
Abstract
The discovery of driver mutations in cancers has raised interest in their suitability as immunotherapeutic targets. A recent study demonstrates that a point mutation in isocitrate dehydrogenase 1 (IDH1R132H), expressed in gliomas and other tumors, is presented on human MHC class II and induces a mutation-specific CD4+ antitumor T cell response in patients and a syngeneic tumor model in MHC-humanized mice.
Next generation sequencing efforts have led to the discovery of a multitude of new mutations, altering our understanding of tumorigenesis and suggesting novel targets for cancer pharmacotherapy. Conceptually, these altered protein sequences may represent neoepitopes suitable as true tumor antigens. With respect to specificity and side effects, tumor antigens are superior to tumor-associated antigens currently targeted in most clinical trials applying cancer vaccines. On the other hand, it is much less likely to identify mutation-specific immunogenicity of mutated antigens. If these antigens are immunogenic, they often are minor epitopes presented on MHC class II rather than class I. We have recently developed and applied a syngeneic sarcoma model in mice which are devoid of mouse MHC and transgenic for human MHC class I (HLA-A2) and class II (HLA-DR1) to test the efficacy of a mutation-specific peptide vaccine targeting mutated isocitrate dehydrogenase 1 (IDH1).Citation1 This study may serve as a proof-of-concept for testing the suitability of mutated tumor antigens for cancer immunotherapy.
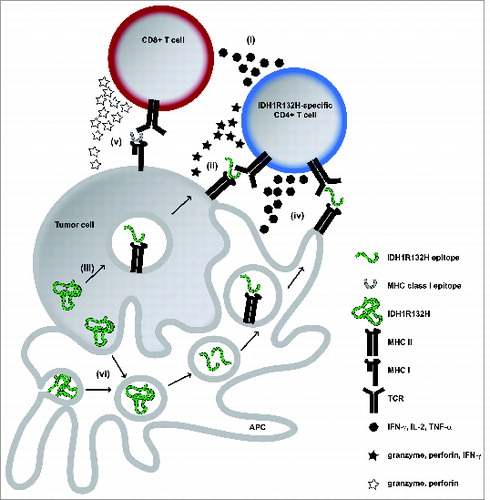
IDH mutations have been identified in a variety of tumor entities such as low grade and anaplastic gliomas and acute myeloid leukemia (AML) and affect an enzymatically critical arginine (Arg, R) residue in the catalytic pocket.Citation2,3 The associated 2-HG accumulation leads to genome-wide hypermethylation, genetic instability and ultimately malignant transformation. In gliomas, more than 70% carry an amino acid exchange from Arg to histidine (His, H) at position 132 (R132H). Since IDH mutations occur early during tumorigenesis, they are homogenously expressed within a tumor, representing an attractive therapeutic target. For instance, malignant transformation is reversed by small molecule inhibition of IDH-mediated 2-HG production.Citation4 In contrast to inhibitor treatment, which does not eliminate IDH-mutated cells, a targeted immunotherapy approach should lead to the eradication of target-expressing tumor cells.Citation1,5 We have shown that IDH1R132H is an immunogenic tumor antigen that induces mutation-specific CD4+ T cell and antibody responses which are capable to control IDH1R132H-expressing tumor growth in vivo in MHC-humanized A2.DR1 mice after vaccination, but not when they spontaneously occur in patients with IDH1R132H-mutated gliomas.Citation1 Thorough analysis of splenocytes from vaccinated A2.DR1 mice revealed that IDH1R132H-responsive T cells were CD4+ and that CD8+ T cells did not respond specifically; furthermore, spontaneous immune responses found in patients were also mediated by CD4+ T cells.Citation1 Several mechanisms can be considered on how CD4+ T cells mediate antitumor immunity and growth suppression (). It has recently been appreciated that CD4+ T cells can in addition to their role as T helper cells (i) directly mediate specific tumor cell killing in an IFNγ and MHC class II-restricted manner (ii) and are suitable for use in adoptive T cell transfer for cancer immunotherapy.Citation6 Tumor cells are able to process MHC class II epitopes by autophagy and present them (iii). IFNγ exposure can lead to upregulation of MHC class II in glioma cells and it has been proposed from preclinical melanoma models that CD4+ T cell-derived IFNγ leads to enhanced MHC class II expression on the tumor, making it susceptible to CD4+ T cell-mediated killing.Citation6 It is reasonable to speculate that IDH1R132H-specific vaccination-induced IFNγ-secreting CD4+ T cells are capable of directly killing established IDH1R132H-mutated tumors in A2.DR1 mice. The most likely scenario, however, is the creation of an inflammatory environment by IDH1R132H-specific CD4+ T cells (iv) and antigen spreading (v). IDH1R132H is released in necrotic tumor areas or by apoptotic tumor cells (vi), phagocytosed, processed, and presented on MHC class II to T helper cells by professional antigen-presenting cells (APC) that are recruited into the tumor. Antigen encounter stimulates cytokine secretion by T helper cells, which in turn activate APC and induce an inflammatory tumor microenvironment, providing co-stimulation for innate and adaptive immune cells e.g. by shifting the tumor-associated macrophage phenotype from an immunosuppressive M2 to a pro-inflammatory M1 phenotype.Citation7 Interestingly, the antitumor effectiveness depended on CD19+ B cells.Citation1 The role of B cells can be attributed to their antigen presentation capacity as well as to antibody production, since clinical responses correlated with IDH1R132H-binding antibodies in vaccinated mice.
