
Abstract
In clinical trials, metronomic cyclophosphamide (CPA) is increasingly being combined with vaccines to reduce tumor-induced immune suppression. Previous strategies to modulate the immune system during vaccination have involved continuous administration of low dose chemotherapy, studies that have posed unique considerations for clinical trial design. Here, we evaluated metronomic CPA in combination with a peptide vaccine targeting HPV16E7 in an HPV16-induced tumor model, focusing on the cytotoxic T-cell response and timing of low dose metronomic CPA (mCPA) treatment relative to vaccination. Mice bearing C3 tumors were given metronomic CPA on alternating weeks in combination with immunization with a DepoVax vaccine containing HPV16E749–57 peptide antigen every 3 weeks. Only the combination therapy provided significant long-term control of tumor growth. The efficacy of the vaccine was uncompromised if given at the beginning or end of a cycle of metronomic CPA. Metronomic CPA had a pronounced lymphodepletive effect on the vaccine draining lymph node, yet did not reduce the development of antigen-specific CD8+ T cells induced by vaccination. This enrichment correlated with increased cytotoxic activity in the spleen and increased expression of cytotoxic gene signatures in the tumor. Immunity could be passively transferred through CD8+ T cells isolated from tumor-bearing mice treated with the combinatorial treatment regimen. A comprehensive survey of splenocytes indicated that metronomic CPA, in the absence of vaccination, induced transient lymphodepletion marked by a selective expansion of myeloid-derived suppressor cells. These results provide important insights into the multiple mechanisms of metronomic CPA induced immune modulation in the context of a peptide cancer vaccine that may be translated into more effective clinical trial designs.
Abbreviations:
- CPA, cyclophosphamide
- CTL, cytotoxic T lymphocyte
- CTLA-4, cytotoxic T lymphocyte-associated protein 4
- DPX, DepoVax
- HPV, human papilloma virus
- IFNγ, interferon γ
- mCPA, metronomic low dose CPA
- MDSC, myeloid-derived suppressor cells
- PD-1/PDCD1, programmed cell death 1
- PO, per os (oral)
- sbCPA, single bolus low dose CPA
- Treg, regulatory T cell
Introduction
Therapeutic intervention of cancer is a complex challenge, and the stage, type of cancer and host immune response may all influence treatment outcome. Effective cancer immunotherapy requires consideration of technical and immune-related issues to optimize vaccine strategies, including: (a) appropriate selection of tumor associated antigens; (b) vaccine immunogenicity (i.e., adjuvant, formulation); and (c) overcoming tumor-induced immune suppression.Citation1,2 Translational studies wherein clinical immune parameters can be emulated in pre-clinical models can permit in-depth evaluation of the immune response to identify potential biomarkers and improve treatment regimens.
We have developed an adjuvanting vaccine platform designed to facilitate a strong and robust immune response to peptide antigens. DepoVaxTM (DPX) is a liposome-in-oil formulation, including an adjuvant and universal T-helper peptide that can be formulated with any epitope or mixture of epitopes. DPX forms a durable and immunogenic depot at the site of immunization which prolongs antigen exposure to the immune system and results in systemic induction of a cytotoxic T lymphocyte (CTL)-mediated immune response.Citation3 We have shown that a single vaccination with DPX results in equivalent or better immune responses than multiple vaccinations with peptides in emulsion formulations.Citation4,5 A DPX based vaccine called DPX-0907 containing 7 peptide epitopes from different tumor associated antigens completed a Phase I clinical trial in breast, ovarian and prostate cancer patients, demonstrating safety and immunogenicity in these advanced tumor-bearing patients.Citation6
Despite the ability to induce antigen-specific T cells, many cancer vaccines are ineffective inducers of tumor immune rejection due to the immunosuppressive mechanisms employed by the tumor to dampen the immune response. To address this, cancer vaccines are increasingly being combined with immune modulators that have the capacity to reverse tumor-imposed immunosuppression and enhance the immune response.Citation7 One example of an immune modulator is cyclophosphamide (CPA) used at low dose. A single bolus intravenous injection of low dose CPA (sbCPA) in mice has been demonstrated to selectively reduce Tregs, increase activation of dendritic cells, and stimulate induction of Type I interferons.Citation8-10 Administration of sbCPA (50–100 mg/kg) 1 to 3 d prior to vaccination of mice also enhanced antigen-specific T-cell responses.Citation11,12 However, sbCPA administration prior to vaccination in humans (100–300 mg/m2) has not demonstrated equivocal results.Citation13,14 One reason for this discrepancy is that the timing of vaccination relative to administration of sbCPA appears to be very sensitive. In mice, if the vaccine is administered too early after sbCPA (i.e., within 6 hours) it could result in increased systemic toxicity, yet when delivered too late (i.e., more than 7 days) the therapy may abrogate any synergistic effects.Citation15,16 This sensitivity may be amplified in clinical studies involving a more immunologically heterogeneous subject population.
In order to prolong the window of immune modulation provided by low dose CPA, a schedule of daily low dose administration of CPA has been proposed as an alternative treatment schedule, which is commonly referred to as metronomic CPA (mCPA).Citation17 Daily mCPA doses are a fraction of the dose commonly used in sbCPA therapy and can be administered orally as a pill. Continuous treatment with mCPA has been associated with an anti-angiogenic effect within the tumor microenvironment.Citation18,19 Recently, Ghiringhelli et al. tested a variation of mCPA scheduling in cancer patients consisting of a daily dose of 50 mg twice a day (∼60 mg/m2) for 1 week on followed by 1 week break.Citation20 After 1 month, this week on/ off regimen was found to provide immune modulation by selectively reducing Tregs and enhancing natural killer (NK) cell activation. However, in comparison to sbCPA, there are few studies that investigate the combination of mCPA and vaccination. One important consideration is the optimal timing between administering the vaccine and providing a continuous treatment with mCPA.
Vaccine studies are currently limited by the lack of models expressing relevant immunogenic epitopes. In this study, we investigated the underlying mechanisms of mCPA-induced vaccine enhancement using a transplantable HPV16 murine tumor model. HPV16 is an oncogenic causative virus of anogenital and oropharyngeal cancers.Citation21,22 Peptide vaccines targeting the HPV16 E6 and E7 proteins have demonstrated strong immunogenicity in clinical trials, yet have failed to consistently provide clinical benefit.Citation23,24 The immunosuppressive tumor environment established by these types of malignancies may be one factor mitigating the efficacy of the vaccine-induced immune response.Citation25 Prior research has established that HPV16 expressing tumor models are sensitive to immune modulation provided by mCPA.Citation26 Here, we conduct an extensive survey of various types of immune cells in the spleen and lymph node, as well as those infiltrating the tumor, following combinatorial treatment with mCPA and vaccination. We have specifically focused on the effects of mCPA treatment on the development of a vaccine-induced cytotoxic T-cell response. We have also investigated various schedules of mCPA and vaccination therapy, which could have important implications for clinical trial designs.
Results
mCPA and sbCPA in combination with a peptide-based vaccine provide enhanced tumor control
We compared immune modulation of low dose CPA administered intravenously (IV) as single bolus (100 mg/kg) or administered orally (PO) as metronomic (20 mg/kg/day) CPA in combination with a DepoVax vaccine containing HPV16E749-57 (R9F) peptide antigen (DPX-R9F). Mice were implanted subcutaneously with HPV16E7 expressing C3 tumors and vaccinated every 3 weeks starting 12 d after implantation. The mCPA schedule was selected to mimic our proposed clinical trial design based on clinical data from Ghiringhelli et al. and was given 1 week on and 1 week off starting on day 5.Citation20 sbCPA was given every 3 weeks, one day before each vaccination, which is common scheduling for this type of low dose CPA therapy.Citation13,14 Starting vaccination on day 12 enabled us to test the combination of low dose CPA with vaccine on established tumors that are difficult to control with either vaccine or low dose CPA monotherapy. As shown in , repeated DPX-R9F vaccine monotherapy delayed tumor growth, as expected, but was ultimately unable to abrogate tumor progression. Both mCPA and sbCPA also delayed tumor growth when given as a monotherapy, but sbCPA provided a longer delay in tumor progression than mCPA. Both mCPA and sbCPA in combination with DPX-R9F vaccine provided durable tumor growth control with no significant differences between the 2 combinations in the long term.
