
Abstract
We recently identified a vicious cycle between granulocyte macrophage colony stimulating factor (GM-CSF) arising from breast cancer cells that have undergone epithelial-mesenchymal transition (EMT) and the tumor-associated macrophage (TAM)-derived chemokine CCL18, a signaling loop that promotes tumor metastasis. Tumor-derived lactate skews GM-CSF-activated macrophages to an anti-inflammatory and immunosuppressive M2 phenotype, suggesting that breaking this cycle in combination with glycolysis inhibitors may inhibit tumor development.
Tumor-associated macrophages (TAMs) are frequently observed at the invasive front of patient tumors, often in conjunction with cancer cells that have undergone epithelial-mesenchymal transition (EMT). This correlation between TAMs and EMT-modified cancer cells suggests a close interaction between these 2 cell populations. We recently reported that EMT-altered breast cancer cells have a superior ability to activate macrophages to an M2 phenotype as compared to their malignant counterparts that have not undergone EMT.Citation1 Mesenchymal-like breast carcinoma cells stimulate macrophages via the production of high levels of granulocyte-macrophage colony-stimulating factor (GM-CSF, also known as CSF-2). Reciprocally, chemokine [C-C motif] ligand 18 (CCL18) from TAMs induces EMT of cancer cells. Furthermore, GM-CSF from cancer cells and CCL18 from TAMs form a positive feedback loop to maintain the EMT and M2-macrophage activation status in co-culture systems and humanized mice. Neutralization of either GM-CSF or CCL18 breaks this vicious cycle and reduces breast cancer metastasis in vitro or in vivo.Citation1 This study has also demonstrated a number of key points that we shall discuss further below.
EMT and Remissive Microenvironment Fostering are Linked by NFκB Signaling
By exploring the mechanism by which cancer cells that undergone EMT massively produce GM-CSF, we have identified nuclear factor κB (NFκB) as a crucial link between malignant cell mobility and their ability to foster a remissive microenvironment for cancer cells.Citation1 The chemokine CCL18 induces NFκB activation via PITPNM3/Pyk2/Src/PI3K/Akt pathway that stabilizes and upregulates Snail (Snai1), a transcription factor underlying EMT. On the other hand, NFκB upregulates a panel of inflammatory cytokines, including GM-CSF that induces TAMs to adopt an immunosuppressive M2 phenotype .
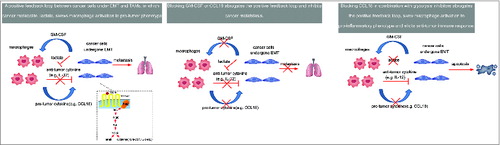
Figure 1. Targeting a positive feedback loop between EMT-modified cancer cells and TAMs in breast cancer. The chemokine (C-C motif) ligand 18 (CCL18) produced by tumor-associated macrophages (TAMs) induces epithelial-mesenchymal transition (EMT) of tumor cells and enhances granulocyte-macrophage colony stimulating factor (GM-CSF) secretion in a PITPNM3-Pyk2-Src-Raf/PI3K-NFκB dependent manner. Reciprocally, GM-CSF from EMT-altered cancer cells activates monocytes to differentiate into a TAM-like phenotype that secrets CCL18. Neutralization of either GM-CSF or CCL18 breaks this vicious cycle and reduces breast cancer metastasis. Immunotherapies blocking CCL18 in combination with glycolysis inhibitors may inhibit cancer metastasis and attenuate immunosuppression, thereby unleashing anticancer immune responses.
Humanized Mouse Models: Bridging Rodent Models to Human Tumor Microvironment
Various positive feedback loops between cancer cells and macrophages have been identified in mouse models, including the macrophage colony stimulating factor (Csf-1, also known as M-CSF) and epidermal growth factor (EGF) paracrine loop.Citation2 However, the chemokine profiles of immune cells are species specific,Citation3 and many chemokines upregulated in human M2 macrophages are either absent or not upregulated in mice.Citation4 CCL18, a key metastasis promoting and TAM-derived cytokine in humans,Citation5 does not have a mouse counterpart. Therefore, we adopted a humanized mouse model by engrafting irradiated and severely immunodeficient mice with CD34+ haematopoietic stem cells (HSCs)Citation3 to bridge the gap between rodent models and patient disease by mimicking the human breast tumor microenvironment. We have reported that neutralizing antibodies that break the vicious cycle between GM-CSF and CCL18 abrogate lung and liver metastasis in these mice.Citation1 These findings are consistent with previous reports that depletion of macrophages by M-CSF knockout in breast cancer susceptible polyoma middle T (PyMT) mice abolished lung metastasis, without altering carcinogenesis and primary tumor growth.Citation2 Therefore, TAMs preferentially affect breast cancer metastatic spread. Furthermore, transplantation of human HSCs generates abundant human CD14+ monocytes/macrophages and CD19+ B cells in the peripheral blood of the humanized mice, whereas human CD3+ T cells and CD56+ natural killer (NK) cells, which are primarily responsible for the antitumor immune response, are barely detectable. Thus, humanized non-obese diabetic severe combined immunodeficiency disease (NOD/SCID) mice represent an excellent model to explore the interaction between human macrophages and cancer cells ongoing during tumor development in vivo, without the influence of anticancer immunity. On the other hand, models in the NOD/SCID/IL2Rγ-/- background that support human T-cell reconstitution, may be suitable to study the interaction between human inflammatory cells and cancer cells, as well as the resultant antitumor immune responses.
