
Abstract
Several human diseases, including Huntington's disease (HD), are associated with the expression of mutated, misfolded, and aggregation-prone amyloid proteins. Cardiac disease is the second leading cause of death in HD, which has been mainly studied as a neurodegenerative disease that is caused by expanded polyglutamine repeats in the huntingtin protein. Since the mechanistic basis of mutant HD-induced cardiomyopathy is unknown, we established a Drosophila heart model that exhibited amyloid aggregate-induced oxidative stress, resulting in myofibrillar disorganization and physiological defects upon expression of HD-causing PolyQ expression in cardiomyocytes. Using powerful Drosophila genetic techniques, we suppressed mutant HD-induced cardiomyopathy by modulating pathways associated with folding defects and oxidative stress. In this addendum, we describe additional potential molecular players that might be associated with HD cardiac amyloidosis. Drosophila, with its high degree of conservation to the human genome and many techniques to manipulate its gene expression, will be an excellent model for the suppression of cardiac amyloidosis linked to other polyglutamine expansion repeat disorders.
Abbreviations
| HD | = | Huntington's disease |
| PolyQ | = | poly-glutamine |
| HTT | = | huntingtin |
| UAS | = | upstream activating sequence |
| GFP | = | green fluorescent protein |
| UPR | = | unfolded protein response |
| SOD | = | superoxide dismutase |
| ROS | = | reactive oxygen species |
Commentary
HD is a rare neurodegenerative disorder resulting from expanded poly-glutamine (PolyQ) repeats in the huntingtin (HTT) protein, leading to HTT misfolding and the formation of pathogenic aggregates in neuronal tissue.Citation1 Several epidemiological studies have suggested a link between HD and cardiac amyloidosis, a group of diseases characterized by protein misfolding and the accumulation of aggregates in the heart.Citation2-4 Heart failure is the second leading cause of death in HD patients, accounting for over 30% of HD deaths compared to 2% of age-matched controls.Citation2-6 However, the mechanisms of PolyQ-induced cardiac defects are unknown and currently, no treatments exist to ameliorate heart failure due to HD. Since HTT is expressed in cardiac muscle and PolyQ aggregates have been reported in non-CNS tissues,Citation3 we postulated that cardiac expression of mutant PolyQ leads to pathogenic HTT aggregate formation in cardiac tissue and subsequent heart dysfunction. To study this, we generated a model for cardiac amyloidosis caused by expanded polyglutamine repeats in HTT using the genetically tractable Drosophila melanogaster system.Citation7
Drosophila has long been used as a model for studying congenital heart diseases, since many of their genes controlling heart function and development are conserved (reviewed by Bier and Bodmer).Citation8 More recently the physiological basis of cardiac dysfunction in adult Drosophila due to aging and disease has been characterized using semi-intact heart preparations in conjunction with high speed digital imaging,Citation9 as well as in vivo by optical coherence tomography.Citation10 For example, cardiac expression of mutant HTT using the upstream activating sequence (UAS)-Gal4 system in conjunction with the Hand driver has allowed us to determine the deleterious effects of mutant PolyQ on cardiac structure and function.Citation7 Thus, Drosophila is a useful model for studying functional abnormalities associated with HD cardiac pathogenesis.
