
Abstract
Metastasis accounts for approximately 90% of breast cancer-related deaths. Therefore, novel approaches which prevent or control breast cancer metastases are of significant clinical interest. Interleukin-12 (IL-12)-based immunotherapies have shown promise in controlling metastatic disease, yet modest responses and severe toxicities due to systemic administration of IL-12 in early trials have hindered clinical application. We hypothesized that localized delivery of IL-12 co-formulated with chitosan (chitosan/IL-12) could elicit tumor-specific immunity and provide systemic protection against metastatic breast cancer while minimizing systemic toxicity. Chitosan is a biocompatible polysaccharide derived primarily from the exoskeletons of crustaceans. In a clinically relevant resection model, mice bearing spontaneously metastatic 4T1 mammary adenocarcinomas received intratumoral injections of chitosan/IL-12, or appropriate controls, prior to tumor resection. Neoadjuvant chitosan/IL-12 immunotherapy resulted in long-term tumor-free survival in 67% of mice compared to only 24% or 0% of mice treated with IL-12 alone or chitosan alone, respectively. Antitumor responses following chitosan/IL-12 treatment were durable and provided complete protection against rechallenge with 4T1, but not RENCA renal adenocarcinoma, cells. Lymphocytes from chitosan/IL-12-treated mice demonstrated robust tumor-specific lytic activity and interferon-γ production. Cell-mediated immune memory was confirmed in vivo via clinically relevant delayed-type hypersensitivity (DTH) assays. Comprehensive hematology and toxicology analyses revealed that chitosan/IL-12 induced transient, reversible leukopenia with no changes in critical organ function. Results of this study suggest that neoadjuvant chitosan/IL-12 immunotherapy prior to breast tumor resection is a promising translatable strategy capable of safely inducing to tumor-specific immunity and, in the long term, reducing breast cancer mortality due to progressive recurrences.
Abbreviations:
- ALT, alanine aminotransferase
- CBC, complete blood count
- CTL, cytotoxic T lymphocyte
- DTH, delayed-type hypersensitivity
- ELISPOT, enzyme-linked immunosorbent spot
- Gy, gray
- IFNγ, interferon-γ;. IL-12, interleukin-12
- i.p., intraperitoneal
- i.t., intratumoral
- RENCA, renal cell adenocarcinoma
- s.c., subcutaneous
- TNBC, triple-negative breast cancer
Introduction
Metastasis is responsible for more than 90% of the estimated 40,430 breast cancer deaths that will occur in the United States this year.Citation1 Despite the high mortality associated with metastatic disease, only about 4% or 9,402 of the 235,030 new cases of breast cancer will be metastatic (Stage IV) at diagnosis.Citation2 These data indicate that the majority of metastasis-related breast cancer deaths result from recurrence and progression of regional disease and not from the comparatively few cancers initially diagnosed at Stage IV. Given that there are no curative therapies for metastatic breast cancer, the most effective strategy for reducing breast cancer mortality is to limit the emergence of progressive recurrences following resection.
Adjuvant chemotherapy, radiotherapy, and hormonal therapy, are effective at reducing breast cancer recurrences, however, there is significant room for improvement. For instance, adjuvant chemotherapy increases the 15 y survival rate by only 4.7% in patients younger than 50, and only 2.6% in patients older than 50.Citation3 Even with adjuvant therapy, 7%, 11%, and 13% of Stage I, II, and III patients, respectively, will experience a tumor recurrence within 5 y. The 10-y recurrence rate for Stage I–III breast cancers is about 20%.Citation4 Furthermore, disadvantages of existing adjuvant therapies include well-known toxicities and an increased risk of developing second malignant neoplasms.Citation5
Cancer vaccines and other active immunotherapies are currently being explored as novel adjuvant and neoadjuvant therapies for breast cancer. A key advantage of active immunotherapies is their elaboration of tumor-specific immune responses capable of training a patient's immune system to recognize and eliminate occult tumor cells,Citation6-Citation8 thus preventing their re-emergence at a later time. Another advantage is the durability of a tumor-specific immune response that potentially persists long after administration of the immunotherapy. In contrast, adjuvant chemotherapies and hormonal therapies are only effective during a finite window of administration.
Among cancer immunotherapies, IL-12-based approaches have demonstrated remarkable antitumor and anti-metastatic activity in numerous preclinical models.Citation9-Citation11 In particular, IL-12-based immunotherapies have been shown to generate durable, tumor-specific immune responses capable of preventing recurrence and inhibiting metastasis.Citation12,13 Unfortunately, severe toxicities in early clinical trials, including two on-study deaths,Citation14 have somewhat dampened enthusiasm for IL-12-based therapies. Yet, in a recent NCI-sponsored workshop, IL-12 was recently ranked third in a list of 20 immunotherapeutic agents with high potential for use in treating cancer.Citation15
We and others have noted that cytokine therapeutics would be more effective and less toxic if delivered and maintained in a tissue of interest, i.e. the tumor microenvironment.Citation16,17 As a result, cell-based, virus-based, and plasmid-based paracrine IL-12 delivery strategies are under development and have been reviewed elsewhere.Citation18,19 In particular, non-viral IL-12-based approaches, with or without assistance from liposomes or electroporation, have shown antitumor efficacy in preclinical studiesCitation20,21 and safety in clinical studies.Citation22,23 Because plasmid-based methods require translation to produce IL-12, the amount or dose of IL-12 in the tumor microenvironment from patient to patient will vary depending on the number and type of cells that are transfected.
Strategies which focus on the paracrine delivery of recombinant IL-12 protein are the most direct and quantifiable in terms of ensuring the accuracy and reproducibility of a delivered dose. Several sustained, local release platforms, including IL-12 encapsulation in polymeric microspheres, have shown promise in preclinical studies.Citation24,25 Our previous studies demonstrated that simple mixtures of IL-12 with solutions of chitosan (chitosan/IL-12) when administered intratumorally (i.t.) or intravesically, can significantly enhance local IL-12 retention and safely induce complete tumor regression and protective immunity in several preclinical tumor models.Citation26,27 Chitosan is a biocompatible, unbranched copolymer of glucosamine and N-acetylglucosamine derived primarily from the exoskeletons of crustaceans. Recombinant IL-12 co-formulated with chitosan is retained in the tumor microenvironment for at least 5–6 d following i.t. injection. In contrast, IL-12 injected alone is undetectable between 24 and 48 h.Citation18
In this study, we evaluated a clinically relevant paradigm in which a neoadjuvant immunotherapy is administered i.t. prior to breast tumor resection. We hypothesized i.t. chitosan/IL-12 immunotherapy would initiate a multivalent, tumor-specific immune response capable of eliminating distant metastases and providing durable protection from recurrence. Because tumor resection is indicated for the vast majority of breast cancer patients, neoadjuvant i.t. immunotherapy is a widely applicable approach that can be used in conjunction with standard-of-care. In particular, patients with Stage II and III breast cancers who have 5-y recurrence rates of 11.2 and 12.7%, respectively,Citation4 would benefit from novel treatment approaches. In addition, patients diagnosed with triple-negative breast cancer (TNBC), with 5-y recurrence rates of up to 60% for Stage III TNBCCitation28 and limited treatment options may also benefit from this treatment strategy.
