
Abstract
Variations of post-translational modifications are important for stability and in vivo behavior of therapeutic antibodies. A recombinant humanized anti-cocaine monoclonal antibody (h2E2) was characterized for heterogeneity of N-linked glycosylation and disulfide bonds. In addition, charge heterogeneity, which is partially due to the presence or absence of C-terminal lysine on the heavy chains, was examined. For cocaine overdose therapy, Fab fragments may be therapeutic, and thus, a simplified method of generation, purification, and characterization of the Fab fragment generated by Endoproteinase Lys-C digestion was devised. Both the intact h2E2 antibody and purified Fab fragments were analyzed for their affinities for cocaine and 2 of its metabolites, benzoylecgonine and cocaethylene, by fluorescence quenching of intrinsic antibody tyrosine and tryptophan fluorescence resulting from binding of these drugs. Binding constants obtained from fluorescence quenching measurements are in agreement with recently published radioligand and ELISA binding assays. The dissociation constants determined for the h2E2 monoclonal and its Fab fragment are approximately 1, 5, and 20 nM for cocaethylene, cocaine, and benzoylecgonine, respectively. Tryptophan fluorescence quenching (emission at 330 nm) was measured after either excitation of tyrosine and tryptophan (280 nm) or selective excitation of tryptophan alone (295 nm). More accurate binding constants are obtained using tryptophan selective excitation at 295 nm, likely due to interfering absorption of cocaine and metabolites at 280 nm. These quenching results are consistent with multiple tryptophan and tyrosine residues in or near the predicted binding location of cocaine in a previously published 3-D model of this antibody's variable region.
Abbreviations:
- mAb, monoclonal antibody
- h2E2, humanized monoclonal antibody against cocaine
- Fab, fragment
- antigen-binding
- Fc, fragment, crystallizable
- ADCC, antibody-dependent cell mediated cytotoxicity
- PNGase-F, peptide N-glycosidase F
- Endo H, endoglycosidase H
- TBP, tributylphosphine
- ABD-F, 7-fluorobenz-2-oxa-1, 3-diazole-4-sulfonamide
- IEF, isoelectric focusing
- NEPHGE, non-equilibrium pH gel electrophoresis
- MOPS, 3-(N-morpholino)propanesulfonic acid
- MES, 2-(N-morpholino)ethanesulfonic acid
- TBS, tris-buffered saline
- Endo Lys-C, lysyl endoproteinase
- BE, benzoylecgonine
- CE, cocaethylene
- HPLC, high performance liquid chromatography
- SCX, strong cation exchange
- CHO, Chinese hamster ovary
- CDR, complementarity determining regions
- ELISA, enzyme-linked immunosorbent assay
- KD, dissociation constant
Introduction
There is an unmet need for developing novel treatments for drug abuse. Biological therapies, including the use of monoclonal antibodies, are a promising direction for new therapies for many indications, including drug addiction. Monoclonal antibodies account for an increasing percentage of newly approved drugs. They have specificities and affinities that can seldom be matched by small molecule drugs. However, unlike small molecule drugs, large protein therapeutics are more complicated in terms of their generation, purification, and characterization.Citation1 Another complication is that there are several well-established variations in post-translational modifications that occur in monoclonal antibodies that are used as therapeutic drugs.Citation2 Thus, it is useful, and required by the FDA in the US, to characterize these antibodies for these post-translational modifications which result in protein heterogeneity, and to subsequently evaluate their in vivo ramifications.
Common post-translational modifications, which introduce heterogeneity in monoclonal antibodies, have recently been reviewed,Citation1 and include charge variants (e.g., variations in the removal of heavy chain C-terminal lysine, N-terminal pyroglutamate formation, asparagine deamidation), oxidation variants (e.g., methionine and tryptophan), disulfide bond scrambled/mispaired variants, glycosylation variants (e.g., immature glycosylation and the presence or absence of fucose and sialic acid on the glycan chains), and differential glycation variants. Although antibody glycosylation commonly increases solubility and stability, while decreasing aggregation,Citation3 Fc glycosylation is also functionally relevant due to modulation of antibody binding to Fcγ receptors, with increased binding leading to increased antibody-dependent cell-mediated cytotoxicity (ADCC).Citation4 Immature glycosylation (high mannose glycan chains) can result in shorter plasma half-lives for the antibodies.Citation5 Asparagine deamidation and aspartic acid isomerization can influence storage stability and shelf life of monoclonal antibodies.Citation6 Tryptophan is commonly found in the antibody complementarity determining regions (CDRs), and its oxidation can result in an attenuation or loss of antigen affinity.Citation7 In addition, aggregation of monoclonal antibodies can occur during production and purification, and is therapeutically relevant, affecting their efficacy and safety, since aggregates tend to increase plasma clearance, systemic side effects, and the likelihood of undesired host immunogenic responses.
A chimeric murine/human anti-cocaine monoclonal antibody (2E2) has been generated,Citation8 using technology described by Lonberg.Citation9 The murine/human chimeric anti-cocaine 2E2 monoclonal antibody was previously shown to inhibit the distribution of cocaine to the brain in mice.Citation10 A re-engineered version of the 2E2 antibody incorporating a humanized light chain (h2E2) was recently shown to have in vitro binding properties and in vivo behavior in rats consistent with its likely utility as a therapy for human cocaine abuse.Citation11 The affinity for cocaine of the recombinant h2E2 antibody was shown to be 3.9 nM by radioligand binding, and the desirable relative selectivity of the re-engineered antibody for cocaine over the major inactive metabolites of cocaine was not adversely affected by the humanization process.Citation11 Thus, the h2E2 monoclonal antibody is a lead candidate for advancement to clinical trials as a therapeutic agent for cocaine abuse.
