:17077164; http://dx.doi.org/10.1189/jlb.0206109
- Puente LG, He JS, Ostergaard HL. A novel PKC regulates ERK activation and degranulation of cytotoxic T lymphocytes: Plasticity in PKC regulation of ERK. Eur J Immunol 2006; 36:1009-18; PMID:16552708; http://dx.doi.org/10.1002/eji.200535277
- Pardo J, Aguiló J, Anel A, Martin P, Joeckel L, Borner C, Wallich R, Müllbacher A, Froelich C, Simon M. The biology of cytotoxic cell granule exocytosis pathway: granzymes have evolved to induce cell death and inflammation. Microbes & Infection 2009; 11:452-9; PMID:19249384; http://dx.doi.org/10.1016/j.micinf.2009.02.004
- Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity 2007; 26:798-811; PMID:17540585; http://dx.doi.org/10.1016/j.immuni.2007.04.010
- Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007; 26:503-17; PMID:17398124; http://dx.doi.org/10.1016/j.immuni.2007.03.006
- Gustafsson K, Ingelsten M, Berggvist L, Nyström J, Andersson B, Karlsson-Parra A. Recruitment and activation of natural killer cells in vitro by a human dendritic cell vaccine. Cancer Res 2008; 68:5965-71; PMID:18632652; http://dx.doi.org/10.1158/0008-5472.CAN-07-6494
- Robertson M. Role of chemokines in the biology of natural killer cells. J Leukoc Biol 2002; 71:173-83; PMID:11818437
- Vujanovic L, Ballard W, Thorne SH, Vujanovic NL, Butterfield LH. Adenovirus-engineered human dendritic cells induce natural killer cell chemotaxis via CXCL8/IL-8 and CXCL10/IP-10. OncoImmunol 2012; 1:448-57; http://dx.doi.org/10.4161/onci.19788
- Sánchez-Martínez D, Krzywinska E, Rathore MG, Saument A, Cornilon A, López-Royuela N, Martínez-Lostao L, Ramírez-Labrada A, Lu Z, Rossi J, et al. All-trans retinoic acid (ATRA) induces miR-23a expression, decreases CTSC expression and granzyme B activity leading to impaired NK cell cytotoxicity. Int J Biochem Cell Biol 2014; 49:42-52; http://dx.doi.org/10.1016/j.biocel.2014.01.003
- Bancerek J, Poss Z, Steinparzer I, Sedlyarov V, Pfanffenwimmer T, Mikulic I, Dölken L, Strobl B, Müller M, Taatjes D, et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013; 38:250-62; PMID:23352233; http://dx.doi.org/10.1016/j.immuni.2012.10.017
- Putz E, Gotthardt D, Hoemann G, Csiszar A, Wirth S, Berger A, Straka E, Rigler D, Wallner B, Jamieson A, et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep 2013; 4:437-44; PMID:23933255; http://dx.doi.org/10.1016/j.celrep.2013.07.012
- Kiessling R, Klein E, Wigzell H. ``Natural" killer cells in the mouse. I. Cytotoxic cells with specificity for Moloney leukemia cells. Specificity and distribution according to genotype. Eur J Immunol 1975; 5:112-7; PMID:1234049; http://dx.doi.org/10.1002/eji.1830050208
- Murali-Krishna K, Altman J, Suresh M, Sourdive D, Zajac A, Miller J, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a re-evaluation of bystander activation during viral infection. Immunity 1998; 8:177-87; PMID:9491999; http://dx.doi.org/10.1016/S1074-7613(00)80470-7
- Pardo J, Wallich R, Ebnet K, Iden S, Zentgraf H, Martin P, Ekiciler A, Prins A, Müllbacher A, Huber M, et al. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ 2007; 14:1768-79; PMID:17599099; http://dx.doi.org/10.1038/sj.cdd.4402183
- Charni S, de Bettignies G, Rathore MG, Aguiló JI, van den Elsen PJ, Haouzi D, Hipskind RA, Enriquez JA, Sanchez-Beato M, Pardo J, et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J Immunol 2010; 185:3498-503; PMID:20729331; http://dx.doi.org/10.4049/jimmunol.1001250