Therefore, the overall aim of this study was to assess the anti-metastatic potential of i.t. chitosan/IL-12 immunotherapy administered prior to resection in an aggressive preclinical model. The chitosan/IL-12 co-formulation was compared to individual components, chitosan, and IL-12 alone, as well as resection alone in terms of overall survival and enumeration of spontaneous pulmonary metastases. The ability of neoadjuvant chitosan/IL-12 immunotherapy to induce a tumor-specific immune response was assessed by cytotoxic T lymphocyte activity, enzyme-linked immunosorbent spot (ELISPOT), and DTH response assays. Finally, comprehensive hematology and blood chemistry analyses were performed to evaluate the safety of i.t. chitosan/IL-12 immunotherapy.
Results
Intratumoral chitosan/IL-12 immunotherapy vs. primary breast tumors
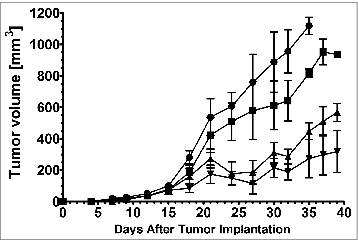
The goal of these initial studies was to determine a dose of IL-12 that, when co-formulated with chitosan, did not significantly impact primary tumor size prior to resection. Thus, mice bearing s.c. 4T1 tumors were treated i.t. with chitosan/IL-12 containing 1, 2, or 5 μg IL-12 on days 6, 9, 12, and 15 after tumor implantation. Saline injections at the same schedule were used in a control cohort. There were no differences in tumor volumes among the different treatments up to day 15 (). After day 15, mean tumor volumes among treatment groups were statistically different (p< 0.001). A Dunnett's post-hoc test confirmed that each of the chitosan/IL-12 treatments was significantly different than control (saline) from day 18 to the conclusion of the experiment (p < 0.05). Bonferroni corrected comparisons of tumor volumes from mice treated with chitosan/IL-12 (1 μg) and chitosan/IL-12 (2 μg) showed significant differences after 21 d (p < 0.001). In contrast, chitosan/IL-12 (2 μg) and chitosan/IL-12 (5 μg) were statistically indistinguishable up to day 35 (p > 0.05). Therefore, chitosan/IL-12 (2 μg) was used in subsequent anti-metastasis studies. Although mice treated with chitosan/IL-12, in particular mice receiving 2 μg or 5 μg IL-12, experienced significant tumor inhibition, no complete regressions were observed.
Figure 1. Effect of chitosan/IL-12 on primary tumor growth. Balb/c mice (n = 5 per group) bearing 4T1 primary tumors were treated i.t. with chitosan (1.5% w/v) co-formulated with 1μg (▪), 2μg (▴) or 5μg (▾) IL-12 on days 6, 9, 12, and 15 following implantation of 1 × 105 4T1 tumor cells in the right flank. Saline (•) was administered as a control. Tumor volumes were measured twice weekly.
Neoadjuvant chitosan/IL-12 vs. breast cancer metastasis
Mice bearing s.c. 4T1 tumors were treated i.t. with saline (control), chitosan solution, IL-12 (2 μg) alone or chitosan/IL-12 (2 μg). All treatments were given on days 6, 9, and 12 prior to tumor resection on day 15. The 4T1 murine mammary adenocarcinoma line spontaneously metastasizes to liver, lung, and bone within one week of inoculation.Citation29,30 Thus, it is likely that implanted tumors had already metastasized prior to i.t. injections and resection. As a result, tumor-bearing mice receiving saline i.t. injections succumbed to metastasis very rapidly, within 38 d of resection (). Similarly, i.t. injections of chitosan alone, which we had previously shown to have no antitumor activity, Citation14,15 was not able to extend survival. The median survival times for saline and chitosan treatment groups were identical at 24 d. The median survival of mice treated with IL-12 alone was 46 d, which was significantly greater than saline-treated controls (p < 0.0001). Six of 25 IL-12-treated mice were rendered tumor-free with no sign of recurrence at the primary site or of metastasis for up to 80 d after resection. Median survival was not reached in mice treated with chitosan/IL-12 as 16 of 24 mice became tumor-free. In terms of survival, chitosan/IL-12 was superior to IL-12 alone (p = 0.0057).
Figure 2. Neoadjuvant chitosan/IL-12 reduces mortality and incidence of distant site metastasis. (A) Balb/c mice bearing 4T1 primary tumors were treated i.t. with chitosan/IL-12 (2 μg) (thick gray line), IL-12 alone (dashed line), chitosan alone (dotted line), or saline (solid black line) on days 6, 9, and 12 post-implantation. Primary tumors were resected on day 15. Mice were followed for the development of lung metastasis. Any mouse exhibiting obvious signs of distress was euthanized and lungs were examined to confirm presence of metastatic disease. Survival was tracked for at least 90 d after resection. (B) In a separate study, mice were euthanized five weeks post-resection. Lungs were removed, infused with India ink, and enumerated under a stereomicroscope. (C) Representative images of lungs from mice receiving neoadjuvant chitosan/IL-12, IL-12 alone or saline five weeks after resection. Lungs from an age-matched naïve, healthy mouse is included for reference.
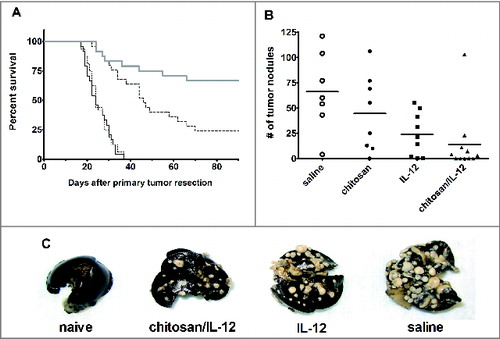
In a separate study, cohorts of 11 tumor-bearing mice were once again treated with saline, chitosan, IL-12, or chitosan/IL-12 prior to resection. Mice surviving 5 weeks after resection were euthanized for enumeration of tumor nodules in the lungs (). The seven surviving saline-treated mice exhibited a wide range of lung lesions with an average of 66.1 ± 39.0 nodules (). It was noted that one of the saline-treated mice had only four nodules, however, nodules appeared to have grown together which caused an underestimation of metastatic lesions. Eight of 11 mice receiving neoadjuvant chitosan averaged 44.4 ± 38.0 nodules. There was no difference in the number of lung nodules between saline and chitosan treated mice (p > 0.05 via ANOVA with Dunnett's post-test). Eight of 11 mice receiving neoadjuvant IL-12 averaged 23.8 ± 21.6 nodules. Two mice had no visible lesions. Finally, 11 of 11 mice receiving neoadjuvant chitosan/IL-12 had 13.7 ± 30.4 nodules on average. Once again, five mice had no visible lesions. Both IL-12 and chitosan/IL-12 treated mice were significantly different from control (p < 0.05 via ANOVA with Dunnett's post-test). The group of mice treated with chitosan/IL-12 had more tumor-free lungs and a lower mean number of lung nodules, than the group treated with IL-12 alone. These differences were not, however, statistically significant (p > 0.05 via ANOVA with a Bonferroni corrected pairwise comparison).
