
Abstract
2Iq23.1 microdeletion syndrome is a recently described rare disease that includes intellectual disability, motor delay, autistic-like behaviors, and craniofacial abnormalities. Dosage insufficiency of the methyl-CpG-binding domain protein 5 (MBD5) gene was suggested as the genetic cause, since all the described patients carry a partial or total heterozygous deletion of MBD5. We reported the generation and characterization of a mouse model with haploinsufficiency for Mbd5 that confirmed this hypothesis. As in human 2q23.1 microdeletion syndrome, the MBD5+/GT mouse model exhibited abnormal social behavior, cognitive impairment, and motor and craniofacial abnormalities, supporting a causal role for MBD5 in 2q23.1 microdeletion syndrome. The use of mouse neuronal cultures uncovered a deficiency in neurite outgrowth, suggesting the participation of MBD5 in neuronal processes. The study of the MBD5+/GT mouse advanced our understanding of the abnormal brain development associated with behavioral and cognitive symptoms.
Abbreviations
| MBD5 | = | methyl-CpG-binding domain protein 5 |
| BrdU | = | Bromodeoxyuridine |
| PR-DUB | = | Polycomb repressive deubiquitinase complex |
2q23.1 Microdeletion Syndrome
Copy number variations have been associated to many neurodevelopmental disorders, such as the recently described 2q23.1 microdeletion syndrome. First identified in 2009,Citation1 the syndrome was further defined in 2010.Citation2 The typical presentation of patients with 2q23.1 microdeletion includes intellectual disability, behavioral difficulties such as autistic spectrum disorder or attention-deficit/hyperactivity disorder, seizures, sleep disturbances, ataxia/unusual gait, failure to thrive, and specific craniofacial abnormalities.
The 2q23.1 locus contains several genes associated with genetic disorders. Analysis of the minimal region of deletion overlap in patients with 2q23.1 microdeletion syndrome identified MBD5 as a candidate causal gene.Citation3
Interestingly, microduplications in the same 2q23.1 region have been observed in cases with autistic or Angelman-like features and developmental delay, indicating that either decreases or increases in expression of wild-type MBD5 can result in clinical manifestations.Citation4-6
MBD5
Methylation of DNA is an epigenetic mark essential for mammalian development, with functional impact on tissue-specific gene expression, X chromosome inactivation, genomic imprinting, and suppression of transposable elements.Citation7,8
MBD5 is a newly recognized member of a family of proteins carrying a defining domain, methyl-CpG-binding domain (MBD), which binds preferentially to methylated DNA. Other members of this group of methyl-CpG binding proteins include MeCP2, MBD1, MBD2, MBD3, MBD4, MBD6, setdb1 and setdb2, and BAZ2A, and BAZ2B.Citation9
In spite of having a recognizable MBD, the preferential binding of MBD5 to methylated DNA has yet to be proved. The MBD domain of MBD5 was not able to bind to methylated DNA in vitro.Citation10 Thus, although the presence of MBD in MBD5 suggests it might play a role as mediator of DNA methylation outcomes,Citation11 its functions need to be experimentally tested.
Our data suggest that the main isoforms of MBD5 in the mouse brain contain a proline and tryptophan-rich domain called PWWP domain, in addition to the MBD domain. The PWWP domain was initially described as a DNA binding domainCitation12 and more recently identified as a recognition domain for histone marks predominantly associated with actively transcribed chromatin.Citation13 Mammalian proteins that contain a PWWP domain are mostly chromatin binding proteins, including DNMT3A, DNMT3B, bromodomain-containing protein 1 (BRD1), bromodomain and PHD finger-containing protein 1 (BRPF1), 2 (BRPF2) and 3 (BRPF3), and DNA mismatch repair protein MSH6.