For some non-clinical studies and clinical uses of therapeutic monoclonal antibodies, it may be desirable to generate Fab fragments, which bind antigens, but lack the constant region and therefore, antibody effector functions. The Fab fragment is smaller and has better tissue and tumor penetration than intact monoclonal antibodies. In the case of an anti-cocaine antibody, it is not unreasonable to assume that the Fab fragment of the h2E2 monoclonal antibody may be preferable to the intact mAb for treatment of acute cocaine overdose, since the Fab fragment has no Fcγ binding region, resulting in much reduced chances for ADCC, and, as shown in this study, the Fab fragment has identical affinity for cocaine as the intact h2E2 antibody. The h2E2 Fab fragment is also very likely to have a substantially shorter plasma half-life, reducing the likelihood of unwanted side effects when used for acute cocaine overdose treatment. In addition, for future, more detailed structural characterization of the antibody and its variants, including crystallization for X-ray structure determination of the antigen-antibody complex, the preparation and purification of the Fab fragment will be very useful as part of a “middle-up” strategy of generating large fragments of antibodies for more detailed structural and post-translational modification analysis.Citation12 Historically, large antibody fragments have been generated using papain or pepsin to generate Fab or F(ab’)2 fragments, respectively. However, Endoproteinase Lys-C has also been used to generate Fab fragments, and has some advantages over the use of papain for this purpose.Citation13
It is necessary to perform thorough preclinical testing and characterization of this antibody prior to applying for approval for testing in human beings. Thus, in this work we have preliminarily characterized both the intact h2E2 monoclonal antibody and its Fab fragment for post-translational modification mediated heterogeneities. A simple, rapid, and high-yield protocol for generating and purifying Fab fragments from this antibody was developed. We also developed a simple, rapid, and quantitative method for measuring binding affinities for cocaine and cocaine metabolites for this antibody based on fluorescence quenching. Notably, the developed fluorescence technique involves only intrinsic protein fluorescence quenching, and does not require derivatization of either the antibody or cocaine with a fluorophore, which would most likely change the affinity and possibly other structural and binding characteristics of the antibody. The developed methods of h2E2 monoclonal antibody characterization, Fab fragment generation, and ligand binding measurements described in this study will facilitate the full characterization of this antibody, which will support its advancement to human clinical trials.
Results
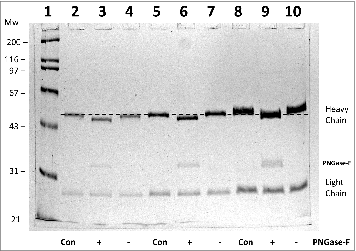
In the present study, the heterogeneity of a humanized anti-cocaine monoclonal antibody, h2E2,Citation11 was characterized. First, the deglycosylation enzyme, PNGase-F, was used to remove all N-linked glycosylation. From the known amino acid sequence of the heavy and light chain of antibody, there is one conserved N-linked glycosylation site on each heavy chain. Thus, the intact antibody should have 2 N-linked glycan chains. As seen in , PNGase-F treatment of the h2E2 monoclonal antibody results in a shift for the heavy chain toward a lower apparent molecular weight of about 2 kDa. Multiple sample volumes containing differing amounts of the treated antibody in the control (Con) as well as a sham treated (−) and PNGase-F treated (+) antibody were loaded onto the gel seen in (the dashed line was added to show reproducibility and aid visualization of the small mobility shift). What is observed in is a reproducible, discernable shift in electrophoretic mobility, indicative of most or all of the h2E2 antibody heavy chain molecules having one N-linked glycan chain. Due to the focus of this preliminary characterization of the h2E2 mAb on the functionality of the antibody (i.e., its affinity for cocaine), we did not perform additional glycan structural analyses.
Figure 1. 10% SDS-PAGE analysis of monoclonal antibody h2E2 with or without treatment with peptide N-glycosidase-F (PNGase-F) to remove all N-linked glycans. After overnight treatment with PNGase-F, either 5 (lanes 2-4), 10 (lanes 5-7), or 20 μg (lanes 8-10) of h2E2 antibody were reduced and denatured in SDS and loaded onto a 10% acrylamide gel, followed by staining with Coomassie Blue R-250. Control, untreated h2E2 (“Con”) along with PNGase-F (“+”) or sham treated (“−”) monoclonal antibody are shown, with the dashed line added to aid visualization of the small differences in electrophoretic mobility observed after N-glycan removal by PNGase-F. The migration positions of the heavy chain, light chain, and PNGase-F enzyme are indicated on the right hand side.
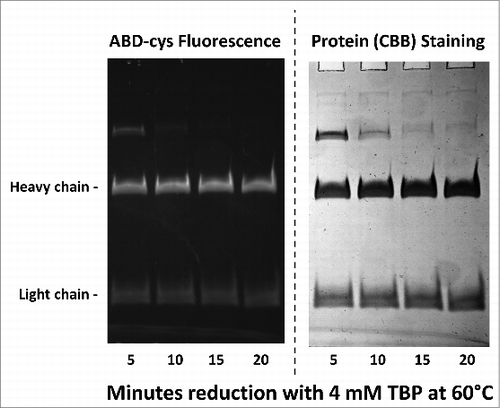
To determine if the disulfide bonds were correctly and completely formed in the Chinese hamster ovary (CHO) cell-expressed h2E2 monoclonal antibody, disulfide and cysteine analyses were performed using the reductant tributyl phosphine (TBP) and the alkylating agent 7-fluorobenz-2-oxa-1,3-diazole-4-sulfonamide (ABD-F). The time course necessary to completely reduce and alkylate the monoclonal antibody protein with these reagents at 60°C was investigated. Data, some of which are shown in , indicate that at least 15 minutes are required to produce complete reduction and alkylation of the antibody under these conditions at 60°. The total cysteine plus half cystine determined using both the reductant tributyl phosphine and the alkylating agent ABD-F was 33.27 per mole monoclonal antibody protein. The theoretical value based on the protein sequence is 32 moles of cysteine per mole monoclonal antibody, all of which are paired in disulfide bonds. By subtracting the small amount of ABD-F labeling seen in the absence of reduction (0.38 moles cysteine per mole monoclonal antibody) one obtains 32.9 ± 0.8 moles of cysteine per mole of h2E2 monoclonal antibody.
Figure 2. 10% SDS-PAGE gel analysis of TBP reduced and ABD-F labeled monoclonal antibody h2E2 heavy and light chains. 0.2 mg/ml antibody was first reduced with 4 mM TBP at 60°C for the times indicated in the figure, followed by alkylation with 4 mM ABD-F for 15 minutes at 22°C. Aliquots (5 μg) of the resultant samples were diluted in SDS-PAGE sample buffer and run on a 10% gel. Following photography under UV light to detect incorporated ABD-cys fluorescence (left hand side); the gel was then stained to measure total protein, and re-photographed (right hand side). Migratory positions of the heavy and light chains are indicated.