Tumor-specific, protective immunity following neoadjuvant chitosan/IL-12 immunotherapy
A cohort of mice receiving neoadjuvant i.t. chitosan/IL-12 immunotherapy followed by resection and surviving without tumor recurrence or morbidity for more than 60 d were rechallenged on the contralateral flank with either 4T1 or RENCA cells. Naïve mice challenged with 4T1 cells served as a control. All mice rendered tumor-free via neoadjuvant i.t. chitosan/IL-12 immunotherapy were completely protected from rechallenge with 4T1, but not RENCA cells ().
Figure 3. Cured mice exhibit enhanced tumor-specific responses. (A) Mice (n = 5 per group) demonstrating complete regression of 4T1 tumor following neoadjuvant chitosan/IL-12 immunotherapy with resection and exhibiting no signs of metastasis at day 60, were challenged with either 1 × 105 4T1 (▴) or RENCA cells (▪). Naïve mice challenged with 1 × 105 4T1 cells (•) served as control. Tumor volume was measured every 2 d. (B) Splenocytes isolated from cured mice (n = 3) were divided into halves and stimulated with either 5 × 105 irradiated 4T1 (solid symbols) or RENCA (open symbols) cells. Matching shapes represent splenocytes isolated from the same mouse. After 6 d, splenocytes were collected and CTL activity against 5 × 103 4T1 (solid symbols) or RENCA (open symbols) targets was assessed. (C) IFNγ production was assessed via ELISPOT. Splenocytes harvested from cured mice (n = 3) were stimulated with irradiated 4T1 or RENCA cells for one week. CD8+ lymphocytes were then isolated and assayed for the number of IFNγ producing cells when co-incubated with the same 1 × 103 irradiated 4T1 (•) or RENCA (▪) cells. (D) Similarly, the number of IFNγ producing CD8+ splenocytes that were isolated directly from cured mice (n = 3), i.e. without stimulation, following co-incubation with either 1 × 103 irradiated 4T1 (•) or RENCA (▪) cells was assessed via ELISPOT. (E) Cured (n = 5) and naïve (n = 3) mice were given intradermal injections of 1 × 105 irradiated 4T1 cells in one ear and saline in the opposite ear. Swelling was measured at 24 h with a spring-loaded dial gauge. Error bars represent standard deviations
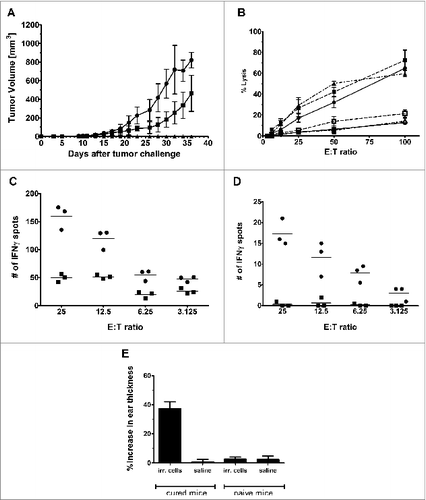
A separate cohort of tumor-free mice was used to assess CTL activity. After 6 d of in vitro stimulation with irradiated 4T1 cells, harvested splenocytes were found to lyse approximately 60–70% of 4T1 targets at an E:T ratio of 100:1 (). In contrast, splenocytes harvested from tumor-free mice and stimulated with irradiated RENCA cells lysed approximately 10–20% of RENCA targets at 100:1 (). Paired t-tests indicated that the 4T1 and RENCA groups were significantly different for all E:T ratios above 12.5:1 (p < 0.05).
ELISPOT analyses to assess tumor-specific IFNγ release was performed using splenocytes that were either stimulated in vitro with irradiated 4T1 cells for 6 d or used immediately after euthanasia. In vitro stimulation with irradiated 4T1 cells increased the numbers of IFNγ producing cells exposed to both 4T1 and RENCA cells (). However, regardless of in vitro stimulation, more splenocytes produced IFNγ when exposed to 4T1 cells than RENCA cells at all E:T ratios (p < 0.05) ().
A DTH study was performed as a potentially relevant clinical indicator of tumor-specific cell-mediated immunity. Tumor-free mice received injections of 4T1 cell lysate and saline in opposite pinnae. Twenty-four hours after injection, thicknesses of ear receiving 4T1 lysates increased by 40% (). Induration did not occur in ear injected with saline. Additionally, naive mice exposed to 4T1 lysate did not experience a significant DTH response.
Toxicology of i.t. chitosan/IL-12
Systemic toxicity remains a serious concern of IL-12-based immunotherapies. Adverse events associated with systemic exposure to IL-12 include, but are not limited to, elevated hepatic enzymes, leukopenia, neutropenia, hypoalbuminemia, intestinal perforations, pleural effusion, and pneumonia. For this study, we were primarily interested in clinically relevant measures of toxicity that could be monitored in the blood. As expected, injections of IL-12 induced significant leukopenia and neutropenia (). Specifically, cou-nts of white blood cells, lymphocytes, and neutrophils, but not monocytes, were suppressed at the 24-h time point. Treatment with chitosan/IL-12 had less of an impact on leukocytes, lymphocytes, and neutrophils than treatment with consecutive daily injections of IL-12. None of the treatments affected hematocrit, platelet or red blood cell counts. Although chitosan/IL-12-induced leukopenia was significant, it was also transient as suppressed cell counts rebounded within 48 h after injection.
Figure 4. Toxicology of i.t. chitosan/IL-12. Tumor bearing mice were treated with either chitosan/IL-12 or saline on day 6. Mice receiving four consecutive daily doses of IL-12 via i.p. injections were used as positive control for toxicity. Blood was collected at 24, 48 or 72 h after injection. In the group receiving consecutive i.p. injections, blood was sampled 24 h after the final injection. (A) Measured hematological parameters included white blood cells (WBC), lymphocytes (LYM), monocytes (MON), neutrophils (NEU), red blood cells (RBC), hematocrit (HCT), and platelets (PLT). (B) Blood chemistry analytes included albumin (ALB), alkaline phosphatase (ALP), globulin (GLOB), alanine aminotransferase (ALT), and glucose (GLU).