Figure 1. mCPA and sbCPA are equally effective at enhancing tumor control in combination with DPX-R9F peptide vaccine. (A and B). Mice (n = 8) were implanted with C3 tumors on day 0 and treated with 20 mg/kg/day metronomic cyclophosphamide (mCPA) orally starting on day 5 in a one week on/one week off schedule (A) or received a 100 mg/kg single bolus (sbCPA) intravenously one day preceding each vaccination (B). Vaccinations with DepoVax containing 10 μg R9F-PADRE antigen (DPX-R9F) commenced on day 12 and repeated every 3 weeks (days 33 and 54). Results are representative of 2 separate experiments. Eight days after the last vaccination (day 62), mice in remaining groups were terminated and vaccine-draining lymph nodes were collected for immune analysis by interferon γ (IFNγ) ELISPOT assay. (C) Lymph node cells were stimulated with syngeneic dendritic cells unloaded (DC-E) or loaded with R9F peptide (DC-R9F); data are pooled from 2 separate experiments. (D) Lymph node cells were also analyzed for the presence of CD8+ T cells and R9F-specific CD8+ T cells using immunostaining with fluorophore-conjugated anti-CD8 antibody and R9F-dextramer. Data are from one experiment only. Statistical analysis was performed by one-way ANOVA with Tukey's post-hoc test; **P < 0.01, ***P < 0.001.
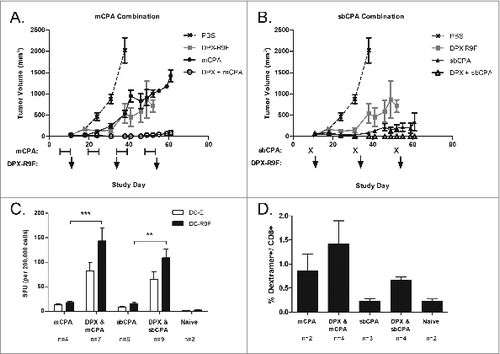
We confirmed the presence of an active immune response 8 d after the final vaccination (day 62 of the study) in each group by performing IFNγ ELISPOT assay using lymph node cells isolated from the vaccine draining inguinal lymph node. As shown in , the mice implanted with C3 tumors and treated with DPX-R9F vaccination in combination with either mCPA or sbCPA generated a strong antigen-specific immune response. It is noteworthy that there is a non-specific IFNγ background in these groups as well, which has been reported by others after low dose CPA treatment and appears to be associated with increased proliferation of T cells.Citation29 Dextramer staining for R9F-specific CD8+ T cells also confirmed the presence of antigen-specific CD8+ T cells among lymph node cells from mice treated with DPX-R9F vaccine combined with mCPA or sbCPA (). Similar immunogenic effects were seen among splenic lymphocytes responding to the combinatorial treatment regimens (data not shown).
mCPA can be administered one week before or one week after vaccine with no detriment to immunogenicity
A disadvantage to sbCPA treatment is that the timing of vaccination relative to CPA administration is very sensitive.Citation15,16 To test whether the timing of mCPA relative to vaccination affected the efficacy of treatment, we performed a tumor challenge experiment wherein mice were consistently vaccinated at either the end or the beginning of one week mCPA administration. To determine whether treatment could be initiated at a later time point after tumor challenge and still provide protection, treatment was begun either one week (day 7) or 2 weeks (day 14) after tumor implantation. All groups were part of one study and were treated in parallel, allowing comparison between treatment groups ( and S1).
Figure 2. Early vaccination provides optimum immunity and tumor control independently of timing of cyclosphosphamide treatment. (A and B) Mice (n = 10) were implanted with C3 tumors on study day 0. Groups were treated with metronomic cyclophosphamide (mCPA) only, DepoVax vaccine (DPX-R9F) only, a combination of mCPA and vaccine, or PBS as a negative control. All groups were part of the same experiment and were treated in parallel. Treatment commenced one week (day 7; A and B) or 2 weeks (day 15; C and D) after implantation. mCPA was administered every other week for 7 consecutive days and vaccine was given corresponding to the first day of each mCPA treatment (A) or the last day (B). Mice were terminated humanely when tumor size reached 2000 mm3. Statistical analysis of differences in survival were analyzed by log-rank (Mantel-Cox) test, comparing vaccine only with combination therapy; *P <0.05, ****P < 0.0001. (C) Mice (n = 5) were implanted with C3 tumors on day 0 and then vaccinated with DPX-R9F on day 21. mCPA was administered for one week before vaccination (days 14–21) or one week after vaccination (days 21–28). Mice were terminated on day 29 and immune responses of immune cells derived from vaccine draining lymph nodes to dendritic cells loaded with R9F peptide (versus unloaded controls) were assessed by IFNγ ELISPOT assay. Statistical analysis was performed by Student's t-test; *P < 0.05.
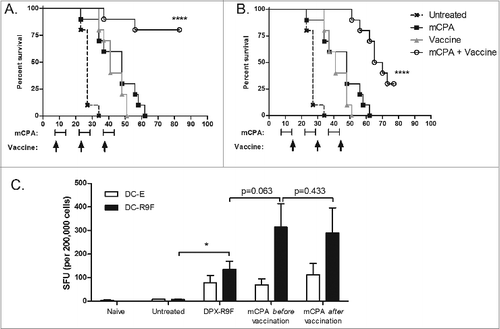
In this survival study, combination therapy provided significantly better protection from tumor growth compared to monotherapy when started early (i.e., day 7; ). When treatment was started later (i.e., day 14) combination therapy provided no additional therapeutic benefit in comparison to either treatment alone (Fig. S1). The comparative efficacy of the combination therapy when vaccination was provided at either the start () or the end () of each mCPA cycle indicates that providing mCPA before or after vaccination does not reduce the immunogenicity of the vaccine. We further confirmed that the timing of the mCPA treatment (i.e., one week before or after vaccination) did not impact vaccine immunogenicity by IFNγ ELISPOT assay using lymph node cells from treated mice ().
mCPA enriches antigen-specific CD8+ T cells in the lymph node
We next sought to investigate the cellular effects on the immune system induced by the combination of mCPA with DPX-R9F in C3 tumor-bearing mice. To ensure the subjects in all groups were still alive at the time of analysis, we tested mice implanted with tumors and treated for one week with mCPA starting on day 14 followed by a single vaccination with DPX-R9F on day 21 (). On day 29, 8 d after vaccination, mice were terminated and vaccine-draining inguinal lymph nodes and spleens were collected, dissociated and immune cells counted (). At this time point, total lymph node counts were significantly reduced in most groups treated with mCPA relative to the non-mCPA control groups (P < 0.05). mCPA did not significantly reduce lymph node size in tumor-bearing mice as compared to untreated tumor-bearing mice (P = 0.683). The lymph nodes in the non-tumor bearing mice treated with mCPA only were too small for analysis.
Figure 3. mCPA enhancement of immune response in lymph node due to enrichment of antigen-specific CD8+ T cells. (A) Description of treatment mice used for assays in (B–D). Mice were implanted with C3 tumors on day 0 and then treated with metronomic cyclophosphamide (mCPA) for one week over days 14–21, or DepoVax containing 10 μg R9F-PADRE (DPX-R9F) on day 21, or the combination. Controls included untreated tumor-bearing mice and non-tumor bearing mice treated in parallel. On day 29, 8 d after vaccination, mice were terminated and vaccine-draining lymph nodes were collected. Lymph nodes were dissociated and constituent cells counted (B), used in ELISPOT assay (C) and analyzed by flow cytometry (D). (B) Total lymph node cell counts, n = 12–20. (C) Lymph node cells were stimulated overnight in interferon γ (IFNγ) ELISPOT plate with unloaded dendritic cells (DC-E) or R9F-loaded dendritic cells (DC-R9F); n = 12–20. (D) Immunofluorescent staining of lymph node cells using fluorophore-conjugated anti-CD8 antibody and R9F-dextramers and cytofluorimetric analysis to determine the percentage of CD8+ cells and antigen-specific CD8+ cells; total number of cells calculated from cell counts. Statistical analysis was performed by one-way ANOVA with Tukey post-hoc test; *P < 0.05, **P < 0.01, ***P < 0.001; ND: not detected (lymph nodes too small for analysis). Results are pooled from 5 separate experiments.