Mutant PolyQ Can Aggregate in the Heart, Causing Cardiac Dysfunction and Myofibrillar Degradation
In our recent publication,Citation7 we sought to explore the cardiac defects associated with expression of aggregation-prone mutant PolyQ, which is known to cause HD at PolyQ repeat lengths greater than 35. Although epidemiological studies have suggested cardiac defects in HD patients,Citation2-5 it remains unclear if these defects result from primary (autonomous) defects in the cardiomyocytes or if expression of mutant HTT in neurons contributes to secondary (non-autonomous) cardiac pathogenesis. Expression of the aggregation prone exon-1 Htt fragment has been used to explore the mechanisms of HD in other tissues, including skeletal muscles.Citation11-13 In our Drosophila model, we observed severe cardiac dilation, contractile defects, and cardiac arrhythmias in a PolyQ length-dependent manner due to cardiac expression of mutant PolyQ.Citation7 It is unknown whether these defects are due to cell autonomous expression of mutant PolyQ since the Hand gene is expressed in pericardial nephrocytes and lymph gland haematopoietic progenitors in addition to cardioblasts.Citation14 Mutant PolyQ hearts also exhibited structural abnormalities, including myofibrillar disarray and loss of actin and myosin-containing myofibrils. These cardiac defects were accompanied by reductions in the lifespan of flies expressing mutant PolyQ. Interestingly, green fluorescent protein (GFP)-tagged mutant PolyQ protein aggregates were detected in the form of GFP+ puncta in hearts expressing pathogenic lengths of PolyQ repeats compared to non-pathogenic PolyQ-25. Thus, cardiac expression of toxic PolyQ is sufficient to cause both functional and structural cardiac defects and may be associated with aggregation of mutant PolyQ. In support of these findings, a mouse model for mutant PolyQ resulted in heart failure by 12 weeks of age and the formation of aggregates that were immunoreactive for anti-amyloid in the heart.Citation15 Unlike our Drosophila model,Citation7 R6/2 mice expressing mutant PolyQ did not exhibit cardiac remodeling or cell death.Citation16 It is possible that the GFP tag used in our model may have increased the susceptibility for aggregation of mutant PolyQ and resulted in more severe defects, including cardiac dilation and non-contractile regions of the heart. However, aggregates were not observed in control hearts expressing GFP-tagged non-pathogenic PolyQ-25, suggesting that the GFP tag alone does not cause aggregation of HTT. It remains unclear if aggregation of PolyQ was directly causative of cardiac dysfunction in miceCitation16 and our Drosophila model.Citation7 However, amyloid formation has been observed in other cardiac amyloid models, including a murine model for desmin related cardiomyopathy (mutant alphaB crystallin R120G).Citation15 Based upon these findings, we predict that pathogenic aggregates accumulate in cardiac tissue of HD patients and contribute to cardiomyopathy. It remains unknown if the aggregates observed in our model contain amyloid deposits.Citation7 Novel therapeutic strategies to reduce protein aggregation may prove useful in clinical settings to treat PolyQ-induced cardiac amyloidosis.
Aggregation of mutant PolyQ is suspected to play a major role in HD cardiac pathogenesis by interfering with several cellular pathways, including the unfolded protein response (UPR) and the oxidative stress response.Citation17-19 Mouse models have shown that expression of mutant HTT or expression of pre-amyloid oligomers is sufficient to cause cardiac defects by inducing protein misfolding, oxidative stress, and autophagy.Citation16,20,21 However, the precise mechanisms of PolyQ-induced cardiomyopathy remain unknown. Thus, we examined the mechanism by which aggregate accumulation caused abnormal heart function.Citation7 As detailed below, we found that cardiac expression of mutant PolyQ produced myofibrillar degeneration and ultrastructural defects in a PolyQ length-dependent manner.
Mutant PolyQ and Protein Misfolding
Molecular chaperones reduce the toxicity of protein aggregation by facilitating folding of aggregation-prone mutant HTT.Citation1 Several chaperones, including Hsp-70, Hsp-40, and Hsp-110, have been shown to modulate HD phenotypes in neurons.Citation22,23 However, it remains unclear if expression of mutant PolyQ results in the misfolding of contractile proteins in the human heart. Although mutant PolyQ leads to cardiac failure in mice,Citation16,24 it was previously unknown if PolyQ directly targets accumulation of key contractile proteins, e.g., myosin and actin. As mentioned in our manuscript,Citation7 a majority of PolyQ mutant hearts contains non-contractile regions, indicative of cell death or mislocalization of contractile proteins. Since the proper folding of such proteins is essential for cardiac