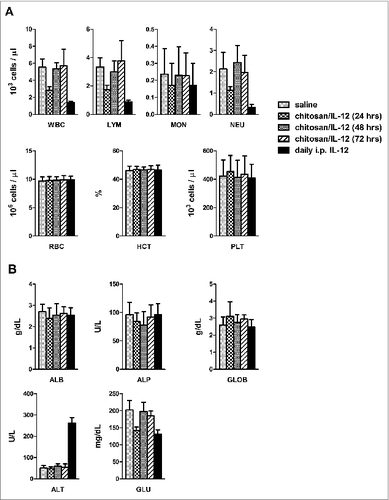
With regard to blood chemistry, treatment-related differences were found in alanine aminotransferase (ALT) and glucose levels. Specifically, when mice were given 4 consecutive daily injections of IL-12, ALT levels increased more than five-fold (). In contrast, mice treated with chitosan/IL-12 did not experience elevated ALT. Glucose levels varied with treatment group in a pattern similar to white blood cell counts. In other words, glucose levels were significantly lower 24 h, but not 48 or 72 h, after treatment with chitosan/IL-12 and 24 h after the daily i.p. injections of IL-12. There were no differences among treatment groups for the remaining 12 analytes ( and supplementary Fig. S1).
Discussion
This study demonstrates that neoadjuvant chitosan/IL-12 immunotherapy is an effective, translatable approach for controlling breast cancer metastasis. Our long-term cure rate of 67% compares favorably with other neoadjuvant immune-based approaches in the highly aggressive 4T1 model. Guiducci et al. demonstrated that i.t. injections of 4T1 primary tumors with an adenovirus encoding CCL16 followed by 4T1 resection resulted in 63% long-term survival.Citation31 Separately, Gil et al. showed that systemic administration of oncolytic vaccinia viruses encoding a CXCR4 agonist followed by 4T1 resection resulted in 20% long-term survival.Citation32 To our knowledge, these are the only studies that have demonstrated long-term survival in the 4T1 model using the clinically feasible approach of neoadjuvant immunotherapy prior to tumor resection.
In a related recent study, IL-12 encapsulated in polylactic acid (PLA) microspheres was administered i.t. as a monotherapy in mice bearing 4T1 tumors.Citation33 IL-12-encapsulated microspheres slowed primary and metastatic tumors but did not appear to promote long-term survival. Co-encapsulation of additional cytokines, interleukin-18 and tumor necrosis factor-α, was shown to improve antitumor activity.Citation33 Another related study investigated the effect of neoadjuvant immunotherapy with IL-12-loaded microspheres in the Line-1 lung cancer model.Citation34 Administration of IL-12-loaded microspheres prior to resection resulted in decreased local recurrence and pulmonary metastases. The percentage of mice with pulmonary nodules was reduced from 60% following resection alone to 20% with neoadjuvant IL-12-loaded microspheres and resection. While the Line-1 model is not as aggressive as the 4T1 model, these results demonstrate that localized IL-12 can be effective at controlling metastases. Furthermore, the mechanisms by which i.t. injected IL-12-encapsulated micropheres induce antitumor immunity has been described in several elegant studies by Egilmez et al. Citation35-Citation39 Mechanisms include: (1) de novo priming of CD8+ effectors in the tumor draining lymph nodes followed by rapid tumor infiltration; (2) restoration of cytotoxic function of tumor-resident CD8+ T cells; (3) induction of apoptosis of tumor-supporting regulatory T cells; (4) delayed tumor infiltration and cytotoxicity by NK cells; and (5) conversion of tumor associated macrophages from tumor supporting to pro-immunogenic. It is not clear if previously reported concerns regarding cytokine stability in microspheres, i.e., up to 80% of the bioactivity of IL-12 was lost when PLA/IL-12 microspheres were stored for three weeks, have been overcome.Citation40
Neoadjuvant chitosan/IL-12 immunotherapy, in addition to improving long-term survival, was found to induce tumor-specific immunity as demonstrated in three immunological studies. CTL and ELISPOT analyses revealed that splenocytes from cured mice contain highly active 4T1-specific lymphocytes (). ELISPOT assays, in particular, are clinically relevant as they are routinely used to enumerate frequencies of antigen-specific lymphocytes among peripheral blood mononucleocytes in clinical studies evaluating cancer vaccines.Citation41 As expected, IFNγ spot formation by CD8+ lymphocytes was significantly higher when exposed to 4T1 (specific) cells versus RENCA (non-specific) cells. In vitro stimulation amplified both specific and non-specific IFNγ spot formation, however, between-group differences remained significant. CTL studies revealed brisk, up to 80%, lysis of 4T1 cells following in vitro stimulation. To rule out the possibility that in vitro stimulation with irradiated tumor cells was capable of inducing an adaptive immune response de novo, splenocytes from mice cured of 4T1 tumors were stimulated with irradiated RENCA cells. The resulting 10–20% lysis of RENCA targets was appreciable but far lower than 4T1-specific killing.
Mice cured following neoadjuvant chitosan/IL-12 immunotherapy also exhibited elevated DTH responses (). DTH studies are particularly useful in the development of cancer vaccines as they measure cell-mediated immune memory directly in the host. In this more complex setting, any immune suppression experienced by the host is also taken into account to provide a more realistic account of antitumor immunity. Not surprisingly, positive DTH responses consistently correlate with favorable prognosis in clinical studies.Citation42,43 Another benefit of DTH responses is the ability to repeat measurements longitudinally to evaluate tumor-specific immunity over time, e.g., every 6 mo. Such data are clinically meaningful in determining if antitumor immunity is maintained in the long-term.
Our data indicate that DTH responses can be achieved with whole tumor cell lysates. Whole cell lysates created by exposing tumor cells to repeated freeze-thaw cycles are not as immunogenic as irradiated tumor cells.Citation44 However, lysates avoid the serious, albeit minimal, risk of injecting patients with live cells that may have survived irradiation.
Tumor protection studies provide another indication that neoadjuvant chitosan/IL-12 immunotherapy induces tumor-specific immunity. The fact that all cured mice were completely protected from a rechallenge with live 4T1 cells was somewhat unexpected. The 4T1 tumor is well known to be non-immunogenic. We (data not shown) and others have found that prophylactic vaccinations with irradiated 4T1 cells do not protect mice from a live tumor challenge.Citation45 Therefore, chitosan/IL-12 injections into a live tumor appear to create a more robust adaptive immune response despite the presence of tumor-derived immune suppressive signals. Egilmez et al. have shown that i.t. injections containing IL-12 can re-activate anergic tumor-specific T cells and reverse immunosuppression.Citation46 It is also possible that local production of IFNγ due to high levels of IL-12 maintained within the tumor microenvironment induced phenotypic changes in tumor cells, such as upregulation of MHC and costimulatory molecules, which may have increased their immunogenicity. The mechanism of enhanced adaptive immunity following chitosan/IL-12 immunotherapy is the subject of ongoing studies.