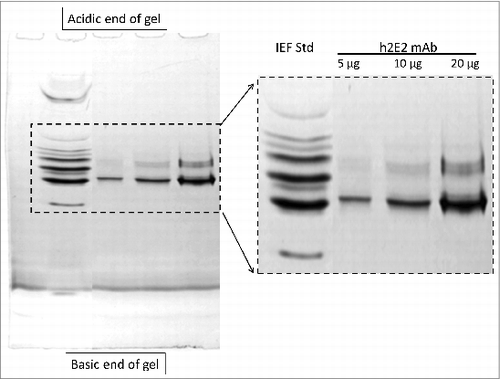
To investigate the charge heterogeneity of this h2E2 antibody, isoelectric focusing (IEF) was originally used. However, the results were not satisfactory, due to the problem of cathodic drift seen in IEF gels when analyzing a monoclonal antibody with a pI that is very basic like this antibody (calculated pI = 8.37 for h2E2). Good separations and reproducible results were obtained using the non-equilibrium pH gel electrophoresis (NEPHGE) technique to analyze this h2E2 antibody, as can be seen in . It is evident that there is substantial charge heterogeneity in this monoclonal antibody based on this non-equilibrium pH gel electrophoresis analysis. Such charge heterogeneity is due to several possible post-translational modifications, and is commonly seen in therapeutic monoclonal antibodies.
Figure 3. 5% acrylamide non-equilibrium pH gel electrophoresis (NEPHGE) analysis of monoclonal antibody h2E2. NEPHGE was performed as described in Methods, with the gel run for 90 minutes at 200 V. Aliquots of 5, 10 and 20 μg of h2E2 antibody were analyzed, and the region of the gel containing the separated variants of the monoclonal antibody is expanded on the right side of the figure. Note the separation of several groups of charged variants of the antibody by this technique. BioRad IEF gel standards are shown in the left lane of the gel.
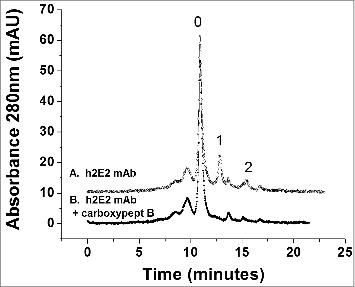
To further investigate the charge heterogeneity of h2E2, high-performance strong cation exchange chromatography was employed. As seen in the upper trace of , the pure, intact monoclonal antibody analyzed by this technique is separated into multiple peaks, indicating charge heterogeneities. One of the major reasons for charge heterogeneity in therapeutic monoclonal antibodies is the differential presence or absence of the C-terminal lysine residue on each heavy chain. To analyze for this, h2E2 was treated with carboxypeptidase B, and compared to the untreated antibody using the same column, the same conditions, and the same gradient. Thus, as seen in the lower trace of , after carboxypeptidase treatment, several peaks have disappeared and coalesced into the main peak, labeled peak “0.” As seen in the Figure, the h2E2 antibody has relatively little lysine present at the C-terminus of both (peak “2”) or one (peak “1”) of the heavy chain C-termini, in contrast to some other monoclonal antibodies. As is also evident from the data in , there are additional charge heterogeneities present in this monoclonal antibody that are not related to the presence or absence of the C-terminal lysine residue on the heavy chains.
Figure 4. High performance strong cation exchange chromatography of the h2E2 antibody before and after treatment with carboxypeptidase B. 100 μg of h2E2 antibody was injected, and eluted with a gradient of NaCl in MES buffer. Note the disappearance of peaks 1 and 2 after removal of the C-terminal lysine residues. These peaks represent h2E2 antibody molecules containing 1 or 2 lysine residues on the C-termini of the 2 heavy chains in the antibody (which, after removal of lysine by carboxypeptidase elute with the main peak labeled ‘0” eluting at approximately 11 minutes).
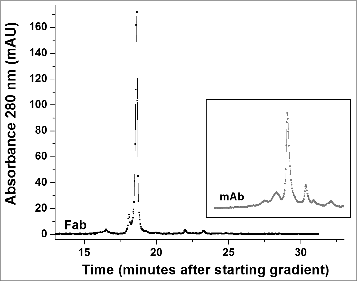
Fab antibody fragments are useful for experimental as well as therapeutic reasons. Therefore, we generated and characterized the Fab fragment of h2E2. The classic way to do this is with papain proteolysis treatment, followed by removal of the Fc fragment by Protein A or G chromatography. In this work, we devised a more efficient and very rapid method for the isolation of very pure Fab fragment. This protocol is described in detail in the Methods section. The procedure was very reproducible, and 7.0 ± 0.4 mg of pure Fab fragment was obtained starting with 20 mg monoclonal antibody (n = 3). demonstrates the purity of the h2E2 Fab fragment, which is shown in lane 2, compared to the starting Endo-Lys-C digest shown in lane 4 and the intact h2E2 monoclonal antibody shown in lane 3. The purity and relative lack of charge heterogeneity of the resultant Fab fragment is also demonstrated by the high-performance strong cation exchange chromatography column analysis shown in . The elution profile and elution position of the intact monoclonal antibody using this column and the same conditions is shown as an inset to this figure for comparative purposes, and to indicate the lack of undigested antibody contamination of the Fab preparation.
Figure 5. Reducing 10% SDS-PAGE analysis of h2E2 Fab, mAb and the Endo Lys-C digest. 5 μg of Fab, 6 μg of h2E2 mAb and 6 μg of Endo-Lys-C digested mAb were loaded onto lanes 2-4, respectively. Note the purity and lack of high molecular weight impurities in the Fab preparation. Migration positions of the reduced heavy and light chains, as well as the reduced Fab and Fc fragments, are indicated.
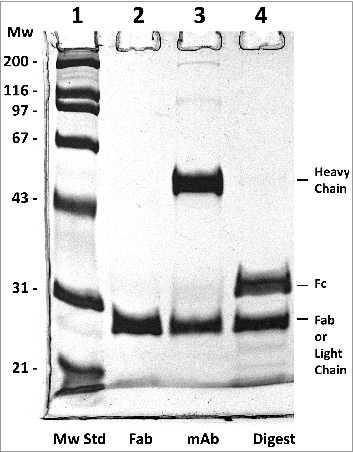
Figure 6. High performance strong cation exchange chromatography of the Fab fragment derived from the h2E2 antibody. 100 μg purified Fab fragment was injected, and eluted with a gradient of NaCl in MES buffer at 22°C. Note the relative lack of charge heterogeneity compared to the intact h2E2 antibody ( and the inset), indicating much of the charge heterogeneity resides in the Fc portion of the h2E2 antibody. For comparative purposes, the position and pattern of the intact antibody on the same column using the same buffers and gradient is shown in the inset (offset in the y-direction for clarity).