Thus, the presence of MBD and PWWP domains in MBD5 suggests a possible role of this protein in transcriptional regulation. We found that, when fused to the DNA-binding domain of GAL4, MBD5 activates transcription of a reporter gene containing a GAL4 binding sequence. Notably, a truncated version lacking the PWWP domain showed higher transcriptional activation capabilities. Thus, in these in vitro assays, the presence of a PWWP domain usually associated with active transcription does not seem to contribute to the activator function of MBD5. In agreement with our data, Tao et al reported that the PWWP domain was unessential for MBD5's activation of the Fth1 promoter, a purported endogenous target of MBD5.Citation14 Interestingly, MBD5 was shown to interact with the human Polycomb group protein complex PR-DUB through their MBD in a manner that is independent of the PWWP domain. Thus, the functionality of the PWWP domain in MBD5 remains to be identified.
Mbd5-haploinsufficient Mice
We have recently showed that mice carrying an insertional mutation in the Mbd5 gene generated through gene-trap mutagenesis (MBD5+/GT) constitute a model for Mbd5 haploinsufficiency.Citation15 These mice express a mutant Mbd5 transcript composed of the first 2 exons of Mbd5 fused to a lacZ reporter gene. We showed that the MBD5+/GT mice express around 60% of endogenous MBD5. In concordance with a recent report of Mbd5-null mice,Citation16 the homozygous MBD5GT/GT mutant animals die soon after birth, suggesting and essential role for MBD5 in postnatal survival. Whether deficiency of MBD5 in humans is also inconsistent with survival is not clear yet, but, to our knowledge, no homozygous inactivating mutations in MBD5 have been reported.
Mbd5- haploinsufficient Mice Recapitulates Critical Aspects of The 2q23.1 Deletion Syndrome
Neurobehavioral characterization of the MBD5+/GT mice revealed impaired learning and memory, and altered social behavior, reminiscent of 2 of the most characteristic clinical features of the 2q23.1 microdeletion patients: developmental delay/intellectual disability, and autistic-like manifestations. The MBD5+/GT mice exhibited deficiencies in both hippocampus and amygdala-dependent fear conditioning, suggesting that the general ability to learn basic associations is impaired. These mice also showed increased self-grooming, a stereotyped behavior in mice that is often compared to the restrictive repetitive and stereotyped patterns of behavior seen in autism.Citation17 Of interest is the observation that, upon introduction of an object into their home cage, the excessive self-grooming of the MBD5+/GT mice was replaced by a compulsive interaction with the novel object. We also observed that MBD5+/GT mice spent more time interacting with stranger mice than with their wild-type littermates. Thus, these data suggested to us that the MBD5+/GT mice demonstrated an abnormal insistence on sameness evocative of human autistic features.
Strength, balance, and movement coordination were affected in MBD5+/GT mice, in line with the diminished motor skills observed in the majority of the 2q23.1 microdeletion syndrome patients.Citation1-4,Citation18
2q23.1 microdeletion syndrome patients are often described as having craniofacial anomalies,Citation3,6,19 however, these are not completely consistent. This was generally supposed to be the result of different deletion size and location and the possible presence of polymorphic cis- and trans-acting modifiers. MBD5+/GT mice present craniofacial abnormalities mimicking the alterations found in 2q23.1 microdeletion syndrome patients. Notably, in spite of these mice having a common mutation and genetic background, we observed deviation of the snout toward the left or the right side in the same proportion in the MBD5+/GT mice. We therefore speculated that the craniofacial phenotype might result from delayed ossification combined with an environmental effect. This may explain the inconsistency of the craniofacial anomalies of the 2q23.1 microdeletion syndrome patients.Citation3,6,19 The mutant mice have reduced abdominal fat, consistent with the failure to thrive/growth retardation observed in 2q23.1 microdeletion syndrome.