Because of severe toxicities in clinical studies due to daily systemic injections of IL-12, any novel strategy employing IL-12 must demonstrate safety prior to being considered for clinical translation. Our data show that, while systemic IL-12 toxicity is detected after i.t. chitosan/IL-12 immunotherapy, the severity is significantly lower than toxicity induced by daily doses of IL-12. This was anticipated, not only because the daily i.p injections utilized a higher cumulative dose of 8μg IL-12, but also because frequent IL-12 administrations are known to be primarily responsible for severe toxicities in human studies. In fact, large boluses at the maximum tolerated dose or higher have been well tolerated if administered weekly or twice-weekly instead of daily.Citation47,48 We anticipate that neoadjuvant i.t. chitosan/IL-12 immunotherapy in breast cancer patients would be scheduled as two or three weekly injections. Our previous study demonstrated that most, if not all, of the co-formulated IL-12 disappeared from an i.t. injection after one week. Taken together, weekly injections would be safer and more feasible than daily injections while providing high, local levels of IL-12 better than biweekly or monthly injections.
Furthermore, our data demonstrate that leukopenia, lymphopenia, and neutropenia are transient with cell counts returning to basal levels as soon as 48 h following treatment. ALT levels, which are often elevated following systemic IL-12 administration,Citation49 were substantially increased in mice receiving daily i.p. IL-12 but not a single i.t. chitosan/IL-12 injection. Again, while this may be considered an unfair comparison due to the difference in cumulative IL-12 dose, the fact that ALT levels in chitosan/IL-12-treated mice were similar to controls is encouraging.
It should be noted that toxicity was only assessed after one chitosan/IL-12 injection. Clinical side effects and IFNγ induction have been shown to decrease with repeated weekly or biweekly IL-12 administrations.Citation14,50 Thus, toxicities are most severe with the first dose of IL-12. Subsequent IL-12 administrations cause a blunted response due to down-regulation of IL-12 signaling and/or increased IL-10 production.
Lastly, a significant advantage of neoadjuvant chitosan/IL-12 immunotherapy in the treatment of breast cancer is its utilization of the host's own tumor as a source of antigen. IL-12-based immunotherapies induce the production of IFNγ and other pro-inflammatory cytokines which have direct cytotoxic effects on tumor cells.Citation51 Antigen presenting cells phagocytose tumor cell debris and traffic to lymph nodes for presentation to T cells in an IL-12-induced Th1 polarized environment. Therefore, unlike most other breast cancer therapies, the efficacy of chitosan/IL-12 immunotherapy is not expected to depend on estrogen receptor (ER), progesterone receptor (PgR) or human epithelial growth factor receptor-2 (HER-2) status. In fact, it has been shown that there is no correlation between antitumor lymphocytic immunity and ER or PgR status.Citation52 HER-2 is a potential target for breast cancer immunotherapy.Citation53 However, other tumor-associated antigens elaborated during chitosan/IL-12 immunotherapy can be expected to compensate for the absence of HER-2.
That efficacy is likely to be independent of receptor status is particularly important as breast cancer is a complex, highly heterogeneous disease with tremendous intra- and inter-patient variability. For instance, only 30% of tumor cells need to stain positive for HER-2 for the tumor to be considered HER2-positive and therefore eligible for trastuzumab.Citation54 This receptor independence also implies a potential opportunity for chitosan/IL-12 in the management of TNBC. Patients with TNBC have fewer treatment options due to incompatibility of endocrine or HER2-directed therapies, lower 5-y survival rates and higher rates of distant recurrence in the first 5 y after diagnosis (hazard ratio = 2.6).Citation55 Thus, neoadjuvant chitosan/IL-12 immunotherapy may provide an effective option for a patient population in need of novel treatments. 4T1 tumors do not express ER, PR or HER2/neu (Supplementary Fig. S2) and are thus a good model of TNBC.
In conclusion, i.t. chitosan/IL-12 neoadjuvant immunotherapy is effective in controlling metastasis in a highly aggressive preclinical model of breast cancer. This active immunotherapy uses the host's own tumor as the source of antigen for the generation of a robust tumor-specific adaptive immune response. The utilization of localized, infrequent delivery of IL-12 minimizes toxicity concerns typically associated with daily, systemic IL-12-based approaches. Chitosan/IL-12 immunotherapy is readily combined with standard-of-care resection to support rapid clinical translation.
Materials and Methods
Animals, cell lines, and reagents
Female BALB/cByJ mice (8–12 weeks old) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were housed and maintained under pathogen-free conditions in microisolator cages. Animal care was in compliance with the recommendations of The Guide for Care and Use of Laboratory Animals (National Research Council).
Dulbecco's modified Eagle's Medium (DMEM), Roswell Park Memorial Institute (RPMI) 1640 medium and Fetal bovine serum (FBS) were obtained from Hyclone Laboratories (Logan, UT). Trypsin-versene EDTA mixture, penicillin-streptomycin, nonessential amino acid solution, L-Glutamine, sodium pyruvate, HEPES, and ACK lysis buffer were purchased from Lonza (Walkersville, MD). Dulbecco's phosphate-buffered saline solution (DPBS) without calcium and magnesium was provided by Mediatech (Manassas, VA). Histopaque 1077 was obtained from MP Biomedicals (Solon, OH, USA). Recombinant murine IL-12 was purchased from Peprotech (Rocky Hill, NJ). Chitosan glutamate (Protosan G213) was purchased from NovaMatrix (Sandvika, Norway).
4T1 murine mammary adenocarcinoma and RENCA renal carcinoma cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cell lines were maintained in vitro at 37°C with 5% CO2 in DMEM medium (4T1 cells) or RPMI 1640 medium (RENCA cells) supplemented with 10% FBS, 100 IU/mL penicillin, 100 μg/mL streptomycin, 1 mM sodium pyruvate, 10 mM HEPES, and 4 mM L-Glutamine.
IL-12 dosing studies
Tumors were generated via subcutaneous (s.c.) injection of 1 × 105 low passage 4T1 tumor cells in the shaved left flank of each mouse. Flank implantation was preferred to orthotopic mammary gland implantation due to the tendency of the latter tumors to ulcerate in our hands. Ten minutes prior to treatment, mice were anesthetized using ketamine (75 mg/kg)/xylazine (15 mg/kg) anesthesia. Mice bearing 4T1 tumors were treated i.t. with 1.5% (w/v) chitosan solutions containing 1, 2, or 5 μg IL-12 on days 6, 9, 12, and 15 after tumor implantation. DPBS injections at the same schedule were used in a control cohort. All injection volumes were 50 μL. Tumor volumes were recorded twice weekly.