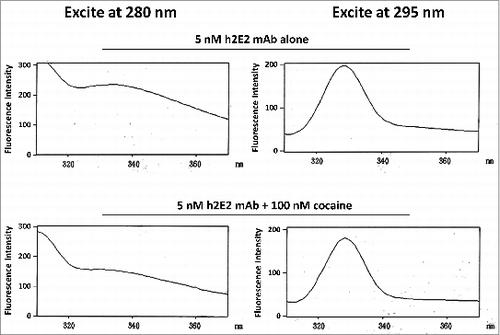
We measured the effect of cocaine and 2 of its metabolites (an active metabolite, cocaethylene (CE) and an inactive metabolite, benzoylecgonine (BE)) on the intrinsic tyrosine and tryptophan fluorescence of both the pure h2E2 monoclonal antibody and the h2E2 Fab fragment. As shown in , we demonstrated intrinsic protein fluorescence quenching by cocaine. The emission spectrum and the emission maximum did not change much for either the Fab fragment or the intact monoclonal antibody either quenched or unquenched, i.e., the emission maximum varied only slightly between 328 nm and 332 nm using either 280 or 295 nm excitation. Therefore, we used a constant emission analysis wavelength of 330 nm. Quenching was measured both using 280 nm excitation and 295 nm excitation. At 280 nm, the approximate absorption maximum for the antibody and Fab fragment, both tyrosine and tryptophan residues are excited, and are expected to contribute to the fluorescence. At 295 nm, excitation should be selective for tryptophan excitation and fluorescence. The absorption spectrum of cocaine and its metabolites demonstrated that these drugs have small but significant absorption at 280 nm, but negligible absorption at 295 nm (for cocaine, the ratio of 280 to 295 nm absorption is approximately 24, data not shown). The emission profiles of the h2E2 mAb with excitation at either 295 or 280 nm in the presence and absence of cocaine are shown in .
Figure 7. Intrinsic h2E2 antibody tryptophan and tyrosine fluorescence quenching by cocaine binding. Shown are emission spectra of 5 nM h2E2 antibody both before and after addition of 100 nM cocaine, with excitation at either 280 nm (tyrosine and tryptophan) or 295 nm (tryptophan). Note the decrease in emission (quenching) caused by cocaine, but little if any change in the emission maximum (near 330 nm in both cases).
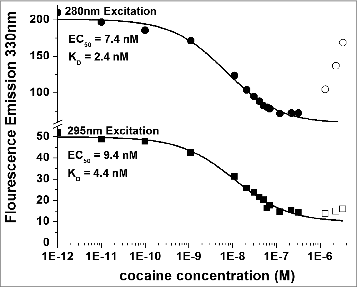
Titrations of both the h2E2 monoclonal antibody and its Fab fragment using cocaine as well as benzoylecgonine (BE) and cocaethylene (CE) were performed. An example of such titrations for cocaine binding to the intact monoclonal antibody is shown in , measuring fluorescence emission at 330 nm and exciting at either 280 nm or 295 nm. The dissociation constant or KD can be calculated from such data and is proportional to the EC50 values derived from sigmoidal fitted curves describing the data. As can also be seen in (open symbols), at the highest concentrations of cocaine used (micromolar) there is an increase in apparent fluorescence emission at 330 nm with increasing concentration of cocaine, especially seen using 280 nm excitation. Thus, the 3 highest concentrations of cocaine used in all these experiments were not included in the fitting of the curves.
Figure 8. Titration of 5 nM h2E2 antibody with cocaine, with excitation at both 280 nm and 295 nm. Only data represented by filled symbols were used for fitting to obtain the indicated sigmoidal curves to derive the EC50 values. Note the y-axis brake in scale to allow visualization of the quality of the fitted curves for both sets of data on a single plot. KD values were calculated from the EC50 values and the antibody or Fab concentration as described in Methods. For the experiment shown in the figure (using the intact h2E2 mAb and 295 nm excitation), KD = EC50 – (0.5 × 2 sites/mAb × 5.0 nM mAb) = 9.4 nM – 5.0 nM = 4.4 nM = KD for cocaine binding to the h2E2 monoclonal antibody.
All the fluorescence quenching data obtained by this method are summarized in . As can be seen from (295 nm excitation data), the active metabolite cocaethylene has the highest affinity for the h2E2 monoclonal antibody and for the Fab fragment of approximately 1 nM (0.53 nM for the intact mAb, 1.26 nM for the Fab fragment), cocaine has an affinity of about 5 nM (5.90 nM for the intact mAb, 3.75 nM for the Fab fragment), and the inactive metabolite benzoylecgonine has an affinity for the antibody of approximately 20 nM (20.3 nM for the intact mAb, 20.2 nM for the Fab fragment). If the sites on both variable regions of the antibody are independent as is expected, then the Fab fragment affinity and the intact monoclonal antibody affinity should be the same for each ligand. This is in fact observed in the 295 nm excitation data for cocaine and benzoylecgonine.
Table 1. Summary of cocaine and cocaine metabolites binding to both the intact antibody and the Fab fragment of the h2E2 mAb, as measured by quenching of intrinsic protein fluorescence.
Discussion
Glycosylation can have an impact on antibody stability and in vivo pharmacokinetics and, therefore, is an important characteristic for therapeutic monoclonal antibodies. Glycosylation varies with the expression system used to produce the monoclonal antibody,Citation14 and can also vary with growth conditions used to produce the antibody with a given expression system. Deglycosylation of the h2E2 antibody with PNGase-F, which removes all N-linked glycosylation, resulted in a small shift in the electrophoretic mobility of the heavy chain of about 2 kDa. This is consistent with one conserved glycosylation site present on each heavy chain molecule, which is predicted based on the sequence of the heavy chain of this antibody (present in the CH2 region of the heavy chain, in the sequence …EEEQYNSTYRV…). Experiments using Endoglycosidase H (Endo H) treatment of the intact monoclonal antibody, which removes only high mannose, incompletely processed, glycan chainsCitation15 revealed no change in electrophoretic mobility upon treatment, indicating no detectable incompletely processed glycan chains were present on the monoclonal antibody (data not shown). However, this analysis does not exclude the possibility that a small percentage of the mAb molecules could have high mannose glycan chains.