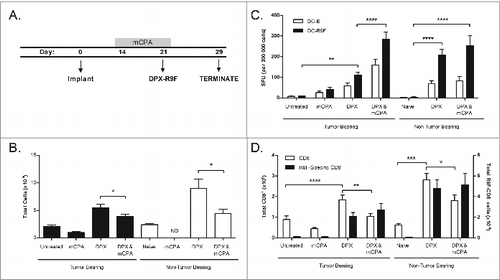
Immune responses in the lymph nodes measured by IFNγ ELISPOT assay showed that the vaccine induced an antigen-specific immune response enhanced by the combination treatment, despite having fewer total lymph node cells (). We determined the absolute numbers of CD8+ T cells and R9F-specific CD8+ T cells by immunostaining and fluorescence cytometry and found that although the mCPA treatment caused a decrease in total CD8+ T cells, the number of antigen-specific CD8+ T cells induced by the vaccine was not affected (), resulting in an overall enrichment of this cell population in the vaccine draining lymph node in both tumor-bearing and non-tumor bearing mice.
mCPA in combination with vaccination increases CTL activity in the spleen
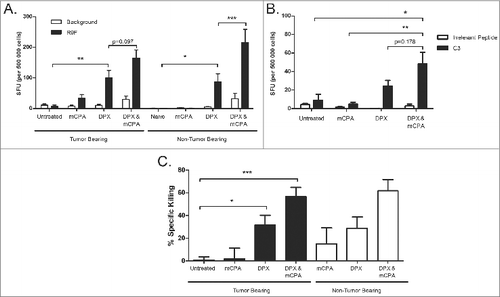
Next, we performed IFNγ ELISPOT assay using splenic cell preparations from mice treated as in . Similar to the draining lymph node, we detected increases in antigen-specific immune response to R9F peptide stimulation among lymphocytes from the spleens of mice treated with mCPA and vaccine as compared to vaccine alone, although this difference was not significant (p = 0.097; ). Additionally, the splenocytes of vaccinated tumor-bearing mice generated a strong response to C3 cell stimulation as determined by the IFNγ ELISPOT assay (). To measure the functional activity of the cytotoxic T cells induced by the vaccine, we performed a CTL in vivo assay (). Mice were treated as in and injected with target cells on day 28 then terminated on day 29. Correlating with the increase in antigen-specific immunogenicity as determined by the IFNγ ELISPOT, the antigen-specific killing of R9F-loaded target cells was significantly increased by DPX-R9F vaccination in both tumor bearing and non-tumor bearing mice, a cytotoxic effect further enhanced by mCPA combination. In contrast, control mice vaccinated with an irrelevant epitope did not display killing of R9F-loaded target cells (data not shown).
Figure 4. mCPA combined with DPX-R9F vaccination stimulates systemic increase in immune response. (A-C) Spleens were removed from mice treated as in with metronomic cyclophosphamide (mCPA), DepoVax containing 10 μg R9F-PADRE (DPX-R9F), or the combination. (A) Interferon γ (IFNγ) ELISPOT using splenocytes stimulated with R9F peptide or unstimulated (background); n = 23–25; mice pooled from 6 separate experiments. (B) IFNγ ELISPOT using splenocytes stimulated with an irrelevant control peptide or C3 tumor cells; n = 5. (C) Cytotoxic T lymphocyte (CTL) in vivo assay, mice vaccinated with DepoVax containing 5 μg R9F and 5 μg F21E; n = 3–12; mice pooled from 3 separate experiments. Statistical analysis was performed by ANOVA; *P < 0.05, **P < 0.01, ***P < 0.001.
Combination therapy increases cytotoxic T cell gene signature in the tumor microenvironment
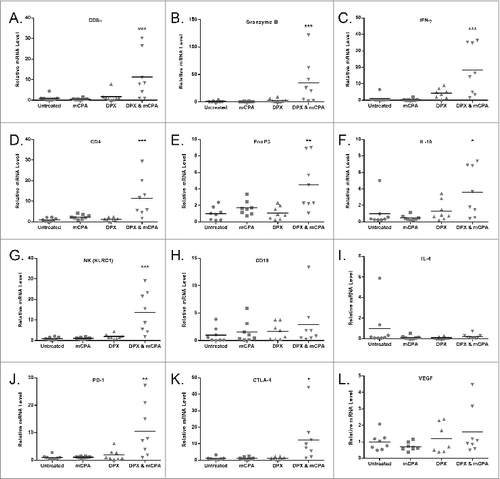
To measure several markers at once within the tumor microenvironment, we extracted total RNA from tumors of mice treated as in on day 29 and analyzed the transcript levels of immune-related genes by quantitative RT-PCR (). We found elevated levels of CD8α (Cd8a) and CD4 (Cd4) mRNA in the combination treated group compared to all other groups, indicating increased T-cell infiltration of the tumor. Coinciding with increased T-cell gene signatures were increased expression of the CTL cytokine markers IFNγ (Ifng) and Granzyme B (Gznb), while levels of IL-4 (Il4) were low across all groups, indicating a skew toward a T helper type 1 (Th1) phenotype within the tumor microenvironment. Expression of the regulatory T (Treg) cell markers IL-10 (Il10) and FoxP3 (Foxp3) were both increased in the combination treated group. B cell levels, as measured by CD19 (Cd19) expression were low in all groups, suggesting that tumor-infiltrating B cell levels were minimal in animals treated with the combinatorial therapy. The expression of co-inhibitory markers, PD-1 (Pdcd1) and CTLA-4 (Ctla4), were also significantly increased in tumors of mice treated with the bimodal mCPA and vaccine therapy.
Figure 5. Treatment with mCPA and vaccination increases the intratumoral expression of genes associated with activated cytotoxic T cells. Mice were treated as in with metronomic cyclophosphamide (mCPA), DepoVax containing 10 μg R9F-PADRE (DPX-R9F), or the combination. On day 29, tumors were removed for quantitative RT-PCR analysis and mRNA expression levels determined relative to Tbp (TATA box-binding protein) control. Relative levels of transcript are shown for: (A) CD8α (Cd8a), (B) Granzyme B (Grnb), (C) IFNγ (Ifng), (D) CD4 (Cd4), (E) FoxP3 (Foxp3), (F) IL-10 (Il10), (G) NKG2A (Klrc1), (H) CD19 (Cd19), (I) IL-4 (Il4), (J) PD-1 (Pdcd1), (K) CTLA-4 (Ctla4), (L) VEGF (Vegf). Data shown are the relative expression level in tumors of individual mice (n = 8), with the mean indicated by the bar; results pooled from 2 separate experiments. Statistical analysis was performed by ANOVA with Tukey's multiple comparisons post-hoc test compared to untreated; *P < 0.05, **P < 0.01, ***P < 0.001.
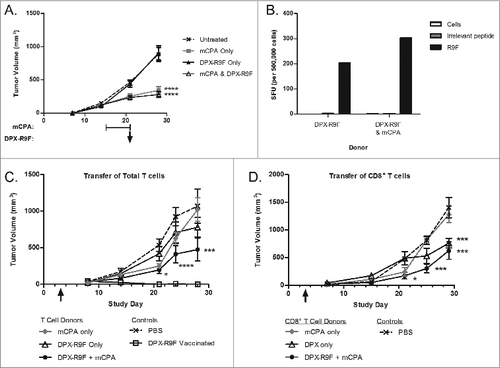
Antigen-specific cytotoxic T cell activity induced by vaccination is enhanced by mCPA
We found that even transient and delayed treatment resulted in significantly smaller tumors in the mCPA only or combination treated groups in treated mice by the end of the study period (i.e., on day 29; ). To determine if immunity could be transferred using T cells derived from treated animals, we dissociated spleens from tumor-bearing treated mice, enriched total T cells, and then transferred the T lymphocytes into recipient mice bearing 3-day old tumors. We confirmed the antigenic reactivity of the donor splenocytes by IFNγ ELISPOT assay () prior to isolating total T cells. T-cell purity was assessed by immunofluorescence staining and flow cytometry and determined to be >85%, with <5% NK T cells (data not shown). As a positive control, one group of recipient mice did not receive T cells but rather was directly immunized with DPX-R9F on day 3, and a negative control group was treated with PBS. Two groups of recipient mice received T cells isolated from donors treated with either DPX-R9F vaccination only, or alternatively, mCPA in combination with DPX-R9F (). Only the T cells isolated from the combinatorial mCPA and DPX-R9F treated donors significantly reduced tumor growth in recipient mice (P < 0.001 on day 29; ). We repeated this experiment but transferred purified CD8+ T cells from either mCPA or DPX-R9F monotherapy or mCPA and DPX-R9F combined treated donors (). In this study, the CD8+ T cells isolated from both the DPX-R9F monotherapy and the combination treated donors significantly reduced tumor volume throughout the duration of the study.