function, we sought to determine if the presence of non-contractile regions in the hearts of PolyQ mutant flies is due to mislocalization of contractile proteins.Citation7 We showed that cardiac expression of mutant PolyQ leads to disruptions in the accumulation of myosin and actin in cardiac myofibrils and that these changes are associated with severe defects in contractility. It is not known if mutant PolyQ leads to myofibrillar abnormalities by interacting with these contractile proteins or by interfering with the unfolded protein response, which is necessary to maintain ER homeostasis and contractile protein turnover (reviewed by Groenendyk et al).Citation25 We showed that GFP-tagged HTT protein aggregates co-localize with both actin and myosin,Citation7 suggesting a possible link between aggregate formation and defects in assembly or maintenance of actin and myosin-containing myofibrils. Alternatively, the formation of pathogenic aggregates may also inhibit the unfolded protein response by directly interfering with chaperone activity. In an amyloid model for desmin-related cardiomyopathy, mutant alphaB-crystallin (R120G) expression in cardiomyocytes caused defects in chaperone activity and folding of contractile proteins.Citation15,21,26 Expression of both mutant alphaB-crystallin and mutant PolyQ resulted in amyloid-positive aggregate formation in cardiomyocytes and myocyte degeneration,Citation21 suggesting a possible link between protein misfolding and cardiomyocyte death. To determine the role of the unfolded protein response in PolyQ-induced cardiomyopathy, we overexpressed the myosin-associated chaperone UNC-45 in PolyQ mutants as a novel approach to suppress PolyQ-induced cardiac defects.Citation7 We showed that overexpression of UNC-45 suppresses PolyQ-induced formation of aggregates, loss of myofibrils, and ameliorated cardiac defects to a great extent, suggesting that protein misfolding plays a role in the loss of myosin due to expression of mutant PolyQ. Although overexpression of UNC-45 in PolyQ mutant hearts restored the integrity of actin-containing myofibrils as well, it remains unclear if the loss of actin-containing myofibrils due to mutant PolyQ is caused by decreased accumulation of myosin, or if PolyQ aggregates interfere with the accumulation of other contractile proteins such as actin. We found that overexpression of UNC-45 alone could not completely suppress cardiac dysfunction. A nearly complete reversal of PolyQ-induced cardiac phenotypes required modulation of the oxidative stress response in addition to UNC-45 overexpression.
Mutant PolyQ and Oxidative Stress
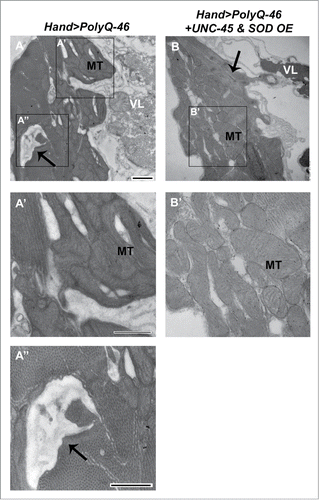
Since the accumulation of misfolded and aggregated proteins is known to induce mitochondrial damage and oxidative stress in several neurodegenerative disorders,Citation27 we hypothesized that oxidative stress plays a role in PolyQ-mediated cardiac dysfunction. In support of our hypothesis, Mihm et al. showed that cardiac expression of mutant PolyQ was associated with mitochondrial morphological defects and enhanced oxidative stress.Citation16 Furthermore, impairment of mitochondrial function and decreased expression of electron transport chain proteins have been linked to the presence of misfolded PolyQ aggregates in neurons of HD patients.Citation28 The majority of reactive oxygen species (ROS) generation is due to leakage of oxidants from the electron transport chain of mitochondria.Citation29 Since mitochondria occupy greater than 25% of cardiomyocyte volume to provide sufficient energy for contractility,Citation30 cardiomyocytes are particularly susceptible to oxidative stress induced by defective mitochondria. Assuming that cardiomyocyte function in Drosophila is heavily dependent on mitochondrial integrity as in mammalian systems,Citation31 we sought to examine if PolyQ-induced cardiac dysfunction is linked to mitochondrial ultrastructural abnormalities. Cardiac expression of mutant PolyQ-46 led to severe mitochondrial cristae fragmentation, which was observed in electron micrographs of thin sections (). In our manuscript,Citation7 we showed that suppression of PolyQ-induced oxidative stress by overexpressing superoxide dismutase (SOD) was critical to restore functional and structural cardiac abnormalities, including mitochondrial ultrastructural defects. It remains unclear whether myofibrillar degeneration due to aggregation of mutant PolyQ is directly inhibiting mitochondrial organization, and/or if oxidative stress produced by PolyQ leads to mitochondrial dysfunction. Mitochondrial organization is meticulously controlled by the cardiomyocyte and, therefore, depends on the proper function and organization of contractile proteins.Citation32 Thus, our studies suggest that HD pathogenesis in the heart is linked to enhanced oxidative stress together with mitochondrial abnormalities.