It is crucial that disulfide bonds are correctly formed during expression and maintained during the purification of the h2E2 mAb. This has been proven to be a problem in some expression systems and with some other monoclonal antibodies.Citation16 The disulfide bond pairing for IgG molecules, and structural variations that can occur resulting from unpaired or mispaired disulfides have been documented,Citation17 and different disulfides contained in IgG have been documented to have varying susceptibilities to reduction and modification.Citation18 Therefore, we performed quantitative cysteine and cystine analysis on the antibody using a phosphine reductant and a fluorescent adduct generated by the ABD-F reagent treatment of cysteine residues. The small amount of labeling detected by 385 nm absorption measurements in the absence of the reducing agent was not observed by fluorescence measurements (not shown), suggesting it may be due to a very small amount of non-selective labeling of amino acids other than cysteine by the ABD-F reagent, which has been reported previously.Citation19 Thus, this analysis was consistent with all cysteine residues in the monoclonal antibody (32 cysteine residues total) being involved in 16 disulfide bonds, as expected, and there is no evidence of mis-paired or unpaired cysteine residues in this h2E2 antibody.
Monoclonal antibody heterogeneities have been summarized,Citation2 and analytical methods for deciphering therapeutic antibody heterogeneities have been recently summarized and reviewed.Citation1 Charge heterogeneity of therapeutic monoclonal antibodies is one relevant variation, which can be derived from several different possible post-translational modifications, including variations in the removal of heavy chain C-terminal lysine, N-terminal pyroglutamate formation, and asparagine deamidation, leading to formation of aspartic acid and isoaspartic acid.Citation1 Charge variations in monoclonal antibodies and derived fragments have been characterized using strong and weak cation exchange HPLC.Citation20 The presence or absence of the C-terminal lysine residue on the heavy chain is a common cause for charge heterogeneity seen in therapeutic monoclonal antibodies. Charge heterogeneity of the pure h2E2 antibody is evident from multiple bands seen in non-equilibrium pH gel electrophoresis (NEPHGE) analysis (), as well as from multiple peaks observed in high performance strong cation exchange chromatography (). From comparison of the 2 HPLC chromatograms shown in , it is evident that some of the charge heterogeneity in this h2E2 antibody indeed derives from differential presence or absence of C-terminal lysine on the heavy chain of the antibody. In this h2E2 mAb, it appears that most of the C-terminal heavy chain lysine has been removed prior to analysis, most likely during the expression of the antibody in the Chinese hamster ovary cell expression system.
Using the historically more common papain fragmentation, followed by purification of the Fab fragment by removal of the Fc fragment using Protein A or Protein G, variable and sometimes poor yields were obtained for h2E2 antibody (data not shown). A method using Endoproteinase Lys-C has been described for generating Fab fragments.Citation21 Therefore, we investigated the use of Endoproteinase Lys-C to generate an h2E2 Fab fragment, followed by batch ion exchange removal of the Fc fragment, and size exclusion chromatography, to remove any undigested antibody and aggregates. Conditions for the Endo Lys-C digestion were optimized in terms of time, temperature, buffer, and ratio of protease to antibody, to achieve nearly complete generation of Fab fragments, with only very small amounts of non-selective over-digestion. Thus, the predicted and desired cleavage point using Endoproteinase Lys-C, which is 2 amino acid residues away from the predicted papain cleavage site, was indeed selectively and nearly quantitatively cleaved. This method of Fab fragmentation and purification led to a very pure Fab fragment () with very little charge heterogeneity (), which can be generated, purified, and partially characterized in one day. The very small amount of charge heterogeneity seen in the Fab fragment () compared to the intact mAb () suggests that most of this heterogeneity resides in the Fc portion of the antibody and that there may be very little charge heterogeneity in the CDRs (which are crucial for cocaine binding).
Several types of optical spectroscopy have been used to quantitatively characterize antibody-antigen binding, as was summarized by Tetin and Hazlett.Citation22 Those authors detailed some of the advantages of fluorescence spectroscopy for such studies, including the sensitivity and simplicity of the measurements. A simple, rapid, and quantitative assay to measure cocaine and cocaine metabolite affinities for this antibody and its Fab fragment was needed. Another group recently reported the ability to measure cocaine binding to a different anti-cocaine monoclonal antibody, using intrinsic protein fluorescence quenching, with excitation at 280 nm.Citation23 Using quenching of intrinsic protein tyrosine and tryptophan fluorescence, we developed a simple, rapid, and quantitative method for measuring affinities for cocaine and its metabolites for both the intact h2E2 antibody and the generated h2E2 Fab fragment. The results obtained, with approximate dissociation constants of 1, 5, and 20 nM for cocaethylene, cocaine, and benzoylecgonine, respectively, are consistent with previously published data obtained with this h2E2 antibody and these cocaine metabolites via ELISA and radioligand binding experiments.Citation11 As expected if the intact antibody has 2 independent binding sites for cocaine and its metabolites, we obtained the same dissociation constants for a given metabolite for both the intact monoclonal antibody and the Fab fragment for cocaine and benzoylecgonine. However, the apparent affinity measured for cocaethylene for the h2E2 mAb was slightly higher than that observed for the Fab fragment (0.53 nM for the intact mAb vs 1.26 nM for the Fab fragment, ). The reason for this difference may be due to working close to the sensitivity limit of this intrinsic fluorescence quenching technique, since lower concentrations of h2E2 antibody were required to measure the higher affinity binding of cocaethylene compared to fluorescence quenching experiments used to measure the lower affinities of cocaine and benzoylecgonine, and this leads to a smaller signal to noise ratio in the data, and therefore more relative error in the determination of the fitted EC50 value and the calculated KD.
The 295 nm emission fluorescence intensity curve shown in appears to change little upon titration with cocaine, compared to the 280 nm data plotted using the same y-axis scaling. However, the EC50 value was accurately and reproducibly determined from the data obtained with excitation at 295 nm. Although the magnitude of the signals and change are greater measured at 280 nm compared to 295 nm, more accurate EC50 values (and therefore KD values) are obtained using the 295 nm excitation data. This is due to a small but significant cocaine absorbance at 280 nm, resulting in the inability to accurately determine the quenched (lower) plateau level at the high concentrations of cocaine needed for a good sigmoidal curve fit of the data. As is evident from , this effect is much smaller at 295 nm, likely because the absorbance of cocaine at 295 nm is negligible. Nonetheless, the 3 highest concentrations of cocaine used in these experiments were not included in the fitting of both of these curves. The net effect of this 280 nm absorption-induced bias is to decrease slightly the EC50 that is measured using the sigmoidal fit of the 280 nm excitation data, resulting in a calculated KD which is smaller than the actual KD. (i.e., resulting in a calculated affinity that is higher than the actual affinity). Thus, it seems likely that previously published measurements using 280 nm excitation to measure cocaine binding by protein fluorescence quenching to a different anti-cocaine antibodyCitation23 were biased due to absorbance of cocaine at 280 nm. Thus, the calculated KD's based on 280 nm excitation data are smaller (and less accurate, since they are biased to yield higher apparent affinities) than the calculated KD values based on 295 nm excitation data (see ).