Studies of Mbd5- haploinsufficient Mice Suggest Mechanisms of Pathogenesis for 2q23.1 Deletion Syndrome
Mbd5 is expressed throughout the brain, as revealed by positive β-gal activity in MBD5+/GT mice, a proxy for Mbd5 expression. Highest expression was observed in neurons of the cortex, olfactory bulb, striatum, hippocampus, and cerebellum. Thus, with the goal of shedding light into the pathogenesis of 2q23.1 deletion syndrome, we searched for neuronal structural deficits by analyzing neurite extension in the early phases of differentiation of cortical neurons in vitro. MBD5+/GT neurons showed decreased neurite length and less branching points compared to wild-type neurons, suggesting an important role for MBD5 in neuronal differentiation. The identification of this cellular phenotype points toward early processes of neuronal differentiation as instrumental in MBD5 brain pathology. Furthermore, the availability of a robust, quantifiable cellular phenotype constitutes a potential platform for future screening of compounds capable of phenotypic modulation.
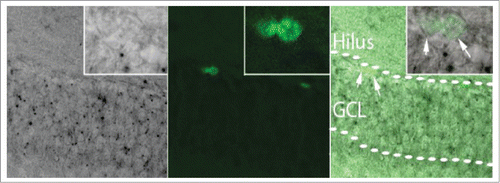
The mechanisms by which MBD5 participates in the regulation of neurite formation, progression, and maintenance remain to be addressed. Expression of MBD5 is not evident in proliferating BrdU-positive cells in the subgranular zone of the dentate gyrus (), suggesting that the onset of expression of MBD5 in neurons might correlate with neuronal differentiation processes.
Figure 1. Proliferating neural stem cells do not express MBD5. 2 month old Mbd5+/GT mice were treated with BrdU 24 h before brain collection and processed for X-gal (left panel) and BrdU immunolabeling (center panel). BrdU positive cells (arrows, merged right panel and magnified inset) in the subgranular zone do not show X gal staining, suggesting that newly generated cells in the brain do not express MBD5. GCL: granule cell layer.
Concluding Remarks
The study of the consequences of Mbd5 haploinsufficiency in mice confirmed a causal role for this gene in most of the clinical manifestations presented by 2q21.3 microdeletion syndrome. Our report established the MBD5+/GT mouse model as a valuable model for social disturbances and intellectual disabilities in patients with 2q21.3 deletions and will be useful for delineation of mechanisms of pathogenesis. The extent of involvement of transcriptional functions of MBD5 in pathogenic mechanisms is unknown and will be subject to future investigations.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest have been disclosed.
Acknowledgments
We acknowledge all authors of the original study.
Funding
We appreciate the financial support of the Le Jerome Lejeune Foundation.
References
- Jaillard S, Dubourg C, Gerard-Blanluet M, Delahaye A, Pasquier L, Dupont C, Henry C, Tabet AC, Lucas J, Aboura A, et al. 2q23.1 microdeletion identified by array comparative genomic hybridisation: An emerging phenotype with angelman-like features? J Med Genet 2009; 46:847-55; PMID:18812405; http://dx.doi.org/10.1136/jmg.2008.058156
- van Bon BW, Koolen DA, Brueton L, McMullan D, Lichtenbelt KD, Ades LC, Peters G, Gibson K, Moloney S, Novara F, et al. The 2q23.1 microdeletion syndrome: Clinical and behavioural phenotype. Eur J Hum Genet 2010; 18:163-70; PMID:19809484; http://dx.doi.org/10.1038/ejhg.2009.152
- Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, Gastier-Foster J, Thrush DL, Kathiresan S, Ruderfer DM, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet 2011; 89:551-63; PMID:21981781; http://dx.doi.org/10.1016/j.ajhg.2011.09.011