Neoadjuvant immunotherapy and resection model
Mice bearing 4T1 tumors received i.t. injections of DPBS, IL-12 (2 mg) alone, 1.5% (w/v) chitosan solution alone, or 1.5% (w/v) chitosan admixed with IL-12 (2 mg) on days 6, 9, and 12 after tumor implantation. All injection volumes were 50 μL. Tumors were resected on day 15 as described previously.Citation30 Briefly, mice were anesthetized as above and the area around the tumor was shaved using a finisher trimmer. The surgical area was then disinfected using betadine solution followed by 70% ethanol solution. The primary tumor was then excised by lifting the tumor with forceps and cutting around the base using a surgical blade. Blood vessels feeding the primary tumor were cauterized and the resection site was closed with wound clips.
Following primary tumor resection, mice were monitored daily for primary tumor recurrence and morbidity. Any mouse experiencing a primary tumor recurrence was excluded from the study. Any mouse experiencing significant weight loss, obvious distress, or labored breathing was euthanized and lungs were removed to confirm death due to metastatic disease. Mice surviving > 80 d following resection were deemed cured.
Additional cohorts of mice were treated and resected as above. Five weeks following primary tumor resection, mice were euthanized. Lungs were perfused with 15% (v/v) India ink and fixed in Fekete's solution for enumeration of tumor nodules under a stereomicroscope.
Tumor rechallenge studies
Mice exhibiting complete regression of 4T1 tumors following chitosan/IL-12 treatment for at least 60 d were challenged with 1 × 105 parental 4T1 tumor cells or 1 × 105 irrelevant syngeneic RENCA cells (n = 5 per group) via s.c. injection in the opposite (right) flank. The tumor volumes were recorded every 2 d Mice were euthanized when tumor volumes reached 2000 mm3.
Cytotoxic T lymphocyte (CTL) assay
Spleens from mice cured following chitosan/IL-12 treatment were harvested, mechanically disrupted with a syringe plunger, and passed through a 70-μm nylon mesh strainer. Erythrocytes were lysed with ACK lysing buffer. Unfractionated splenocytes from each mouse were divided into four upright T-25 flasks containing 20 mL complete medium and either 5 × 105 irradiated (100 Gy) 4T1 or RENCA cells. After 6 d, splenocytes were collected on a histopaque gradient and counted. Cytotoxic activity of recovered splenocytes against 5 × 103 4T1 or RENCA cells (targets) was assayed in a 96-well plate using a non-radioactive cytotoxicity assay (CytoTox-Glo; Promega; Madison, WI) per the manufacturer's instructions.
Enzyme-linked immunosorbent spot (ELISPOT) assay
Splenocytes were harvested from cured mice as described above. Approximately half of the splenocytes were stimulated in vitro with irradiated 4T1 cells, as described above, while the remaining half was used without additional stimulation. CD8+ lymphocytes were isolated from splenocyte fractions via negative selection using magnetic beads (Dynabeads Untouched Mouse CD8 kit; Invitrogen; Carlsbad, CA). Isolated CD8+ lymphocytes were used as responder cells in a Mouse Interferon-γ (IFNγ) ELISPOT kit (BD Bioscien ces; San Diego, CA). Briefly, pre-coated ELISPOT plates were blocked with RPMI containing 10% FBS. CD8+ (responder) cells were co-incubated with 5 × 103 4T1 or RENCA cells. Responder cells plated without target cells served as background controls. After 24 h, wells were washed and incubated with biotinylated detection antibody and horseradish peroxidase conjugated streptavidin. Spots, representing IFNγ secreting cells, were developed with AEC substrate set. After allowing plates to dry, spots were counted manually under a stereomicroscope at 300X magnification.
Delayed-type hypersensivity (DTH) assay
Baseline thicknesses of both ears of cured mice were measured with a spring-loaded dial gauge (Mitutoyo Corp., Tokyo, Japan). Prior to antigen challenge, mice were anesthetized. Ten microliters of DPBS or lysate from 1 × 105 4T1 cells were injected into opposite pinnae. Tumor lysates were generated via three freeze-thaw cycles of 4T1 cells. Ear thicknesses were measured 24 h after challenge. The thickness of the ear challenged with tumor lysate was divided by the thickness of the ear challenged with DPBS to obtain percent increase in ear thickness.
Hematology and blood chemistry analyses
Blood composition and chemistry analyses were performed on mice bearing 6-d old 4T1 tumors that were treated with either chitosan/IL-12 or DPBS via intratumoral injection. A group of mice receiving four consecutive daily doses of IL-12 (2 μg) via intraperitoneal (i.p.) injections was used as a positive control for toxicity. Blood (100–200 μL) was collected from the submandibular vein 24, 48, and 72 h after treatment with chitosan/IL-12. No mouse was sampled more than once. Similarly, blood was collected, 24 h after saline injections and 24 h after the final i.p. injection of IL-12 alone. Fourteen blood chemistries including albumin, alkaline phosphatase, ALT, amylase, calcium, creatinine, globulin, glucose, phosphorus, potassium, sodium, total bilirubin, total protein, and blood urea nitrogen were analyzed on a VetScan VS2 (Abaxis; Union City, CA). Complete blood counts (CBCs) with 3-part differential were performed on a hematology analyzer (HM5; Abaxis; Union City, CA).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
968001_Supplementary_Materials.zip
Download Zip (330.5 KB)Funding
This work was supported by grants from the Arkansas Breast Cancer Research Programs (the University of Arkansas for Medical Sciences Translational Research Institute, CTSA Grant Award # UL1TR000039, provided resources during grant review and selection process), the Arkansas Biosciences Institute, the University of Arkansas Women's Giving Circle, The Life Tie Project, and the National Institutes of Health (K22CA131567, R01CA172631, and P30GM103450).