The ability to measure cocaine binding to the h2E2 antibody and its Fab fragment by tyrosine and tryptophan intrinsic fluorescence quenching is consistent with a 3-D model of cocaine binding to the 2E2 monoclonal antibody.Citation8 That 2E2 mAb variable region model predicts 2 tryptophan residues in close contact with the bound cocaine (trp 94 and trp 145), as well as several other tryptophan residues in the variable region which are predicted to be close enough to have their fluorescence yields changed by the binding of cocaine. In addition, there are 3 tyrosine residues in the CDRs of the 2E2 antibody, 1 in the human heavy chain CDR2, and 2 in the mouse light chain variable region (CDR1 and CDR3), which are also in close proximity to the bound cocaine molecule in this theoretical model. The pure Fab fragment generated in our current work may aid in the experimental evaluation of the published theoretical 3-D model, and aid in the interpretation of cocaine quenching of h2E2 intrinsic protein fluorescence, since Fab fragments of antibodies are often more amenable to protein crystallization and X-ray crystal analysis than are intact antibodies.
In conclusion, we have performed preliminary characterization of the properties and heterogeneities of the h2E2 monoclonal antibody, which shows promise for the treatment of cocaine addiction. This antibody has characteristics and a degree of heterogeneity similar to other more completely characterized therapeutic antibodies. The current results are encouraging for continuing efforts aimed at approval for therapeutic use. The Fab fragment generated and characterized will facilitate future structural studies, and may have therapeutic utility for treating acute cocaine overdose. The intrinsic tryptophan fluorescence quenching method for measuring cocaine binding will also facilitate future evaluation of antibody mutants designed to improve its pharmacokinetic properties and cocaine affinity. One specific type of post-translational heterogeneity and modification that was not examined in the present study is oxidation of tryptophan that could occur during expression, purification, or storage of the mAb. It seems likely based on the present study and existing theoretical structural models of this antibody that multiple tryptophan residues could be directly involved in cocaine binding. Thus, oxidation of these tryptophan residues would be expected to lead to attenuated affinity for cocaine, which would be crucial for the therapeutic use of the antibody for cocaine addiction. Thus, although limited by the preliminary nature of the heterogeneity analyses performed, the current study enables future studies crucial for both the therapeutic use of this antibody, as well as for detailed structural analyses and perturbations of the cocaine binding site.
Material and Methods
The generation and production of h2E2 monoclonal antibody was recently described in detail.Citation11 Briefly, stably transfected CHO-S cells containing multiple gene copies of the human sequence γ1 heavy chain of mAb 2E2 and a partially humanized version of its murine λ light chain were generated. The proprietary GPEx gene expression technologyCitation24 of Catalent (Madison, WI) was used to carry out transfections that generated stably transfected CHO-S cells containing multiple transgene copies inserted into transcriptionally active regions of the genome. Thus, h2E2 is a unique monoclonal, with the variable regions of the light chains being derived from mouse sequences, and the remainder of the antibody being derived from human sequences. h2E2 was transfected into CHO-S cells, and the h2E2 mAb was derived from the 3 cell lines with the highest levels of expression and secretion. The recombinant h2E2 mAb was purified by protein A affinity chromatography with initial yields of approximately 0.5 g/l. A 10L production run of antibody was purchased from Catalent, which produced 5.7 g of purified recombinant h2E2 mAb.
Acrylamide, bisacrylamide, and all reagents used to run and stain SDS-PAGE gels were from BioRad. The deglycosylation enzymes, peptide N-glycosidase F (PNGase-F, cat. P0704S) and Endoglycosidase H (Endo H, cat. P0702S) were purchased from New England Biolabs, and the deglycosylation procedures used with these enzymes followed the manufacturer suggested protocols. The reductant, tributylphosphine (TBP, cat. 90830) was obtained from Fluka, and the cysteine-selective alkylation agent, 7-fluorobenz-2-oxa-1,3-diazole-4-sulfonamide (ABD-F, cat. A5597) was from Tokyo Chemical Industries. Ampholines used for IEF and non-equilibrium pH gel electrophoresis (NEPHGE) gels were pHisolytes from Brinkman (pH 6-8, cat. 49 00 250-5 and pH 8-10, cat. 49 00 350-1). IEF and SDS-PAGE molecular weight standard protein mixtures were from BioRad.
Reduction with TBP and alkylation with ABD-F were performed and quantified basically as described previously.Citation25 ABD-F forms a fluorescent adduct after reaction with cysteine residues.25 Quantification of ABD-cys was done using an extinction coefficient of 7800 cm−1M−1.Citation26 Phosphine reducing agents are typically used with the ABD-F reagent, since reduction and alkylation can be performed simultaneously with these reagents, in contrast to using dithiothreitol or β-mercaptoethanol, which react directly with the ABD-F reagent (and other cysteine alkylating reagents). The more water soluble, less toxic phosphine, tris(2-carboxyethyl)phosphine was also evaluated for this purpose, but less complete reduction of the light chain was observed (not shown), possibly because, despite the fact that the reaction is carried out in denaturing SDS, the more hydrophobic TBP penetrated the protein structure better to reduce all the h2E2 disulfides.
Quantification of the cocaine monoclonal antibody was based on the extinction coefficient calculated from the amino acid sequence of h2E2, 219,500 cm−1M−1, as was the quantification of the Fab fragment of the h2E2 antibody generated by Endo Lys-C digestion, 73, 965 cm−1M−1.
The technique of non-equilibrium pH gel electrophoresis (NEPHGE),Citation27 a non-equilibrium technique used to analyze proteins with basic pIs, was performed to analyze this monoclonal antibody. This technique is a variant of IEF, in which the polarity of the electrodes is reversed and the protein is applied at the acidic end of the gel, and electrophoresis is performed for a defined period of time toward the basic end of the gel.