Figure 1. Potential CD4+ IDH1R132H-specific Tcell-mediated antitumor immunity in IDH1 mutated tumors. (i)–(vi), for details refer to main text. APC, antigen presenting cell; IDH1R132H, isocitrate dehydrogenase 1 (R132H); MHC, major histocompatibility complex; TCR, T cell receptor; IFNγ, interferon-γ; IL-2, interleukin-2; TNF-α, tumor necrosis factor-α.
Although antigen-specific CD8+ T cells are widely considered superior, the advantages of CD4+ T cell-mediated responses to tumor antigens over CD8+ T cells are indeed various. As opposed to strictly HLA-type-restricted MHC class I-dependent CD8+ T cell responses, CD4+ T cell-mediated immunity is rather promiscuous in terms of HLA-type. We did not find a correlation of one or more distinct HLA-types with spontaneous immune responses against IDH1R132H in our patient cohort; similarly, IDH1R132H-specific responses were not only inducible in A2.DR1 mice, but also in mice transgenic for HLA-DR4.Citation1 Moreover, CD4+ T cells are independent of antigen presentation by tumor cells, whose presentation machinery often is defective. Lastly, CD4+ T cells exhibit versatile effector functions and represent a plastic cell population capable to “evolve, adapt, and self-regulate”.Citation7
Although the exact mechanisms on how IDH1R132H-specific CD4+ T cells contribute to elimination of the tumor remain to be elucidated, what is clear from our data is that IDH1R132H represents a novel tumor-specific antigen that can be targeted by active immunotherapy. As for all cancer vaccines, particularly for gliomas, which are characterized by a profound immunosuppressive microenvironment,Citation8 translation into clinical trials ought to consider combinations with pharmacological approaches targeting key immunosuppressive pathways in gliomas such as tryptophan catabolism.Citation9
Disclosure of Potential Conflicts of Interest
TS, LB, WW and MP have filded patents related to this work (EPA 14190538.0, PCT/EP2013/050048, WO 2013/102641).
References
- Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, Menn O, Osswald M, Oezen I, Ott M, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014; 512(7514):324-7; PMID:25043048; http://dx.doi.org/10.1038/nature13387
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361(11):1058-66; PMID:19657110; http://dx.doi.org/10.1056/NEJMoa0903840
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321(5897):1807-12; PMID:18772396; http://dx.doi.org/10.1126/science.1164382
- Cairns RA and Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov 2013; 3(7):730-41; PMID:23796461; http://dx.doi.org/10.1158/2159-8290.CD-13-0083
- Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE 2nd, McLendon RE, Mitchell DA, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010; 28(31):4722-9; PMID:20921459; http://dx.doi.org/10.1200/JCO.2010.28.6963
- Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010; 207(3):637-50; PMID:20156971; http://dx.doi.org/10.1084/jem.20091918
- Muranski P and Restifo NP. Adoptive immunotherapy of cancer using CD4(+) T cells. Curr Opin Immunol 2009; 21(2):200-8; PMID:19285848; http://dx.doi.org/10.1016/j.coi.2009.02.004
- Platten M, Ochs K, Lemke D, Opitz C, Wick W. Microenvironmental clues for glioma immunotherapy. Curr Neurol Neurosci Rep 2014; 14(4):440; PMID:24604058; http://dx.doi.org/10.1007/s11910-014-0440-1
- Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res 2012; 72(21):5435-40; PMID:23090118; http://dx.doi.org/10.1158/0008-5472.CAN-12-0569