Figure 6. Protective immunity is partially transferred through donor T cells from mCPA and DPX-R9F treated, tumor-bearing mice. (A-D) Donor mice were treated as in with metronomic cyclophosphamide (mCPA), DepoVax containing 10 μg R9F-PADRE (DPX-R9F), or the combination. (A) C3 tumor growth in donor mice until termination. On day 29, donor mice were terminated and spleens and lymph nodes collected and pooled from each group. Total T cells were purified by magnetic separation and injected intravenously into recipient mice which had been implanted with tumors 3 d prior to adoptive transfer. (B) Antigen-specific immune response of donor cells was confirmed by interferon γ (IFNγ) ELISPOT before transfer to recipient mice. Shown are representative data from a single experiment. (C) C3 tumor growth in recipient mice (n = 5–15) transferred (on day 3, indicated with arrow) with total T cells derived from mCPA and DPX-R9F treated, tumor-bearing mice. (D) C3 tumor growth in recipient mice (n = 5–10) that were transferred (on day 3, indicated with arrow) with CD8+ T cells derived from mCPA and DPX-R9F treated, tumor-bearing mice. Results pooled from 2–3 separate experiments. Statistical analysis was performed by 2-way ANOVA with Bonferroni post test of treatment groups versus the PBS control; ***P < 0.001, ****P < 0.0001.
mCPA alters the immune profile of the spleen of tumor bearing mice
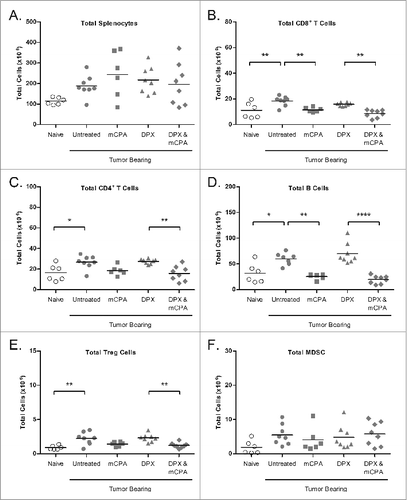
The adoptive transfer of T cells or CD8+ T cells was only able to partially confer immunity to recipient mice, indicating that the mCPA may have other effects on the immune system besides enhancing CTL activity. Low dose sbCPA has been reported to have a transient effect on immune cells, which reach nadir 4 d after treatment before rebounding.Citation8,15 As the spleen is an important reservoir for immune suppressor cells, we looked at the levels of several cell types 4 d and 8 d after treatment by cytofluorimetric analysis using flow cytometry (days 25 and 29 of study, respectively).Citation30
On day 25, 4 d after DPX-R9F vaccination and/or 4 d after mCPA treatment end, we found that while the total number of splenocytes was not different between groups, the total number of CD8+ T cells, CD4+ T cells, and B cells were significantly reduced in groups treated with mCPA compared to non-mCPA treated groups (). No significant differences were detected in total NK cells (data not shown). There was a significant reduction in total Tregs in the combination treated group compared to the vaccine only treated group, but the decline in Tregs observed in the mCPA only treatment group was not significant compared to untreated tumor bearing mice. We also did not detect any differences in the number of myeloid derived suppressor cells (MDSCs) at this time point.
Figure 7. mCPA combined with DPX-R9F vaccination alters the immune cell profile in the spleen. (A-F) C3 tumor-bearing mice (n = 6-8) were treated as in with metronomic cyclophosphamide (mCPA), DepoVax containing 10 μg R9F-PADRE (DPX-R9F), or the combination. On day 25 (4 d after mCPA treatment end), mice were terminated and spleens collected. Total spleen counts (A) were determined and the distribution of splenic immune populations (B-F) quantified by immunofluorescence staining (using the indicated markers) and cytofluorimetric analysis: (A) total splenocytes; (B) total CD8+ T cells (CD8+); (C) total CD4+ T cells (CD4+); (D) total B cells (CD19+); (E) total Treg cells (CD4+/ FoxP3+/ CD25hi); (F) total MDSC (CD11b+/ GR-1hi). Mice were pooled from 2 separate experiments. Statistical analysis was performed by one-way ANOVA with Tukey's post-hoc test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
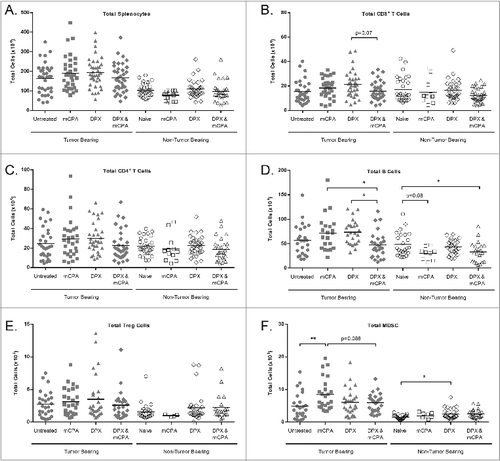
On day 29, 8 d after DPX-R9F vaccination and/or 8 d after mCPA treatment end, we evaluated the spleen populations of tumor bearing and non-tumor bearing mice (). By this time, the total numbers of CD8+ and CD4+ T cells were similar in all groups. Total B cells remained significantly diminished in the combination treated mice as compared to mice treated with mCPA or vaccine alone. In the combination therapy group, however, the total number of Tregs was not significantly different. Notably, total MDSCs in tumor-bearing mice treated with mCPA alone were selectively increased, but not in non-tumor bearing mice treated with mCPA. Of particular relevance, tumor-bearing mice treated with mCPA and DPX-R9F vaccination combinatorial therapy did not display elevated MDSCs levels.
Figure 8. DPX-R9F vaccination attenuates mCPA-induced enrichment of immunosuppressive cells in the spleen. C3 tumor-bearing mice (n = 34–38) were treated as in with metronomic cyclophosphamide (mCPA), DepoVax containing 10 μg R9F-PADRE (DPX-R9F), or the combination. Naïve mice (n = 14–33) without tumor challenge were treated in parallel. On day 29 (8 d after mCPA treatment end), mice were terminated and spleens collected. Total spleen counts (A) were determined and the distribution of splenic immune populations (B-F) quantified by immunofluorescence staining (using the indicated markers) and cytofluorimetric analysis: (A) total splenocytes; (B) total CD8+ T cells (CD8+); (C) total CD4+ T cells (CD4+); (D) total B cells (CD19+); (E) total Treg cells (CD4+/ FoxP3+/ CD25hi); (F) total MDSC (CD11b+/ GR-1hi). Tumor-bearing mice were pooled from 9 separate experiments; non-tumor bearing mice were pooled from 8 separate experiments. Statistical analysis was performed by one-way ANOVA with Tukey's post-hoc test; *P < 0.05, **P < 0.01.
To confirm that these MDSCs had functional activity, they were isolated from the spleens of each tumor-bearing group and used in a suppression assay by co-culturing them at a 2:1 ratio with purified Oregon green-labeled T cells stimulated with anti-CD3/ CD28 beads. The MDSCs from all groups equally suppressed T cell proliferation (Fig. S2).
Discussion
The majority of pre-clinical and clinical studies reported to date evaluating low dose CPA as an immune modulator for vaccination have used single bolus administration of CPA (e.g., 50–100 mg/kg intravenously in mice). Unfortunately, however, this form of administration has displayed poor translational efficacy, which may be in part due to its sensitivity to timing of CPA administration and vaccination.Citation14-16 In contrast, daily low dose metronomic CPA administration (e.g., 20 mg/kg/day orally in mice) may allow more flexibility in the timing of CPA dosing relative to vaccination, as well as the added convenience of pill administration. In our model, both forms of low dose CPA administration enhanced the efficacy of a peptide-based vaccine targeting HPV16-expressing tumors (). In the context of monotherapy, sbCPA provided better protection than mCPA, reflecting inherent differences in these 2 forms of low dose CPA treatment. These results are consistent with the findings of Peng et al. who found that both forms of low dose CPA can enhance the efficacy of a DNA-based HPV16 vaccine as demonstrated by tumor growth and flow cytometric analysis of immune populations.Citation26 In this study, we extended the analysis of metronomic low dose CPA in combination with a peptide vaccine to evaluate immune functionality as well as cell populations.