The high performance strong cation exchange column used for heterogeneity analyses was a 10 μm, 4 × 250 mm MabPac SCX-10 analytical column from Thermo Scientific (cat. 074625). MES buffer for this chromatography was prepared from MES from Sigma (cat. M-8250). The column was run at 1.0 ml/min at room temperature on an Akta Basic HPLC from Amersham Biosciences (pump module P-900; capable of pressures up 25MPa). The Akta HPLC is controlled using UNICORN software version 5.01 all run on Windows XP. The gradient used to analyze the intact h2E2 mAb () was from 20–40% B in 30 minutes. Buffer A = 20 mM MES, 60 mM NaCl, pH = 5.6, Buffer B = 20 mM MES, 300 mM NaCl, pH = 5.6. The gradient used to analyze the Fab fragment () was from 0-35% B from 2 minutes to 52 minutes. Buffer A = 20 mM MES, pH = 5.6, Buffer B = 20 mM MES, 300 mM NaCl, pH = 5.6. Results obtained in this study are consistent with analysis of other therapeutic monoclonal antibodies produced under similar expression conditions and analyzed with this column using similar conditions (as shown in manufacturer data supplied with this HPLC column).
Carboxypeptidase B digestion of the h2E2 antibody was accomplished using Carboxypeptidase B (Worthington, cat. LS05305). 2.0 mg/ml antibody and 0.05 mg/ml carboxypeptidase B were incubated for 1 hr at 22°C in 50 mM Tris-Cl, pH = 8.0, and subsequently diluted 10:1 with 20 mM MES, pH = 5.6, and stored at −80°C, prior to HPLC ion exchange analysis.
Endoproteinase Lys-C (lysyl Endoproteinase, Achromobacter protease I, Endo-Lys-C, cat. 129-02541) was obtained from Wako Pure Chemicals. This procedure begins with proteolytic digestion not with papain, but with Endoproteinase Lys-C (the cleavage site for Endo Lys-C is 2 amino acids away from the papain cleavage site in the heavy chain sequence. … EPKSCDK1TH2TCPP. … where 1 is the Endo Lys-C cleavage site and 2 is the papain cleavage site). Digestion with Endo Lys-C for Fab fragment generation was performed using 2.0 mg/ml antibody plus 0.004 mg/ml protease (1:500 protease to IgG) in 50 mM Tris-Cl, pH = 8.0 for 45 minutes at 22°C. The sample was then dialyzed for 3-4 hours at 4°C vs 20 mm MOPS, pH = 7.4. After incubation with a strong cation exchange resin (for 20 mg antibody digest used 1.5 ml bed volume of BioRad Macro-Prep High S Support (cat. 156-0030)) for 5 minutes at 22°C, the resin beads were poured into a column, washed extensively with 20 mM MOPS buffer, and the Fab fragment eluted in a minimum volume (approximately 3 ml) of 20 mM MOPS/200 mM NaCl, pH = 7.4 (theoretical protein pI values are: intact h2E2 mAb = 8.37, Fab fragment = 8.57, Fc fragment = 7.0). The sample was then applied to a 44 ml bed volume, 28 cm long Sephacryl S-100 column (Amersham Biosciences cat. 17-0612-10) and eluted at 0.5 ml/minute. 1.0 ml fractions were collected and measured for absorbance at 280 nm. Fractions were chosen to eliminate the small amount of contaminating aggregates and undigested antibody, which eluted before the main monomeric Fab fragment peak, and pooled for Fab concentration and analysis. The resultant pure Fab fragment was concentrated and quantified (the extinction coefficient for the Fab fragment calculated from its amino acid sequence is 73,965 cm−1M−1).
Measurements of intrinsic protein fluorescence and quenching of that fluorescence by cocaine and 2 of its metabolites were made using a Hitachi F-2000 fluorescence spectrophotometer at 20°C. 2 ml MAB or Fab solutions in Tris-Buffered Saline (TBS) were analyzed and titrated with ligands, measuring emission at 330 nm with excitation at both 280 nm and 295 nm after each addition of ligand. Blank values (fluorescence measurements using TBS with no added h2E2 mAb or Fab), were subtracted, and the resultant data fitted to a sigmoidal plot using Origin 7.0 software to obtain the EC50 values. Three or 4 independent titrations were performed to determine all drug-protein affinities. All KD values were calculated using the equation KD = EC50 – (0.5 × number of antibody binding sites), i.e., KD = [Free Ligand] at the EC50 of the fitted sigmoidal curve, thus KD = [Total Ligand] – [Bound Ligand] = [Free Ligand], where [Free Ligand] = [Total Ligand] added – [Total antibody binding sites] × 0.5 (since half of the antibody binding sites are occupied at the EC50 value). For example, for the 295 nm data shown by the filled square symbols in , KD = EC50 – (0.5 × 2 sites/mAb × 5.0 nM mAb) = 9.4 nM – 5.0 nM = 4.4 nM = KD for cocaine binding to the intact h2E2 monoclonal antibody.
Disclosure of Potential Conflicts of Interest
Dr. Norman is named as a co-inventor on a patent application for the use of the humanized anti-cocaine monoclonal antibody that is the subject of this manuscript.
Acknowledgments
We thank Dr. Thomas Thompson for the use of the Akta HPLC needed for the high performance cation exchange chromatography experiments, and Ryan Walker in Dr. Thompson's laboratory for instruction and help in its use. We also thank Dr. Jim Ball and Mike Tabet for their helpful comments and critiques of the manuscript.
Funding
This work was supported by the National Institutes of Health National Institute on Drug Abuse Grant DP1DA031386.