References
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9-29; PMID:24399786; http://dx.doi.org/10.3322/caac.21208
- NCDB Analytic Cases: Disease site by American Joint Committee on Cancer Stage. NCDB Benchmark Reports; September 13, 2013. Available from: https://cromwell.facs.org/BMarks/BMCmp/ver10/Docs/. Accessed June 1, 2014
- (EBCTCG) EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 365:1687-717; PMID:15894097; http://dx.doi.org/10.1016/S0140-6736(05)66544-0
- Brewster AM, Hortobagyi GN, Broglio KR, Kau SW, Santa-Maria CA, Arun B, Buzdar AU, Booser DJ, Valero V, Bondy M et al. Residual risk of breast cancer recurrence 5 years after adjuvant therapy. J Natl Cancer Inst 2008; 100:1179-83; PMID:18695137; http://dx.doi.org/10.1093/jnci/djn233
- Dong C, Chen L. Second malignancies after breast cancer: The impact of adjuvant therapy. Mol Clin Oncol 2014; 2:331-6; PMID:24772296
- Hanna MG. Immunotherapy with autologous tumor cell vaccines for treatment of occult disease in early stage colon cancer. Hum Vaccin Immunother 2012; 8:1156-60; PMID:22854664; http://dx.doi.org/10.4161/hv.20740
- Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunol Rev 2011; 239:27-44; PMID:21198663; http://dx.doi.org/10.1111/j.1600-065X.2010.00979.x
- Lu W, Su J, Kim LS, Bucana CD, Donawho C, He J, Fidler IJ, Dong Z. Active specific immunotherapy against occult brain metastasis. Cancer Res 2003; 63:1345-50; PMID:12649197
- Horinaga M, Harsch KM, Fukuyama R, Heston W, Larchian W. Intravesical interleukin-12 gene therapy in an orthotopic bladder cancer model. Urology 2005; 66:461-6; PMID:16040105; http://dx.doi.org/10.1016/j.urology.2005.03.052
- Colombo MP, Trinchieri G. Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13:155-68; PMID:11900991; http://dx.doi.org/10.1016/S1359-6101(01)00032-6
- Boggio K, Nicoletti G, Di Carlo E, Cavallo F, Landuzzi L, Melani C, Giovarelli M, Rossi I, Nanni P, De Giovanni C et al. Interleukin 12-mediated prevention of spontaneous mammary adenocarcinomas in two lines of Her-2neu transgenic mice. J Exp Med 1998; 188:589-96; PMID:9687535; http://dx.doi.org/10.1084/jem.188.3.589
- Pan WY, Lo CH, Chen CC, Wu PY, Roffler SR, Shyue SK, Tao MH. Cancer immunotherapy using a membrane-bound interleukin-12 with B7-1 transmembrane and cytoplasmic domains. Mol Ther 2012; 20:927-37; PMID:22334018; http://dx.doi.org/10.1038/mt.2012.10
- Rakhmilevich AL, Janssen K, Hao Z, Sondel PM, Yang NS. Interleukin-12 gene therapy of a weakly immunogenic mouse mammary carcinoma results in reduction of spontaneous lung metastases via a T-cell-independent mechanism. Cancer Gene Ther 2000; 7:826-38; PMID:10880012; http://dx.doi.org/10.1038/sj.cgt.7700176
- Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 1997; 90:2541-8; PMID:9326219
- Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev 2008; 222:357-68; PMID:18364014; http://dx.doi.org/10.1111/j.1600-065X.2008.00604.x
- Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, Rosenberg SA. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res 2012; 18:1672-83; PMID:22291136; http://dx.doi.org/10.1158/1078-0432.CCR-11-3050
- Simpson-Abelson MR, Purohit VS, Pang WM, Iyer V, Odunsi K, Demmy TL, Yokota SJ, Loyall JL, Kelleher RJ Jr, Balu-Iyer S et al. IL-12 delivered intratumorally by multilamellar liposomes reactivates memory T cells in human tumor microenvironments. Clin Immunol 2009; 132:71-82; PMID:19395317; http://dx.doi.org/10.1016/j.clim.2009.03.516
- Salem ML, Gillanders WE, Kadima AN, El-Naggar S, Rubinstein MP, Demcheva M, Vournakis JN, Cole DJ. Review: novel nonviral delivery approaches for interleukin-12 protein and gene systems: curbing toxicity and enhancing adjuvant activity. J Interferon Cytokine Res 2006; 26:593-608; PMID:16978064; http://dx.doi.org/10.1089/jir.2006.26.593
- Sangro B, Melero I, Qian C, Prieto J. Gene therapy of cancer based on interleukin 12. Curr Gene Ther 2005; 5:573-81; PMID:16457647; http://dx.doi.org/10.2174/156652305774964712
- Cemazar M, Jarm T, Sersa G. Cancer electrogene therapy with interleukin-12. Curr Gene Ther 2010; 10:300-11; PMID:20560875; http://dx.doi.org/10.2174/156652310791823425
- Hallaj-Nezhadi S, Lotfipour F, Dass C. Nanoparticle-mediated interleukin-12 cancer gene therapy. J Pharm Pharm Sci: Pub Can Soc Pharm Sci, Soc Can Des Sci Pharm 2010; 13:472-85; PMID:21092717
- Anwer K, Barnes MN, Fewell J, Lewis DH, Alvarez RD. Phase-I clinical trial of IL-12 plasmidlipopolymer complexes for the treatment of recurrent ovarian cancer. Gene therapy 2010; 17:360-9; PMID:20033066; http://dx.doi.org/10.1038/gt.2009.159
- Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK, Munster PN, Sullivan DM, Ugen KE, Messina JL et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol: Off J Am Soc Clin Oncol 2008; 26:5896-903; PMID:19029422; http://dx.doi.org/10.1200/JCO.2007.13.9048
- Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res 2000; 60:3832-7; PMID:10919657
- Egilmez NK, Kilinc MO, Gu T, Conway TF. Controlled-release particulate cytokine adjuvants for cancer therapy. Endocr Metab Immune Disord Drug Targets 2007; 7:266-70; PMID:18220947; http://dx.doi.org/10.2174/187153007782794335
- Zaharoff DA, Hance KW, Rogers CJ, Schlom J, Greiner JW. Intratumoral immunotherapy of established solid tumors with chitosanIL-12. J Immunother 2010; 33:697-705; PMID:20664357; http://dx.doi.org/10.1097/CJI.0b013e3181eb826d
- Zaharoff DA, Hoffman BS, Hooper HB, Benjamin CJ, Khurana KK, Hance KW, Rogers CJ, Pinto PA, Schlom J, Greiner JW. Intravesical immunotherapy of superficial bladder cancer with chitosaninterleukin-12. Cancer Res 2009; 69:6192-9; PMID:19638573; http://dx.doi.org/10.1158/0008-5472.CAN-09-1114
- Pogoda K, Niwińska A, Murawska M, Pieńkowski T. Analysis of pattern, time and risk factors influencing recurrence in triple-negative breast cancer patients. Med Oncol 2013; 30:388; PMID:23292831; http://dx.doi.org/10.1007/s12032-012-0388-4
- Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 1992; 52:1399-405; PMID:1540948
- Pulaski BA, Ostrand-Rosenberg S. Mouse 4T1 breast tumor model. Curr Protoc Immunol 2001; 39:20.2:20.2.1–20.2.16; PMID:18432775; http://dx.doi.org/10.1002/0471142735.im2002s39