One of the disadvantages with sbCPA/ vaccine combination is that it is very sensitive to timing.Citation15,16 In mice, the therapeutic window appears to be vaccination 1 to 3 d after sbCPA administration. Since mCPA therapy is administered over a longer period of time, it may prolong the therapeutic window and allow more flexibility in timing of vaccination relative to immune modulation. To test this hypothesis, we investigated whether vaccine efficacy was compromised if given at the beginning or end of a weekly mCPA treatment (). The rapid C3 tumor growth rate taken together with tumor treatment sensitivity impacted tumor burden such that it was inherently different when mice were treated at different time points, thus obscuring differences due to temporal/kinetic effects. However, significant protection was provided to the combination group in which mice received vaccination and mCPA starting 7 d after tumor implantation (mCPA after vaccination) as well as the combination group in which mice received mCPA starting on day 7 followed by vaccination on day 14. While early vaccination is a significant contributor to the effect seen in , it is important to note that this effect was enhanced rather than reduced whether mCPA was provided, irrespectively of whether this was before or after vaccination. Supporting this, the immunogenicity of the vaccine tested by IFNγ ELISPOT was the same in either group (). These results indicate that vaccination may benefit from concurrent mCPA treatment with less timing constraints, a considerable improvement on scheduling flexibility compared to sbCPA.
To understand the mechanism of how mCPA was enhancing vaccine efficacy, we dissected the immune response in the vaccine-draining lymph node and spleen of tumor bearing mice after a single round of treatment with mCPA and/or vaccine. Increased antigen-specific IFNγ ELISPOT responses in the lymph node correlated with an enrichment of antigen-specific CD8+ T cells due to mCPA-induced depletion of non-specific CD8+ T cells. Increased IFNγ ELISPOT responses were also detected in the spleen and correlated with increased CTL activity. These observations indicate that mCPA-induced enrichment of antigen-specific CD8+ T cells in the lymph node allows more efficient proliferation and migration of vaccine-induced immune response to the spleen, ultimately resulting in better systemic CTL activity. The fact that mCPA does not reduce antigen-specific CD8+ T cells induced by vaccination substantiates that this form of immune modulation is particularly suitable for combining with vaccine therapy.
We analyzed the mRNA levels of several genes associated with cytotoxic T-cell responses in order to obtain a comprehensive snapshot of immune modulation within the tumor microenvironment. Quantitative RT-PCR was particularly feasible since the tumors of treated mice were small (<500 mm3, ). Although a limitation of this data is the inability to correlate mRNA expression with a given cell type in this mosaic cell population, the increased expression of several genes associated with CTL activity is highly suggestive of increased TIL cell subsets, although malignant cell (or other, non-immune tumor cell) expression remains possible. Furthermore, the increased CTL activity observed in the spleen and lymph node also supports the notion that the combinatorial therapy increases CTL infiltration into the tumor microenvironment. Interestingly, the mRNA encoding the co-inhibitory receptors programmed cell death 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) were also increased in combination treated mice. Previous studies have demonstrated that PD-1 positive CD8+ T cells isolated from tumors correlate with an antigen-specific response.Citation31 This suggests that the combination treatment is inducing a highly functional CTL response that can penetrate the tumor, but which may still be susceptible to tumor-induced immune suppression in the form of PD-1 or CTLA-4 induction. In light of these observations, it would be of interest to combine mCPA with vaccine and monoclonal antibodies toward these receptors to further enhance therapy, even in the case of advanced disease.
The adoptive transfer experiment demonstrated that tumor control can be partially transferred to recipient mice by total T cells or CD8+ T cells from combination treated donor mice (). Lack of complete efficacy could be attributed to mCPA treatment having a direct antitumor effect, or it could be providing immune modulation in some other form. The significant reduction in lymph node sizes in response to mCPA therapy detected 8 d after treatment indicated that mCPA was capable of inducing prolonged lymphodepletion (). Analysis of splenic immune populations provided additional understanding of the unique effects on the immune cells in each treatment group. The differences between the splenic populations in tumor-bearing mice treated with mCPA alone versus mCPA combined with vaccination provided insight, particularly since these 2 groups had comparable tumor sizes at the time of termination (). We found that mCPA treatment, alone or in combination with vaccine, significantly decreased CD8+ T cells, CD4+ T cells and B cells 4 d after completion of mCPA treatment, but by day 8, the T cell populations returned to pre-treatment levels whereas B cells remained scarce in combination treated groups. The sensitivity of B cells toward various doses of cyclophosphamide treatment is well known, but the participation of B cells in vaccine-induced antitumor immune responses is not well defined.Citation32,33 A suppressor subset of B cells, referred to as regulatory B cells or B10 and recently identified as CD19+CD5+CD1dhi, may play a role in fostering tumor progression.Citation34,35 B cells in general may play an underappreciated role in promoting tumor growth, as B-cell deficiency results in slower tumor growth in some cancer models.Citation36,37 Furthermore, the B-cell depleting anti-CD20 monoclonal antibody has been shown to increase subsequent cellular responses to tumors in miceCitation38 and enhance T-cell responses after vaccination.Citation39 Since treatment with combination therapy provided effective tumor control, it may be inferred that B cells in the HPV16 murine tumor model are promoting tumor growth, and therefore reduction by mCPA contributes to immune enhancement.
Low dose CPA is often associated with a selective decrease in Tregs.Citation8,32 In this study we found that Tregs were significantly reduced in combination treated tumor-bearing mice 4 d after mCPA treatment end, but since the total levels of CD4+ T cells were also reduced at this time point we could not conclude that the Treg reduction was selective. Total Tregs were comparable in all groups by day 8 (). Others have also reported this lack of a targeted effect on Treg populations after low dose CPA treatment in both mouse models and human clinical trials.Citation12,14,40 It has also been reported that instead of depleting Tregs, low dose CPA renders them non-functional, a phenomenon that was not addressed in this study.Citation8 Furthermore, it has been reported that low-dose CPA selectively reduces intratumoral levels of Tregs, but the quantitative RT-PCR analysis of tumors for FoxP3 suggests that this immune cell subset is not being reduced in the tumor microenvironment either ().Citation12 We speculate that while the temporary reduction in Tregs may be a factor contributing to the immune enhancement provided by mCPA, this effect is not sufficient to account for the significant increases in antitumor immunity.
On day 29, splenocytes of mice treated with mCPA monotherapy exhibited a selective and significant increase in MDSCs. Increased occurrence of CD11b+/ GR-1hi MDSCs after low dose CPA treatment has been previously reported, leading to the suggestion that low dose CPA may be limited as a monotherapy because it stimulates accumulation of MDSCs that ultimately leads to rebounding of tumor growth.Citation32,41 To compensate, low dose CPA treatment has been combined with drugs that specifically reduce MDSC levels such as gemcitabine or ATRA.Citation42-44 We found that the combination of mCPA plus DPX-R9F did not increase MDSC levels, negating the need for additional drugs. MDSC's are essentially akin to haematopoietic stem/progenitor cells arrested in development, presumably by the inflammatory cytokine milieu created by the tumor.Citation45,46 We found that the MDSC population was not increased in non-tumor bearing mice in response to mCPA treatment, which suggests that the presence of the tumor was causal in promoting their selective expansion (). We found that the MDSCs isolated from mice in each group retained similar immunosuppressive functionality (Fig. S2), which is in contrast to previous studies reporting that CPA-induced MDSCs displayed higher suppressive activity, or alternatively, no suppressive activity.Citation15,43 Differences such as these may be a reflection of biological factors, such as the type of tumor model used, or technical, such as the assay conditions.
This study was performed with the HPV16 mouse tumor model that expresses the highly immunogenic MHC Class I-restricted epitope HPV16E749–57. Other cancer vaccine models, such as B16, express only weakly immunogenic epitopes, such that extending these findings into other models may not be direct. Additionally, the use of a model overexpressing a viral oncoprotein may influence the immunosuppressive nature of the tumors used herein by making the lesions more amenable to mCPA therapy. However, we have recently completed a Phase I clinical trial of a survivin-targeted vaccine that uses immunogenic modified peptide antigens (DPX-Survivac) in combination with mCPA to treat ovarian cancer patients and have found similarities in the immune responses generated in those patients to the results acquired here (manuscript in preparation). In this respect, our findings in regards to the scheduling of vaccination relative to mCPA treatment were helpful in designing the clinical trial and identifying appropriate biomarkers to assess. While the results of this pre-clinical study cannot be expected to be recapitulated fully in other pre-clinical models or human clinical trials, its sensitivity to immune modulation can be used to probe how immunogenicity of a peptide vaccine and subsequent antitumor activity may be influenced by combination therapies.