Cancer Cells Autonomously Produce GM-CSF
Our study is consistent with previous reports that cancer cells can autonomously produce high amounts of GM-CSF, a potent cytokine that promotes the progression of various malignancies. For example, in a variety of experimental tumors, cancer cell derived GM-CSF rapidly induces the generation of myeloid-derived suppressor cells (MDSCs) and tumor immunosuppression in mice.Citation6 Human disease relevancy has been validated in a clinical trial in which a subset of MDSCs were expanded in metastatic melanoma patients administered GM-CSF-based vaccines.Citation7 Interestingly, GM-CSF has been reported to be among the most potent immune adjuvants for administering tumor vaccines. However, clinically, GM-CSF efficacy as a tumor vaccine adjuvant is controversial. Some studies have demonstrated that GM-CSF enhances antitumor immune responses, whereas others have shown no effect, or even an adverse outcome.Citation8 Our data, and those of others, clearly demonstrate the potential for opposing effects of GM-CSF via activating macrophages in various malignancies.
The GM-CSF Signaling Context Decodes the TAM Response
It has been suggested that the existence of concomitant stimulatory factors contribute to the dichotomy of GM-CSF effects in inflammatory tissues. Indeed, we have observed that GM-CSF alone can induce the production of both pro-inflammatory and anti-inflammatory cytokines.Citation1 In the presence of lactate, which is abundant in the tumor microenvironment, GM-CSF-activated macrophages cannot produce pro-inflammatory cytokines, but rather, generate vast quantities of anti-inflammatory cytokines. It is widely accepted that cancer cells produce excessive lactate even under normoxic conditions, a phenomenon termed the Warburg Effect, or aerobic glycolysis. Such high levels of lactate facilitate GM-CSF stimulated TAMs to undergo M2 polarization, highlighting the link between cancer metabolism and tumor-related inflammation. Indeed, recent studies indicate lactate may contribute to cancer cell immune evasion. For example, lactate impairs differentiation of monocytes to dendritic cells and production of pro-inflammatory cytokine by cytotoxic T cells, but does not affect the function of regulatory T cells.Citation9 Therefore, the selective effects of lactate on different immune cell subsets foster an immunosuppressive microenvironment for cancer progression and its underlying mechanism warrants further investigations.
An increasing number of drugs have been developed to tackle cancer metabolism by targeting key enzymes. Many glycolysis inhibitors re-sensitize cancer cells to chemotherapy and radiotherapy.Citation10 In support, we have observed that blocking lactate production by pretreating MDA-MB-231 cells with oxamic acid, an inhibitor of lactate dehydrogenase, skewed the polarization of macrophages stimulated by tumor-derived GM-CSF to a pro-inflammatory phenotype.Citation1 Collectively, our results link tumor metabolism with immunosuppression and suggest that the combination of glycolysis inhibitors with cancer immunotherapy, potentially including vaccines and immune checkpoint targeting agents, may potentiate clinical efficacy and block tumor development .
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Su S, Liu Q, Chen J, Chen F, He C, Huang D, Wu W, Lin L, Huang W, Zhang J, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014; 25:605-20; PMID:24823638; http://dx.doi.org/10.1016/j.ccr.2014.03.021
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006; 124:263-6; PMID:16439202; http://dx.doi.org/10.1016/j.cell.2006.01.007
- Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol 2012; 12:786-98; PMID:23059428; http://dx.doi.org/10.1038/nri3311
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 2009; 27:451-83; PMID:19105661; http://dx.doi.org/10.1146/annurev.immunol.021908.132532
- Chen J, Yao Y, Gong C, Yu F, Su S, Liu B, Deng H, Wang F, Lin L, Yao H, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 2011; 19:541-55; PMID:21481794; http://dx.doi.org/10.1016/j.ccr.2011.02.006
- Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 2010; 32:790-802; PMID:20605485; http://dx.doi.org/10.1016/j.immuni.2010.05.010
- Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol 2007; 25:2546-53; PMID:17577033; http://dx.doi.org/10.1200/JCO.2006.08.5829
- Eggermont AM. Immunostimulation versus immunosuppression after multiple vaccinations: the woes of therapeutic vaccine development. Clin Cancer Res 2009; 15:6745-7; PMID:19903779; http://dx.doi.org/10.1158/1078-0432.CCR-09-2377
- Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res 2011; 71:6921-5; PMID:22084445; http://dx.doi.org/10.1158/0008-5472.CAN-11-1457
- Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol 2012; 30:671-8; PMID:22781696; http://dx.doi.org/10.1038/nbt.2285