- Chung BH, Stavropoulos J, Marshall CR, Weksberg R, Scherer SW, Yoon G. 2q23 de novo microdeletion involving the MBD5 gene in a patient with developmental delay, postnatal microcephaly and distinct facial features. Am J Med Genet A 2011; 155A:424-9; PMID:21271666; http://dx.doi.org/10.1002/ajmg.a.33821
- Bonnet C, Ali Khan A, Bresso E, Vigouroux C, Beri M, Lejczak S, Deemer B, Andrieux J, Philippe C, Moncla A, et al. Extended spectrum of MBD5 mutations in neurodevelopmental disorders. Eur J Hum Genet 2013; 21:1457-61; PMID:23422940; http://dx.doi.org/10.1038/ejhg.2013.22
- Mullegama SV, Rosenfeld JA, Orellana C, van Bon BW, Halbach S, Repnikova EA, Brick L, Li C, Dupuis L, Rosello M, et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet 2014; 22:57-63; PMID:23632792; http://dx.doi.org/10.1038/ejhg.2013.67
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33 Suppl:245-54; PMID:12610534; http://dx.doi.org/10.1038/ng1089
- Bestor TH, Bourc'his D. Transposon silencing and imprint establishment in mammalian germ cells. Cold Spring Harb Symp Quant Biol 2004; 69:381-7; PMID:16117671; http://dx.doi.org/10.1101/sqb.2004.69.381
- Roloff TC, Ropers HH, Nuber UA. Comparative study of methyl-CpG-binding domain proteins. BMC Genomics 2003; 4:1; PMID:12529184
- Laget S, Joulie M, Le Masson F, Sasai N, Christians E, Pradhan S, Roberts RJ, Defossez PA. The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS One 2010; 5:e11982; PMID:20700456; http://dx.doi.org/10.1371/journal.pone.0011982
- Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol 1998; 18:6538-47.
- Laguri C, Duband-Goulet I, Friedrich N, Axt M, Belin P, Callebaut I, Gilquin B, Zinn-Justin S, Couprie J. Human mismatch repair protein MSH6 contains a PWWP domain that targets double stranded DNA. Biochemistry 2008; 47:6199-207; PMID:18484749; http://dx.doi.org/10.1021/bi7024639
- Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010; 142:967-80; PMID:20850016; http://dx.doi.org/10.1016/j.cell.2010.08.020
- Tao Y, Wu Q, Guo X, Zhang Z, Shen Y, Wang F. MBD5 regulates iron metabolism via methylation-independent genomic targeting of Fth1 through KAT2A in mice. Br J Haematol 2014; 166:279-91; PMID:24750026; http://dx.doi.org/10.1111/bjh.12863
- Camarena V, Cao L, Abad C, Abrams A, Toledo Y, Araki K, Araki M, Walz K, Young JI. Disruption of Mbd5 in mice causes neuronal functional deficits and neurobehavioral abnormalities consistent with 2q23.1 microdeletion syndrome. EMBO Mol Med 2014; 6:1003-15; PMID:25001218; http://dx.doi.org/10.15252/emmm.201404044
- Du Y, Liu B, Guo F, Xu G, Ding Y, Liu Y, Sun X, Xu G. The essential role of Mbd5 in the regulation of somatic growth and glucose homeostasis in mice. PLoS One 2012; 7:e47358; PMID:23077600; http://dx.doi.org/10.1371/journal.pone.0047358
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), Text Revision. Washington, D.C: American Psychiatric Press, Inc, 2000.
- Noh GJ, Graham JM,Jr. 2q23.1 microdeletion of the MBD5 gene in a female with seizures, developmental delay and distinct dysmorphic features. Eur J Med Genet 2012; 55:354-7; PMID:22659271; http://dx.doi.org/10.1016/j.ejmg.2012.05.003
- Williams SR, Mullegama SV, Rosenfeld JA, Dagli AI, Hatchwell E, Allen WP, Williams CA, Elsea SH. Haploinsufficiency of MBD5 associated with a syndrome involving microcephaly, intellectual disabilities, severe speech impairment, and seizures. Eur J Hum Genet 2010; 18:436-41; PMID:19904302; http://dx.doi.org/10.1038/ejhg.2009.199