Figure 1. Mitochondrial ultrastructural defects in mutant PolyQ-expressing hearts are suppressed with UNC-45 and SOD overexpression. (A, A′, A″) Cardiac expression of mutant PolyQ-46 resulted in degenerated areas within the cardiomyocyte (arrow), along with fragmentation of mitochondria in cardiomyocytes. MT indicates a fragmented mitochondrion. (B, B’) Combined overexpression of SOD and UNC-45 in PolyQ-46-expressing hearts improved mitochondrial ultrastructure. MT indicates a normally shaped mitochondrion with densely packed cristae. Degeneration (arrow) was reduced to some extent in SOD+UNC-45 overexpressing hearts. VL refers to non-cardiac ventral-longitudinal fibers. Scale bar is 500 nm.
Several studies suggest that certain cardiac amyloidosis-based disorders are associated with oxidative stress and mitochondrial dysfunction. The vulnerability of mitochondria to oxidative stress was shown in a mouse model for oxidative stress-related dilated cardiomyopathy by genetic ablation of the mitochondrial form of superoxide dismutase, SOD2, which resulted in an abnormal “ballooning” of mitochondria in electron micrographs of dendrites and decreased lifespan.Citation33 Additionally, cardiac-specific expression of mutant alphaB-crystallin also resulted in the accumulation of amyloid-containing protein aggregates in cardiomyocytes in a cell-autonomous manner, leading to enhanced oxidative stress.Citation15,32,34 Decreasing SOD production in alphaB-crystallin mutants by pharmacologically inhibiting the activity of the superoxide-generating xanthine oxidase restored mitochondrial function and membrane potential, suggesting a link between mitochondrial dysfunction and oxidative stress due to cardiac amyloidosis.Citation34 Thus, enhanced oxidative stress and mitochondrial dysfunction leading to heart failure may depict a common mechanism among cardiac amyloidosis-based disorders.
Multiple Cellular Pathways are Involved in HD Cardiac Pathogenesis
In our manuscript,Citation7 we manipulated both the oxidative stress and protein misfolding pathways as a novel approach to suppressing PolyQ-induced cardiac phenotypes. We found that overexpression of UNC-45 or SOD alone did not completely suppress mutant PolyQ-induced cardiac phenotypes, suggesting that multiple cellular pathways are involved in HD cardiac pathogenesis. Our studies demonstrated that overexpression of SOD in conjunction with the myosin-associated UNC-45 chaperone led to better suppression of PolyQ-induced cardiac dysfunction in vivo than either treatment alone. Overexpression of both was required to improve fractional shortening and decrease arrhythmias in PolyQ-72 mutants nearly to the extent of driver line controls. Additionally, this combined treatment reduced aggregation of mutant GFP+ PolyQ punctae in PolyQ-72 mutant hearts more than either treatment alone. We did not show whether ultrastructural phenotypes in PolyQ mutant hearts were suppressed with overexpression of UNC-45 and SOD in our manuscript.Citation7 Here, we compared thin sections of hearts from 4 week-old flies in mutant and suppressor lines, using transmission electron microscopy to determine if combined overexpression suppresses ultrastructural defects caused by expression of mutant PolyQ (). Expression of mutant PolyQ-46 resulted in severe disruptions in mitochondrial cristae structure and myofibrillar degeneration, which were suppressed with UNC-45 and SOD overexpression. Hearts expressing both suppressors contained normally shaped mitochondria with dense cristae as well as reduced myofibrillar degeneration (arrow). In support of our findings, PolyQ aggregation in mouse neurons was enhanced in vitro with oxidative stress and suppressed with overexpression of SOD together with chaperones Hsp-70/Hsp-40,Citation35 suggesting that targeting both cellular pathways is efficacious in HD pathogenesis in multiple tissue types. Our results suggest that both the oxidative stress and protein unfolding pathways play a role in PolyQ-induced cardiac pathogenesis, and targeting both pathways is a relevant approach for suppressing PolyQ-induced cardiomyopathy.Citation7 It is unknown whether combined overexpression of UNC-45 and SOD rescues cardiac defects in other cardiac amyloidosis disorders. If the cellular pathways governing other cardiac amyloidosis disorders are similar to those involved in PolyQ-induced cardiomyopathy, our model can provide valuable insight into potential disease mechanisms in other models and relevant therapeutic approaches. However, some aggregation-based cardiac diseases may not involve mitochondrial dysfunction. Thus, further studies are necessary to determine the broad implications of manipulating both pathways to treat other cardiac amyloidosis disorders.