- Guiducci C, Di Carlo E, Parenza M, Hitt M, Giovarelli M, Musiani P, Colombo MP. Intralesional injection of adenovirus encoding CC chemokine ligand 16 inhibits mammary tumor growth and prevents metastatic-induced death after surgical removal of the treated primary tumor. J Immunol 2004; 172:4026-36; PMID:15034014; http://dx.doi.org/10.4049/jimmunol.172.7.4026
- Gil M, Seshadri M, Komorowski MP, Abrams SI, Kozbor D. Targeting CXCL12CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc Natl Acad Sci U S A 2013; 110:E1291-300; PMID:23509246; http://dx.doi.org/10.1073/pnas.1220580110
- Sabel MS, Su G, Griffith KA, Chang AE. Intratumoral delivery of encapsulated IL-12, IL-18 and TNF-alpha in a model of metastatic breast cancer. Breast Cancer Res Treat 2010; 122:325-36; PMID:19802695; http://dx.doi.org/10.1007/s10549-009-0570-3
- Sabel MS, Hill H, Jong YS, Mathiowitz E, Bankert RB, Egilmez NK. Neoadjuvant therapy with interleukin-12-loaded polylactic acid microspheres reduces local recurrence and distant metastases. Surgery 2001; 130:470-8; PMID:11562672; http://dx.doi.org/10.1067/msy.2001.115839
- Kilinc MO, Aulakh KS, Nair RE, Jones SA, Alard P, Kosiewicz MM, Egilmez NK. Reversing tumor immune suppression with intratumoral IL-12: activation of tumor-associated T effectormemory cells, induction of T suppressor apoptosis, and infiltration of CD8+ T effectors. J Immunol 2006; 177:6962-73; PMID:17082611; http://dx.doi.org/10.4049/jimmunol.177.10.6962
- Kilinc MO, Gu T, Harden JL, Virtuoso LP, Egilmez NK. Central role of tumor-associated CD8+ T effectormemory cells in restoring systemic antitumor immunity. J Immunol 2009; 182:4217-25; PMID:19299720; http://dx.doi.org/10.4049/jimmunol.0802793
- Kilinc MO, Rowswell-Turner RB, Gu T, Virtuoso LP, Egilmez NK. Activated CD8 +T-effectormemory cells eliminate CD4+ CD25+ Foxp3+ T-suppressor cells from tumors via FasL mediated apoptosis. J Immunol 2009; 183:7656-60; PMID:19923444; http://dx.doi.org/10.4049/jimmunol.0902625
- Gu T, Kilinc MO, Egilmez NK. Transient activation of tumor-associated T-effectormemory cells promotes tumor eradication via NK-cell recruitment: minimal role for long-term T-cell immunity in cure of metastatic disease. Cancer Immunol, Immunother: CII 2008; 57:997-1005; PMID:18049819; http://dx.doi.org/10.1007/s00262-007-0430-0
- Watkins SK, Egilmez NK, Suttles J, Stout RD. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol 2007; 178:1357-62; PMID:17237382; http://dx.doi.org/10.4049/jimmunol.178.3.1357
- Sharma A, Harper CM, Hammer L, Nair RE, Mathiowitz E, Egilmez NK. Characterization of cytokine-encapsulated controlled-release microsphere adjuvants. Cancer Biother Radiopharm 2004; 19:764-9; PMID:15665625; http://dx.doi.org/10.1089/cbr.2004.19.764
- Slota M, Lim JB, Dang Y, Disis ML. ELISpot for measuring human immune responses to vaccines. Expert Rev Vaccines 2011; 10:299-306; PMID:21434798; http://dx.doi.org/10.1586/erv.10.169
- Yamaguchi Y, Ohshita A, Kawabuchi Y, Hihara J, Miyahara E, Noma K, Toge T. Locoregional immunotherapy of malignant ascites from gastric cancer using DTH-oriented doses of the streptococcal preparation OK-432: treatment of Th1 dysfunction in the ascites microenvironment. Int J Oncol 2004; 24:959-66; PMID:15010836
- Disis ML, Schiffman K, Gooley TA, McNeel DG, Rinn K, Knutson KL. Delayed-type hypersensitivity response is a predictor of peripheral blood T-cell immunity after HER-2neu peptide immunization. Clin Cancer Res 2000; 6:1347-50; PMID:10778962
- Strome SE, Voss S, Wilcox R, Wakefield TL, Tamada K, Flies D, Chapoval A, Lu J, Kasperbauer JL, Padley D et al. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res 2002; 62:1884-9; PMID:11912169
- Takeda K, Yamaguchi N, Akiba H, Kojima Y, Hayakawa Y, Tanner JE, Sayers TJ, Seki N, Okumura K, Yagita H et al. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med 2004; 199:437-48; PMID:14769851; http://dx.doi.org/10.1084/jem.20031457
- Egilmez NK, Kilinc MO. Tumor-resident CD8+ T-cell: the critical catalyst in IL-12-mediated reversal of tumor immune suppression. Arch Immunol Ther Exp (Warsz) 2010; 58:399-405; PMID:20872283; http://dx.doi.org/10.1007/s00005-010-0097-7
- Bajetta E, Del Vecchio M, Mortarini R, Nadeau R, Rakhit A, Rimassa L, Fowst C, Borri A, Anichini A, Parmiani G. Pilot study of subcutaneous recombinant human interleukin 12 in metastatic melanoma. Clin Cancer Res 1998; 4:75-85; PMID:9516955
- Gollob JA, Mier JW, Veenstra K, McDermott DF, Clancy D, Clancy M, Atkins MB. Phase I trial of twice-weekly intravenous interleukin 12 in patients with metastatic renal cell cancer or malignant melanoma: ability to maintain IFN-gamma induction is associated with clinical response. Clin Cancer Res 2000; 6:1678-92; PMID:10815886
- Rodriguez-Galan MC, Reynolds D, Correa SG, Iribarren P, Watanabe M, Young HA. Coexpression of IL-18 strongly attenuates IL-12-induced systemic toxicity through a rapid induction of IL-10 without affecting its antitumor capacity. J Immunol 2009; 183:740-8; PMID:19535628; http://dx.doi.org/10.4049/jimmunol.0804166
- Portielje JE, Lamers CH, Kruit WH, Sparreboom A, Bolhuis RL, Stoter G, Huber C, Gratama JW. Repeated administrations of interleukin (IL)-12 are associated with persistently elevated plasma levels of IL-10 and declining IFN-gamma, tumor necrosis factor-alpha, IL-6, and IL-8 responses. Clin Cancer Res 2003; 9:76-83; PMID:12538454
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3:133-46; PMID:12563297; http://dx.doi.org/10.1038/nri1001
- Elliott RL, Head JF, McCoy JL. Comparison of estrogen and progesterone receptor status to lymphocyte immunity against tumor antigens in breast cancer patients. Breast Cancer Res Treat 1994; 30:299-304; PMID:7981448; http://dx.doi.org/10.1007/BF00665971
- Baxevanis CN, Voutsas IF, Gritzapis AD, Perez SA, Papamichail M. HER-2neu as a target for cancer vaccines. Immunotherapy 2010; 2:213-26; PMID:20635929; http://dx.doi.org/10.2217/imt.09.89
- Wolff AC, Hammond ME, Hayes DF. Re: predictability of adjuvant trastuzumab benefit in N9831 patients using the ASCOCAP HER2-positivity criteria. J Natl Cancer Inst 2012; 104:957-8; PMID:22581974; http://dx.doi.org/10.1093/jnci/djs243
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 2007; 13:4429-34; PMID:17671126; http://dx.doi.org/10.1158/1078-0432.CCR-06-3045