In conclusion, we have demonstrated that mCPA provides immune modulation to enhance the efficacy of a peptide vaccine in an HPV16 tumor model. Timing of mCPA therapy in relation to vaccination appears to be less stringent than sbCPA, and therefore may be emulated better in human clinical trials. Increased immune responses detected in this HPV16 murine tumor model are an accurate reflection of immune monitoring data obtained from a Phase I study combining mCPA and DPX-Survivac vaccination and may facilitate the interpretation of those results as well as guide design of subsequent trials. Overall, our data indicates that metronomic cyclophosphamide causes transient lymphodepletion that is quickly repopulated. In the absence of a strong active immune response, immune reconstitution is influenced by the immunosuppressive nature of the tumor, and, at least in this model, resulted in a selective expansion of MDSCs. However, in the context of an active immune response induced by vaccination, immune reconstitution favors antitumor immunity. This bias results in an enrichment of antigen-specific T cells with cytotoxic capacity and with tumor-infiltrating abilities, specifically upon treatment with the combination of mCPA and vaccine. When treating advanced tumors, additional forms of immune modulation, such as anti-PD1 therapy, may further promote an efficacious immune response conducive to the use of vaccine-based immunotherapy in more aggressive clinical settings.
Materials and Methods
Mice and tumor implantation
Pathogen-free, 6–8 week old female C57BL6 mice were obtained from Charles River Labs (St Constant, PQ, Canada). Mice were housed under filter-top conditions and provided food and water ad libitum. Institutional animal care and use guidelines set by Carleton Animal Care Facility at Dalhousie University (Halifax, NS, Canada) were followed for all experiments. All experimental procedures performed during this study were approved by the Ethics Committee at Dalhousie University and strictly followed the guidelines set by The Canadian Council on Animal Care (CCAC).
The C3 tumor line was provided by Dr Martin Kast (USC, Los Angeles, USA). The C3 cells are derived from C57BL6 mouse embryo cells transfected to express HPV16.Citation27 The C3 tumor line was maintained in Iscove's modified Dulbecco's medium (IMDM; Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone) 2% penicillin-streptomycin (Gibco), 50 mM mercaptoethanol (Gibco) and 2 mM L-glutamine (Gibco). For tumor implantation, mice were subcutaneously injected in the left flank with 5 × 105 C3 tumor cells. Tumor growth was measured with digital calipers twice weekly and tumor volume calculated using the formula [(width2 × length)/2]. For experiments requiring determinations of survival, endpoint was determined to be when mice had tumor volumes of ≥ 2000 mm3, or showed significant signs of ill health, such as wasting, severe dehydration, significant decrease in activity and hunched or prostate posture. When survival endpoint was determined, mice were humanely euthanized per the CCAC guidelines.
Peptides
All peptides were synthesized by NeoMPS (San Diego, CA, USA) at >90% purity. The CD8 epitope HPV16E749–57 (RAHNIVYTIF; R9F) was used in each study. In some studies, the irrelevant peptide TRP2180–188 was used (SVYDFFVWL; S9L). All vaccines contained a universal T helper peptide, either PADRE (AKXVAAWTLKAA) or TT830–843 (FNNFTVSFWLRVPKVSASHLE; F21E), as indicated.
Vaccine preparation and immunization
DepoVax peptides vaccines were formulated with a proprietary adjuvant as previously described.Citation3 Briefly, peptides and adjuvant were solubilized in appropriate buffer and mixed with a dioleoylphosphatidyl choline (DOPC) and cholesterol mixture (Lipoid GmBH) to form liposomes. The aqueous mixture was lyophilized to a dry cake, which was reconstituted with Montanide ISA51 VG (SEPPIC) just prior to injection. Mice were vaccinated subcutaneously on the right flank with 50 μL of vaccine. Each dose of vaccine contained 10 μg R9F fused to PADRE + 20 μg adjuvant or 5 μg R9F + 5 μg F21E + 20 μg adjuvant, where indicated. When multiple vaccinations were administered they were given in the same general area while avoiding previous immunization sites.
Cyclophosphamide treatment
Cyclophosphamide (Sigma-Aldrich) was reconstituted in PBS and given either as a single intravenous injection in the lateral tail vein at 100 mg/kg for IV sbCPA administration or provided for consecutive days in drinking water at 0.133 mg/mL calculated to deliver 20 mg/kg/day based on 3 mL water/ mouse/ day for PO mCPA administration. For oral administration, water was changed every 2–3 d Mice that were treated with cyclophosphamide were monitored daily for signs of ill health indicating adverse reactions to cyclophosphamide treatment.
IFNγ ELISPOT
IFNγ ELISPOT assay was performed as previously described.Citation3 Briefly, mature dendritic cells (DCs) were generated by culturing bone marrow cells from naïve C57BL6 mice in complete RPMI media [RPMI 1640 (Gibco) + 10% FBS, 2% penicillin/ streptomycin (Gibco), 2mM L-glutamine (Gibco), 50 mM β-mercaptoethanol (Sigma-Aldrich), and 5mM HEPES buffer (Gibco)] supplemented with murine GM-CSF (Peprotech). DCs were loaded with 10 μg/mL peptides on d 7. Day 8 DCs were resuspended in complete RPMI at 2 × 105 cells/mL and used as antigen presenting cells for the ELISPOT assay.
Right inguinal lymph nodes were collected from mice upon euthanasia. Single cell suspensions were prepared in complete RPMI media and cell concentration adjusted to 2 × 106 cells/mL. Lymph node cells (100 μL) and DCs (100 μL) were added to IFNγ ELISPOT plates (BD Bioscience). The ELISPOT plate was incubated overnight at 37°C, 5% CO2 and then developed the next day using AEC substrate kit (Sigma-Aldrich). Spots were counted using an ImmunoSpot Analyzer, ELISPOT plate reader (C.T.L. Ltd, Shaker Heights, OH, U.S.A.) and enumerated as number of spot-forming units (SFU) per well.
IFNγ ELISPOT performed using splenocytes had the following modifications. Single cell suspensions of splenocytes were prepared by lysing RBCs with ammonium-chloride-potassium solution and resuspending the cells at 5 × 106 cells/mL in complete RPMI media. A volume of 100 μL cells was added into IFNγ ELISPOT plate and stimulated with 100 μL complete RPMI containing no peptide (background control), 20 μg/mL R9F or irrelevant peptide, or 5 × 105 cells/mL C3 tumor cells.
In vivo cytotoxic lymphocyte assay
CTL in vivo assays were performed as described previously.Citation28 Briefly, RBC-lysed splenocytes from naïve C57BL6 mice were resuspended at 108 cells/ mL in HBSS+HEPES and divided into 2 tubes which were pulsed with R9F peptide at 20 μg/mL or unpulsed for 1.5 hours at 37°C. Both fractions were washed and resuspended in PBS. Each fraction was labeled with Oregon Green 488 (Life Technologies) at 5 μM for peptide-pulsed or 0.5 μM for unpulsed. After labeling, the fractions were washed, resuspended in HBSS at 108 cells/mL and pooled 1:1. Recipient mice received 200 μL of the mixed cells via lateral tail vein injection. Eighteen hours later, the mice were terminated and spleens collected. A single cell splenocyte suspension was prepared and the relative levels of each target population detected by flow cytometry. Percent specific lysis of fluorescent donor spleen cells in each mouse was calculated as follows: 1−(rnaive/rvaccinated) × 100%, where r = number of control cells/ number of peptide-pulsed target cells.
Fluorescence cytometry
Cells were pre-incubated with anti-CD16/CD32 Fc blocking antibody (eBioscience) to block non-specific staining. Antibody cocktails were added and cells incubated at 4°C for 30 min. Intracellular staining for FoxP3 was performed by pretreating with a permeabilization kit from eBioscience. The following fluorochrome-conjugated anti-mouse antibodies were used to stain the cells: anti-CD3, anti-CD4, anti-CD8a, anti-GR1, anti-CD11b, anti-CD11c, anti-CD25, anti-NK1.1, anti-CD19, and anti-FoxP3, all purchased from eBioscience. Phycoerythrin conjugated R9F-dextramer-PE was obtained from Immudex. A FACSCalibur (BD Biosciences) was used for acquisition of flow cytometry data and analysis was performed using WinList 7.0 (Verity Software, Topsham, ME, USA).
Adoptive transfer of immunity
Total T cells or CD8+ T cells were isolated from donor mice via negative magnetic selection using EasySep Mouse Pan T and EasySep Mouse CD8+ T cell isolation kits (Stem Cell Technologies) and T cell purity was assessed by fluorescence cytometry. Cells were prepared in HBSS and 10 × 106 T cells or 4 × 106 CD8+ T cells were injected intravenously into recipient mice that had been challenged with C3 tumor cell injections 3 d prior.