Future Directions
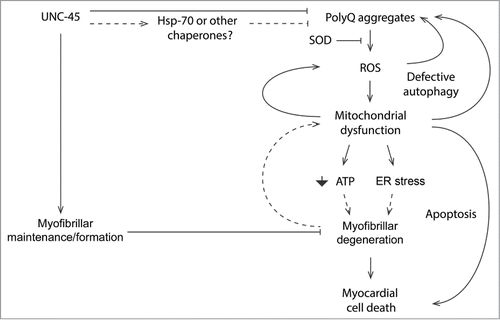
Further investigation will explore the exact molecular players associated with the accumulation of PolyQ-induced cardiomyopathy (). Based on our findings, we propose a model in which mutant PolyQ is associated with aggregate formation in the heart, possibly causing mitochondrial abnormalities associated with enhanced ROS. Although other studies (reviewed by Taverne et al.)Citation36 have also suggested a role for ROS in inducing cardiac dysfunction, it is unknown if accumulation of PolyQ aggregates generates ROS by interfering with mitochondrial function directly, and/or if mitochondrial damage is a result of ROS generation due to PolyQ aggregation. Although not tested in our model, we speculate that some mutant PolyQ-induced cardiac defects are due to mitochondrial dysfunction independent of PolyQ aggregation. Further studies are necessary to determine if cardiac defects occur in the absence of aggregate formation. Additionally, it is unknown whether enhanced oxidative stress and mitochondrial dysfunction are causative of cardiac dysfunction or markers of disease. The exact mechanisms regulating mitochondrial damage in association with ROS will be deciphered in future studies. Although ROS generation is known to promote PolyQ aggregation in HD 150Q cultured cells, causing proteosomal defects and mutant HTT aggregation,Citation35 it is unknown if decreased proteosomal function plays a role in exacerbating PolyQ aggregation in cardiomyocytes.
Figure 2. Mechanisms of PolyQ-induced cardiac amyloidosis. Our findings support a model in which both the oxidative stress and protein unfolding pathways underlie cardiac pathogenesis arising from mutant PolyQ. Future approaches will aim to characterize the exact molecular players associated with the production of PolyQ-induced cardiomyopathy.
The molecular mechanisms whereby UNC-45 suppresses cardiac dysfunction and myofibrillar/mitochondrial abnormalities due to mutant PolyQ remain unclear. However, protein folding to enhance myofibrillar integrity. It is well-established that UNC-45 promotes the assembly and refinement of the contractile apparatus by directly binding and folding myosin to enhance thick filament assembly as well as cardiac contractility.Citation37-40 However, it remains unknown if UNC-45 directly prevents the formation or mediates the clearance of PolyQ aggregates to promote refolding of mutant HTT protein. Although UNC-45 is known to bind other chaperones including Hsp-70 and Hsp-90 in vitro,Citation41,42 it is unknown if these proteins act as co-chaperones to promote refolding of mutant HTT protein. Further studies will determine if UNC-45 directly interacts with contractile proteins to suppress myofibril degeneration or assembly defects due to mutant PolyQ, and/or if UNC-45 can inhibit formation or mediate clearance of PolyQ aggregates through interactions with other chaperones.
We are interested in additional cellular processes involved in PolyQ-induced cardiomyopathy. In PolyQ-72 mutant hearts, Lysotracker+-positive punctae co-localized with GFP+ aggregates, suggesting a possible role of autophagy in the clearance of PolyQ aggregates. In support of our model, a non-HTT mouse cardiac model for PolyQ repeat expansion disorders showed increased autophagy associated with aggregation of mutant PolyQ and heart failure.Citation43 Further studies are necessary to confirm the enhanced formation of autophagosomes in mutant HTT hearts and their potential roles in the clearance of protein aggregates, defective mitochondria, or both. In mice, cardiac expression of mutant PolyQ led to defects in autophagy-mediated clearance of defective mitochondria.Citation21 In our model, mitochondrial ultrastructural defects were observed in PolyQ mutant hearts, which were suppressed with SOD overexpression, suggesting that mitochondrial dysfunction and ROS generation play a role in myofibrillar degeneration.Citation7 Thus, modulating the autophagy-mediated clearance of defective mitochondria may become a useful therapeutic strategy for future approaches to combat PolyQ-induced cardiomyopathy.