References
- Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S. Characterization of therapeutic antibodies and related products. Anal Chem 2013; 85:715-36; PMID:23134362; http://dx.doi.org/10.1021/ac3032355
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426-47; PMID:17828757; http://dx.doi.org/10.1002/jps.21180
- Kayser V, Chennamsetty N, Voynov V, Forrer K, Helk B, Trout BL. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol J 2011; 6:38-44; PMID:20949542; http://dx.doi.org/10.1002/biot.201000091
- Broussas M, Broyer L, Goetsch L. Evaluation of antibody-dependent cell cytotoxicity using lactate dehydrogenase (LDH) measurement. Methods Mol Biol 2013; 988:305-17; PMID:23475728; http://dx.doi.org/10.1007/978-1-62703-327-5_19
- Goetze AM, Liu YD, Zhang Z, Shah B, Lee E, Bondarenko PV, Flynn GC. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011; 21:949-59; PMID:21421994; http://dx.doi.org/10.1093/glycob/cwr027
- Sreedhara A, Cordoba A, Zhu Q, Kwong J, Liu J. Characterization of the isomerization products of aspartate residues at two different sites in a monoclonal antibody. Pharm Res 2012; 29:187-97; PMID:21809161; http://dx.doi.org/10.1007/s11095-011-0534-2
- Qi P, Volkin DB, Zhao H, Nedved ML, Hughes R, Bass R, Yi SC, Panek ME, Wang D, Dalmonte P, et al. Characterization of the photodegradation of a human IgG1 monoclonal antibody formulated as a high-concentration liquid dosage form. J Pharm Sci 2009; 98:3117-30; PMID:19009595; http://dx.doi.org/10.1002/jps.21617
- Lape M, Paula S, Ball WJ, Jr. A molecular model for cocaine binding by the immunotherapeutic human/mouse chimeric monoclonal antibody 2E2. Eur J Med Chem 2010; 45:2291-8; PMID:20185210; http://dx.doi.org/10.1016/j.ejmech.2010.02.004
- Lonberg N. Human antibodies from transgenic animals. Nat Biotechnol 2005; 23:1117-25; PMID:16151405; http://dx.doi.org/10.1038/nbt1135
- Norman AB, Tabet MR, Norman MK, Buesing WR, Pesce AJ, Ball WJ. A chimeric human/murine anticocaine monoclonal antibody inhibits the distribution of cocaine to the brain in mice. J Pharm Exp Ther 2007; 320:145-53; PMID:17023573; http://dx.doi.org/10.1124/jpet.106.111781
- Norman AB, Gooden FC, Tabet MR, Ball WJ. A recombinant humanized anti-cocaine monoclonal antibody inhibits the distribution of cocaine to the brain in rats. Drug Metab Dispos 2014; 42:1125-31; PMID:24733787; http://dx.doi.org/10.1124/dmd.114.057034
- Gadgil HS, Bondarenko PV, Pipes G, Rehder D, McAuley A, Perico N, Dillon T, Ricci M, Treuheit M The LC/MS analysis of glycation of IgG molecules in sucrose containing formulations. J Pharm Sci 2007; 96:2607-21; PMID:17621682; http://dx.doi.org/10.1002/jps.20966
- Ren D, Pipes G, Xiao G, Kleemann GR, Bondarenko PV, Treuheit MJ, Gadgil HS. Reversed-phase liquid chromatography-mass spectrometry of site-specific chemical modifications in intact immunoglobulin molecules and their fragments. J Chromatogr A 2008; 1179:198-204; PMID:18096172; http://dx.doi.org/10.1016/j.chroma.2007.11.088
- Raju TS. Glycosylation variations with expression systems and their impact on biological activity of therapeutic immunoglobulins. BioProcess Int 2003; April:44-53.
- Maley F, Trimble RB, Tarentino AL, Plummer TH, Jr. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem 1989; 180:195-204; PMID:2510544; http://dx.doi.org/10.1016/0003-2697(89)90115-2
- Mullan B, Dravis B, Lim A, Clarke A, Janes S, Lambooy P, Olson D, O'Riordan T, Ricart B, Tulloch AG. Disulphide bond reduction of a therapeutic monoclonal antibody during cell culture manufacturing operations. BMC Proc 2011; 5 Suppl 8:P110; http://dx.doi.org/10.1186/1753-6561-5-S8-P110
- Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. mAbs 2012; 4:17-23; PMID:22327427; http://dx.doi.org/10.4161/mabs.4.1.18347
- Liu H, Chumsae C, Gaza-Bulseco G, Hurkmans K, Radziejewski CH. Ranking the susceptibility of disulfide bonds in human IgG1 antibodies by reduction, differential alkylation, and LC-MS analysis. Anal Chem 2010; 82:5219-26; PMID:20491447; http://dx.doi.org/10.1021/ac100575n
- Husted LB, Sorensen ES, Sottrup-Jensen L. 4-(Aminosulfonyl)-7-fluoro-2,1,3-benzoxadiazole is not specific for labeling of sulfhydryl groups in proteins as it may also react with phenolic hydroxyl groups and amino groups. Anal Bioch 2003; 314:166-8; PMID:12633619; http://dx.doi.org/10.1016/S0003-2697(02)00650-4
- Lau H, Pace D, Yan B, McGrath T, Smallwood S, Patel K, Park J, Park SS, Latypov RF. Investigation of degradation processes in IgG1 monoclonal antibodies by limited proteolysis coupled with weak cation-exchange HPLC. J Chromatogr B Analyt Technol Biomed Life Sci 2010; 878:868-76; PMID:20206584; http://dx.doi.org/10.1016/j.jchromb.2010.02.003
- Kleemann GR, Beierle J, Nichols AC, Dillon TM, Pipes GD, Bondarenko PV. Characterization of IgG1 immunoglobulins and peptide-Fc fusion proteins by limited proteolysis in conjunction with LC-MS. Anal Chem 2008; 80:2001-9; PMID:18293943; http://dx.doi.org/10.1021/ac701629v
- Tetin SY, Hazlett TL. Optical spectroscopy in studies of antibody-hapten interactions. Methods 2000; 20:341-61; PMID:10694456; http://dx.doi.org/10.1006/meth.1999.0927
- Nakayama H, Kenjyou N, Shigetoh N. A novel monoclonal antibody specific for cocaine. Monoclon Antib Immunodiagn Immunother 2013; 32:262-4; PMID:23909419; http://dx.doi.org/10.1089/mab.2013.0002
- Bleck GT. Consistent production of genetically stable mammalian cell lines. BioPharm Int 2012; 25:56-9.
- Kirley TL. Reduction and fluorescent labeling of cyst(e)ine-containing proteins for subsequent structural analyses. Anal Biochem 1989; 180:231-6; PMID:2554752; http://dx.doi.org/10.1016/0003-2697(89)90422-3
- Toyo'oka T, Imai K. Isolation and characterization of cysteine-containing regions of proteins using 4-(Aminosulfonyl)-7-2,1,3-benzoxadiazole and high-performance liquid chromatography. Anal Chem 1985; 57:1931-7; PMID:4037347; http://dx.doi.org/10.1021/ac00286a032
- O'Farrell PZ, Goodman HM, O'Farrell PH. High resolution two-dimensional electrophoresis of basic as well as acidic proteins. Cell 1977; 12:1133-41; PMID:23215; http://dx.doi.org/10.1016/0092-8674(77)90176-3