Quantitative RT-PCR
Total RNA was isolated from using RNeasy Plus Mini Kit (QIAGEN); 5 μg aliquots were treated with DNAse I (Invitrogen) and reverse transcribed using a SuperScript III reverse transcriptase kit (Invitrogen) and oligo(dT) primer (Invitrogen). PCR primers for Cd8a, Gzmb, Ifng, CD4, FoxP3, Il10, Il4, Pdcd1, Ctla4, and Tbp were designed using Primer-BLAST algorithm (Table S1). Amplifications of these transcripts were performed on a Rotor Gene 3000 real-time PCR machine using a QuantiFast SYBR Green PCR kit (QIAGEN). Data were analyzed based on the standard curve method and normalized against levels of Tbp mRNA encoding the housekeeping TATA box binding protein (Tbp). Quantitative amplifications of CD19, Klrc1 and and Tbp transcripts were perform using RT2 qPCR Primer Assays (QIAGEN) and RT2 SYBR Green ROX FAST kit (QIAGEN). Quantifications were performed based on ΔΔCT method as recommended in the provided protocol.
Statistical analyses
Statistical analyses were conducted with GraphPad Prism 6 (La Jolla, CA, USA) software. Data was analyzed by appropriate test as indicated in figure legends, including one-way ANOVA with Tukey's post-hoc test, Student's t test, log-rank test, and 2-way ANOVA with Bonferroni post test. Significant differences were considered and denoted as: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Disclosure of Potential Conflicts of Interest
The authors GW, OH, MS, MK and MM are currently employed by Immunovaccine Inc. NB is a consultant for Immunovaccine Inc. RL has no conflict of interest to declare.
Supplemental Materials
Supplemental materials for this article are available online at the publisher's website.
953407_Supplementary_Materials.zip
Download Zip (65 KB)Acknowledgments
The authors wish to thank Drs Walter Storkus and Martin Kast for critical review of this manuscript. Thanks to V Kaliaperumal, A MacKay, T Quinton, R Rajagopalan and B Ray for technical assistance.
Funding
This work was supported in part by the MITACS Accelerate program as well as the Atlantic Innovation Fund provided by ACOA.
References
- Berzofsky, JA, Terabe, M, Wood, LV. Strategies to use immune modulators in therapeutic vaccines against cancer. Semin Oncol 2012; 39:348-57; PMID:22595057; http://dx.doi.org/10.1053/j.seminoncol.2012.02.002
- Weir, GM, Liwski, RS, Mansour, M. Immune modulation by chemotherapy or immunotherapy to enhance cancer vaccines. Cancers (Basel) 2011; 3:3114-42; PMID:24212948; http://dx.doi.org/10.3390/cancers3033114
- Karkada, M, Weir, GM, Quinton, T, Sammatur, L, MacDonald, LD, Grant, A, Liwski, R, Juskevicius, R, Sinnathamby, G, Philip, R, et al. A novel breast/ovarian cancer peptide vaccine platform that promotes specific type-1 but not Treg/Tr1-type responses. J Immunother 2010; 33:250-61; PMID:20445345; http://dx.doi.org/10.1097/CJI.0b013e3181c1f1e9
- Daftarian, PM, Mansour, M, Pohajdak, B, Fuentes-Ortega, A, Korets-Smith, E, Macdonald, L, Weir, G, Brown, RG, Kast, WM. Rejection of large HPV-16 expressing tumors in aged mice by a single immunization of VacciMax encapsulated CTL/T helper peptides. J Transl Med 2007; 5:26; PMID:17555571; http://dx.doi.org/10.1186/1479-5876-5-26
- Mansour, M, Pohajdak, B, Kast, WM, Fuentes-Ortega, A, Korets-Smith, E, Weir, GM, Brown, RG, Daftarian, P. Therapy of established B16-F10 melanoma tumors by a single vaccination of CTL/T helper peptides in VacciMax. J Transl Med 2007; 5: 20; PMID:17451606; http://dx.doi.org/10.1186/1479-5876-5-20
- Berinstein, NL, Karkada, M, Morse, MA, Nemunaitis, JJ, Chatta, G, Kaufman, H, Odunsi, K, Nigam, R, Sammatur, L, Macdonald, LD, et al. First-in-man application of a novel therapeutic cancer vaccine formulation with the capacity to induce multi-functional T cell responses in ovarian, breast and prostate cancer patients. J Transl Med 2012; 10:156; PMID:22862954; http://dx.doi.org/10.1186/1479-5876-10-156
- Galluzzi, L, Senovilla, L, Zitvogel, L, Kroemer, G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov 2012; 11:215-33; PMID:22301798; http://dx.doi.org/10.1038/nrd3626
- Lutsiak, ME, Semnani, RT, De Pascalis, R, Kashmiri, SV, Schlom, J, Sabzevari, H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005; 105:2862-8; PMID:15591121; http://dx.doi.org/10.1182/blood-2004-06-2410
- Radojcic, V, Bezak, KB, Skarica, M, Pletneva, MA, Yoshimura, K, Schulick, RD, Luznik, L. Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol Immunother 2009; 59:137-48; PMID:19590872; http://dx.doi.org/10.1007/s00262-009-0734-3
- Schiavoni, G, Mattei, F, Di Pucchio, T, Santini, SM, Bracci, L, Belardelli, F, Proietti, E. Cyclophosphamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood 2000; 95:2024-30; PMID:10706870
- Taieb, J, Chaput, N, Schartz, N, Roux, S, Novault, S, Menard, C, Ghiringhelli, F, Terme, M, Carpentier, AF, Darrasse-Jeze, G, et al. Chemoimmunotherapy of tumors: cyclophosphamide synergizes with exosome based vaccines. J Immunol 2006; 176:2722-9; PMID:16493027; http://dx.doi.org/10.4049/jimmunol.176.5.2722
- Wada, S, Yoshimura, K, Hipkiss, EL, Harris, TJ, Yen, HR, Goldberg, MV, Grosso, JF, Getnet, D, Demarzo, AM, Netto, GJ, et al. Cyclophosphamide augments antitumor immunity: studies in an autochthonous prostate cancer model. Cancer Res 2009; 69:4309-18; PMID:19435909; http://dx.doi.org/10.1158/0008-5472.CAN-08-4102
- Audia, S, Nicolas, A, Cathelin, D, Larmonier, N, Ferrand, C, Foucher, P, Fanton, A, Bergoin, E, Maynadie, M, Arnould, L, et al. Increase of CD4+ CD25 +regulatory T cells in the peripheral blood of patients with metastatic carcinoma: a phase I clinical trial using cyclophosphamide and immunotherapy to eliminate CD4 +CD25+ T lymphocytes. Clin Exp Immunol 2007; 150:523-30; PMID:17956583; http://dx.doi.org/10.1111/j.1365-2249.2007.03521.x
- Vermeij, R, Leffers, N, Hoogeboom, BN, Hamming, IL, Wolf, R, Reyners, AK, Molmans, BH, Hollema, H, Bart, J, Drijfhout, JW, et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int J Cancer 2011; 131:E670-80; PMID:22139992; http://dx.doi.org/10.1002/ijc.27388
- Salem, ML, Kadima, AN, El-Naggar, SA, Rubinstein, MP, Chen, Y, Gillanders, WE, Cole, DJ. Defining the ability of cyclophosphamide preconditioning to enhance the antigen-specific CD8+ T-cell response to peptide vaccination: creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J Immunother 2007; 30:40-53; PMID:17198082; http://dx.doi.org/10.1097/01.cji.0000211311.28739.e3
- Machiels, JP, Reilly, RT, Emens, LA, Ercolini, AM, Lei, RY, Weintraub, D, Okoye, FI, Jaffee, EM. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res 2001; 61:3689-97; PMID:11325840
- Le, DT, Jaffee, EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res 2012; 72:3439-44; PMID:22761338; http://dx.doi.org/10.1158/0008-5472.CAN-11-3912
- Browder, T, Butterfield, CE, Kraling, BM, Shi, B, Marshall, B, O’Reilly, MS, Folkman, J. Antiangiogenic scheduling of chemotherapy improves efficacy against experimental drug-resistant cancer. Cancer Res 2000; 60:1878-86; PMID:10766175
- Man, S, Bocci, G, Francia, G, Green, SK, Jothy, S, Hanahan, D, Bohlen, P, Hicklin, DJ, Bergers, G, Kerbel, RS. Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res 2002; 62:2731-5; PMID:12019144
- Ghiringhelli, F, Menard, C, Puig, PE, Ladoire, S, Roux, S, Martin, F, Solary, E, Le Cesne, A, Zitvogel, L, Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 2007; 56:641-8; PMID:16960692; http://dx.doi.org/10.1007/s00262-006-0225-8
- Smith, JS, Lindsay, L, Hoots, B, Keys, J, Franceschi, S, Winer, R, Clifford, GM. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int J Cancer 2007; 121:621-32; PMID:17405118; http://dx.doi.org/10.1002/ijc.22527
- Ragin, CC, Modugno, F, Gollin, SM. The epidemiology and risk factors of head and neck cancer: a focus on human papillomavirus. J Dent Res 2007; 86:104-14; PMID:17251508; http://dx.doi.org/10.1177/154405910708600202
- Welters, MJ, Kenter, GG, Piersma, SJ, Vloon, AP, Lowik, MJ, Berends-van der Meer, DM, Drijfhout, JW, Valentijn, AR, Wafelman, AR, Oostendorp, J, et al. Induction of tumor-specific CD4+ and CD8+ T-cell immunity in cervical cancer patients by a human papillomavirus type 16 E6 and E7 long peptides vaccine. Clin Cancer Res 2008; 14:178-87; PMID:18172269; http://dx.doi.org/10.1158/1078-0432.CCR-07-1880
- Smyth, LJ, Van Poelgeest, MI, Davidson, EJ, Kwappenberg, KM, Burt, D, Sehr, P, Pawlita, M, Man, S, Hickling, JK, Fiander, AN, et al. Immunological responses in women with human papillomavirus type 16 (HPV-16)-associated anogenital intraepithelial neoplasia induced by heterologous prime-boost HPV-16 oncogene vaccination. Clin Cancer Res 2004; 10:2954-61; PMID:15131030; http://dx.doi.org/10.1158/1078-0432.CCR-03-0703
- Conesa-Zamora, P. Immune responses against virus and tumor in cervical carcinogenesis: treatment strategies for avoiding the HPV-induced immune escape. Gynecol Oncol 2013; 131:480-8; PMID:23994536; http://dx.doi.org/10.1016/j.ygyno.2013.08.025
- Peng, S, Lyford-Pike, S, Akpeng, B, Wu, A, Hung, CF, Hannaman, D, Saunders, JR, Wu, TC, Pai, SI. Low-dose cyclophosphamide administered as daily or single dose enhances the antitumor effects of a therapeutic HPV vaccine. Cancer Immunol Immunother 2012; 62:171-82.