Since mitochondria occupy a large portion of cardiomyocyte volume to provide sufficient energy for contractility,Citation30,44 myofibrillar degeneration and disorganization in PolyQ mutant hearts may occur due to defective ATP generation by dysfunctional mitochondria or enhanced ER stress. Alternatively, mitochondrial dysfunction may trigger activation of apoptotic pathways, causing cell death in PolyQ mutant hearts. The presence of non-contractile regions in mutant PolyQ hearts suggests that myocardial cell death plays a role in cardiac pathogenesis, and further studies will determine if cell death occurs through defects in energy generation or by activation of apoptotic pathways.
In conclusion, we demonstrated that modulating both the protein unfolding and oxidative stress pathways suppress PolyQ-induced cardiac defects in a Drosophila model. Additional cellular pathways that underlie cardiac failure in HD patients have not been explored and will provide a basis for developing further therapeutic interventions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by NIH R21RR032100 and a Beginning Grant-in-Aid grant from American Heart Association (13BGIA17260057) to G.C.M., NIH R01AR055958 to S.I.B., NIH R01 HL54732 and P01 AG033456 to R.B. and a Scientist Development Grant from the American Heart Association (AHA0835243N) to K.O. R.R. was supported by the NIH/NIGMS SDSU MARC Program, grant T34GM008303.
References
- Arrasate M., Finkbeiner S. Protein aggregates in Huntington's disease. Exp Neurol 2012; 238:1-11; PMID:22200539; http://dx.doi.org/10.1016/j.expneurol.2011.12.013
- Lanska DJ, Lavine L, Lanska MJ, Schoenberg BS. Huntington's disease mortality in the United States. Neurology 1988; 38:769-72; PMID:2966305; http://dx.doi.org/10.1212/WNL.38.5.769
- Bradford JW, Li S, Li XJ. Polyglutamine toxicity in non-neuronal cells. Cell Res 2010; 20:400-07; PMID:20231860; http://dx.doi.org/10.1038/cr.2010.32
- Chiu E, Alexander L. Causes of death in Huntington's disease. Med J Aust 1982; 1:153; PMID:6210834
- Heemskerk AW, Roos RA. Aspiration pneumonia and death in Huntington's disease. PLoS Curr 2012; 4:RRN1293; PMID:22307361; http://dx.doi.org/10.1371/currents.RRN1293
- Sorensen SA, Fenger K. Causes of death in patients with Huntington's disease and in unaffected first degree relatives. J Med Genet 1992; 29:911-4; PMID:1479606; http://dx.doi.org/10.1136/jmg.29.12.911
- Melkani GC, Trujillo AS, Ramos R, Bodmer R, Bernstein SI, Ocorr K. Huntington's disease induced cardiac amyloidosis is reversed by modulating protein folding and oxidative stress pathways in the Drosophila heart. PLoS Genet 2013; 9:e1004024; PMID:24367279; http://dx.doi.org/10.1371/journal.pgen.1004024
- Bier E, Bodmer R. Drosophila, an emerging model for cardiac disease. Gene 2004; 342:1-11; PMID: 15527959; http://dx.doi.org/10.1016/j.gene.2004.07.018
- Ocorr K, Akasaka T, Bodmer R. Age-related cardiac disease model of Drosophila. Mech Ageing Dev 2007; 128: 112-6; PMID:17125816; http://dx.doi.org/10.1016/j.mad.2006.11.023
- Wolf MJ, Amrein H, Izatt JA, Choma MA, Reedy MC, Rockman HA. Drosophila as a model for the identification of genes causing adult human heart disease. Proc Natl Acad Sci U S A 2006; 103:1394-9; PMID: 16432241; http://dx.doi.org/10.1073/pnas.0507359103
- Li SH, Schilling G, Young WS 3rd, Li XJ, Margolis RL, Stine OC, Wagster MV, Abbott MH, Franz ML, Ranen NG, et al. Huntington's disease gene (IT15) is widely expressed in human and rat tissues. Neuron 1993; 11:985-93; PMID:8240819; http://dx.doi.org/10.1016/0896-6273(93)90127-D
- Waters CW, Varuzhanyan G, Talmadge RJ, Voss AA. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc Natl Acad Sci U S A 2013; 110:9160-5; PMID: 23671115; http://dx.doi.org/10.1073/pnas.1220068110
- Strand AD, Aragaki AK, Shaw D, Bird T, Holton J, Turner C, Tapscott SJ, Tabrizi SJ, Schapira AH, Kooperberg C, et al. Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Hum Mol Genet 2005; 14:1863-76; PMID:15888475; http://dx.doi.org/10.1093/hmg/ddi192
- Han Z, Yi P, Li X, Olson EN. Hand, an evolutionarily conserved bHLH transcription factor required for Drosophila cardiogenesis and hematopoiesis. Development 2006; 133:1175-82; PMID:16467358; http://dx.doi.org/10.1242/dev.02285
- Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, Robbins J. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A 2004; 101:10132-6; PMID:15220483; http://dx.doi.org/10.1073/pnas.0401900101
- Mihm MJ, Amann DM, Schanbacher BL, Altschuld RA, Bauer JA, Hoyt KR. Cardiac dysfunction in the R6/2 mouse model of Huntington's disease. Neurobiol Dis 2007; 25:297-308; PMID:17126554; http://dx.doi.org/10.1016/j.nbd.2006.09.016
- Tashiro E, Zako T, Muto H, Itoo Y, Sörgjerd K, Terada N, Abe A, Miyazawa M, Kitamura A, Kitaura H, et al. Prefoldin protects neuronal cells from polyglutamine toxicity by preventing aggregation formation. J Biol Chem 2013; 288:19958-72; PMID:23720755; http://dx.doi.org/10.1074/jbc.M113.477984
- Albers DS, Beal MF. Mitochondrial dysfunction and oxidative stress in aging and neurodegenerative disease. J Neural Transm Suppl 2000; 59:133-54; PMID: 10961426
- Cheng B, Gong H, Xiao H, Petersen RB, Zheng L, Huang K. Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochim Biophys Acta 2013; 1830:4860-71; PMID:23820032
- Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G, Lim D, Malorni W, Davies KJ, Carafoli E, Scorrano L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington's disease to apoptotic stimuli. EMBO Mol Med 2010; 2:490-503; PMID:21069748; http://dx.doi.org/10.1002/emmm.201000102
- Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation 2008; 117:2743-51; PMID:18490523; http://dx.doi.org/10.1161/CIRCULATIONAHA.107.750232
- Kobayashi, Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 2000; 275:8772-8; PMID:10722721; http://dx.doi.org/10.1074/jbc.275.12.8772
- Kuo Y, Ren S, Lao U, Edgar BA, Wang T. Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis 2013; 4:e833; PMID:24091676; http://dx.doi.org/10.1038/cddis.2013.351
- Kiriazis H, Jennings NL, Davern P, Lambert G, Su Y, Pang T, Du X, La Greca L, Head GA, Hannan AJ, et al. Neurocardiac dysregulation and neurogenic arrhythmias in a transgenic mouse model of Huntington's disease. J Physiol 2012; 590:5845-60; PMID:22890713; http://dx.doi.org/10.1113/jphysiol.2012.238113
- Groenendyk J, Sreenivasaiah PK, Kim do H, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res 2010; 107:1185-97; PMID:21071716; http://dx.doi.org/10.1161/CIRCRESAHA.110.227033
- Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, Hewett T, Robbins J. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res 2001; 89:84-91; PMID:11440982; http://dx.doi.org/10.1161/hh1301.092688
- Rodolfo C, Ciccosanti F, Giacomo GD, Piacentini M, Fimia GM. Proteomic analysis of mitochondrial dysfunction in neurodegenerative diseases. Expert Rev Proteomics 2010; 7:519-42; PMID:20653508; http://dx.doi.org/10.1586/epr.10.43
- Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum Mol Genet 2011; 20:1438-55; PMID:21257639; http://dx.doi.org/10.1093/hmg/ddr024
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552:335-44; PMID:14561818; http://dx.doi.org/10.1113/jphysiol.2003.049478