- Feltkamp, MC, Smits, HL, Vierboom, MP, Minnaar, RP, de Jongh, BM, Drijfhout, JW, ter Schegget, J, Melief, CJ, Kast, WM. Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur J Immunol 1993; 23:2242-9; PMID:7690326; http://dx.doi.org/10.1002/eji.1830230929
- Wonderlich, J, Shearer, G, Livingstone, A, Brooks, A. Induction and measurement of cytotoxic T lymphocyte activity. Curr Protoc Immunol 2006; 3: Unit 3.11.
- Guo, Q, Lv, Z, Fu, Q, Jiang, C, Liu, Y, Lai, L, Chen, Q, Shen, J, Wang, Q. IFN-gamma producing T cells contribute to the increase of myeloid derived suppressor cells in tumor-bearing mice after cyclophosphamide treatment. Int Immunopharmacol 2012; 12:425-32; PMID:22226750; http://dx.doi.org/10.1016/j.intimp.2011.12.016
- Ugel, S, Peranzoni, E, Desantis, G, Chioda, M, Walter, S, Weinschenk, T, Ochando, JC, Cabrelle, A, Mandruzzato, S, Bronte, V. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell Rep 2012; 2:628-39; PMID:22959433; http://dx.doi.org/10.1016/j.celrep.2012.08.006
- Inozume, T, Hanada, K, Wang, QJ, Ahmadzadeh, M, Wunderlich, JR, Rosenberg, SA, Yang, JC. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother 2010; 33: 956-64; PMID:20948441; http://dx.doi.org/10.1097/CJI.0b013e3181fad2b0
- Salem, ML, Al-Khami, AA, El-Nagaar, SA, Zidan, AA, Al-Sharkawi, IM, Marcela Diaz-Montero, C, Cole, DJ. Kinetics of rebounding of lymphoid and myeloid cells in mouse peripheral blood, spleen and bone marrow after treatment with cyclophosphamide. Cell Immunol 2012; 276:67-74; PMID:22560674; http://dx.doi.org/10.1016/j.cellimm.2012.03.010
- Smith, PC Sladek, NE. Sensitivity of murine B- and T-lymphocytes to oxazaphosphorine and non-oxazaphosphorine nitrogen mustards. Biochem Pharmacol 1985; 34:3459-63; PMID:3876834; http://dx.doi.org/10.1016/0006-2952(85)90718-X
- Balkwill, F, Montfort, A, Capasso, M. B regulatory cells in cancer. Trends Immunol 2013; 34(4):169-73.
- Yanaba, K, Bouaziz, JD, Haas, KM, Poe, JC, Fujimoto, M, Tedder, TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity 2008; 28:639-50; PMID:18482568; http://dx.doi.org/10.1016/j.immuni.2008.03.017
- Brodt, P, Gordon, J. Natural resistance mechanisms may play a role in protection against chemical carcinogenesis. Cancer Immunol Immunother 1982; 13:125-7; PMID:6984356
- Zhang, Y, Eliav, Y, Shin, SU, Schreiber, TH, Podack, ER, Tadmor, T, Rosenblatt, JD. B lymphocyte inhibition of anti-tumor response depends on expansion of Treg but is independent of B-cell IL-10 secretion. Cancer Immunol Immunother 2012; 62:87-99; PMID:22772949
- Abes, R, Gelize, E, Fridman, WH, Teillaud, JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 2010; 116:926-34; PMID:20439625; http://dx.doi.org/10.1182/blood-2009-10-248609
- Manzur, S, Cohen, S, Haimovich, J, Hollander, N. Enhanced therapeutic effect of B cell-depleting anti-CD20 antibodies upon combination with in-situ dendritic cell vaccination in advanced lymphoma. Clin Exp Immunol 2012; 170:291-9; PMID:23121670; http://dx.doi.org/10.1111/j.1365-2249.2012.04658.x
- Ellebaek, E, Engell-Noerregaard, L, Iversen, TZ, Froesig, TM, Munir, S, Hadrup, SR, Andersen, MH, Svane, IM. Metastatic melanoma patients treated with dendritic cell vaccination, Interleukin-2 and metronomic cyclophosphamide: results from a phase II trial. Cancer Immunol Immunother 2012; 61:1791-804; PMID:22426890; http://dx.doi.org/10.1007/s00262-012-1242-4
- Angulo, I, de las Heras, FG, Garcia-Bustos, JF, Gargallo, D, Munoz-Fernandez, MA, Fresno, M. Nitric oxide-producing CD11b(+)Ly-6G(Gr-1)(+)CD31(ER-MP12)(+) cells in the spleen of cyclophosphamide-treated mice: implications for T-cell responses in immunosuppressed mice. Blood 2000; 95:212-20; PMID:10607705
- Tongu, M, Harashima, N, Monma, H, Inao, T, Yamada, T, Kawauchi, H, Harada, M. Metronomic chemotherapy with low-dose cyclophosphamide plus gemcitabine can induce anti-tumor T cell immunity in vivo. Cancer Immunol Immunother 2012; 62:383-91; PMID:22926062; http://dx.doi.org/10.1007/s00262-012-1343-0
- Mikyskova, R, Indrova, M, Pollakova, V, Bieblova, J, Simova, J, Reinis, M. Cyclophosphamide-induced myeloid-derived suppressor cell population is immunosuppressive but not identical to myeloid-derived suppressor cells induced by growing TC-1 tumors. J Immunother 2012; 35:374-84; PMID:22576342; http://dx.doi.org/10.1097/CJI.0b013e318255585a
- Song, X, Guo, W, Cui, J, Qian, X, Yi, L, Chang, M, Cai, Q, Zhao, Q. A tritherapy combination of a fusion protein vaccine with immune-modulating doses of sequential chemotherapies in an optimized regimen completely eradicates large tumors in mice. Int J Cancer 2010; 128:1129-38; http://dx.doi.org/10.1002/ijc.25451
- Bunt, SK, Clements, VK, Hanson, EM, Sinha, P, Ostrand-Rosenberg, S. Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J Leukoc Biol 2009; 85:996-1004; PMID:19261929; http://dx.doi.org/10.1189/jlb.0708446
- Ostrand-Rosenberg, S Sinha, P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499-506; PMID:19342621; http://dx.doi.org/10.4049/jimmunol.0802740