- Barth E, Stammler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 1992; 24:669-81; PMID:1404407; http://dx.doi.org/10.1016/0022-2828(92)93381-S
- Zang Q, Maass DL, White J, Horton JW. Cardiac mitochondrial damage and loss of ROS defense after burn injury: the beneficial effects of antioxidant therapy. J Appl Physiol 1985; 102:103-12 (2007); PMID:16931562; http://dx.doi.org/10.1152/japplphysiol.00359.2006
- Maloyan A, Sanbe A, Osinska H, Westfall M, Robinson D, Imahashi K, Murphy E, Robbins J. Mitochondrial dysfunction and apoptosis underlie the pathogenic process in alpha-B-crystallin desmin-related cardiomyopathy. Circulation 2005; 112:3451-61; PMID: 16316967; http://dx.doi.org/10.1161/CIRCULATIONAHA.105.572552
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet 1998; 18:159-63; PMID:9462746; http://dx.doi.org/10.1038/ng0298-159
- Maloyan A, Osinska H, Lammerding J, Lee RT, Cingolani OH, Kass DA, Lorenz JN, Robbins J, et al. Biochemical and mechanical dysfunction in a mouse model of desmin-related myopathy. Circ Res 2009; 104:1021-8; PMID:19299643; http://dx.doi.org/10.1161/CIRCRESAHA.108.193516
- Goswami A, Dikshit P, Mishra A, Mulherkar S, Nukina N, Jana NR. Oxidative stress promotes mutant huntingtin aggregation and mutant huntingtin-dependent cell death by mimicking proteasomal malfunction. Biochem Biophys Res Commun 2006; 342:184-90; PMID:16472774; http://dx.doi.org/10.1016/j.bbrc.2006.01.136
- Taverne YJ, Bogers AJ, Duncker DJ, Merkus D. Reactive oxygen species and the cardiovascular system. Oxid Med Cell Longev 2013; 2013:862423; PMID:23738043; http://dx.doi.org/10.1155/2013/862423
- Wohlgemuth SL, Crawford BD, Pilgrim DB. The myosin co-chaperone UNC-45 is required for skeletal and cardiac muscle function in zebrafish. Dev Biol 2007; 303:483-92; PMID:17189627; http://dx.doi.org/10.1016/j.ydbio.2006.11.027
- Lee CF, Melkani GC, Yu Q, Suggs JA, Kronert WA, Suzuki Y, Hipolito L, Price MG, Epstein HF, Bernstein SI. Drosophila UNC-45 accumulates in embryonic blastoderm and in muscles, and is essential for muscle myosin stability. J Cell Sci 2011; 124:699-705; PMID:21285246; http://dx.doi.org/10.1242/jcs.078964
- Gazda L, Pokrzywa W, Hellerschmied D, Löwe T, Forné I, Mueller-Planitz F, Hoppe T, Clausen T. The myosin chaperone UNC-45 is organized in tandem modules to support myofilament formation in C. elegans. Cell 2013; 152:183-95; PMID:23332754; http://dx.doi.org/10.1016/j.cell.2012.12.025
- Melkani GC, Bodmer R, Ocorr K, Bernstein SI. The UNC-45 chaperone is critical for establishing myosin-based myofibrillar organization and cardiac contractility in the Drosophila heart model. PLoS One 2011; 6:e22579; PMID:21799905; http://dx.doi.org/10.1371/journal.pone.0022579
- Srikakulam R, Liu L, Winkelmann DA. Unc45b forms a cytosolic complex with Hsp90 and targets the unfolded myosin motor domain. PLoS One 2008; 3:e2137; PMID:18478096; http://dx.doi.org/10.1371/journal.pone.0002137
- Barral JM, Hutagalung AH, Brinker A, Hartl FU, Epstein HF. Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science 2002; 295:669-71; PMID:11809970; http://dx.doi.org/10.1126/science.1066648
- Pattison JS, Robbins J. Protein misfolding and cardiac disease: establishing cause and effect. Autophagy 2008; 4:821-3; PMID:18612262; http://dx.doi.org/10.4161/auto.6502
- Kim H-D, Kim CH, Rah B-J, Chung H-I, Shim T-S. Quantitative study on the relation between structural and functional properties of the hearts from three different mammals. The Anatomical Record 1994; 238:199-206; PMID:8154606; http://dx.doi.org/10.1002/ar.1092380206