
Abstract
Huntington's disease (HD) is a rare genetic neurodegenerative disorder for which there is currently no cure. Early hyperkinetic motor symptoms are consistent with reduced activity of indirect pathway striatal projection neurons (iSPNs) responsible for suppression of unwanted actions. Our recent work suggests that one of the factors contributing to this deficit is impaired brain-derived neurotrophic factor (BDNF) signaling that regulates the strength of iSPN excitatory synapses. Specifically, we found that BDNF-dependent corticostriatal synaptic long-term potentiation (LTP) was lost in iSPNs from 2 genetic models of HD, just as they began to robustly manifest motor symptoms. This deficit was not attributable to problems in BDNF production, delivery or receptor binding. Rather, the plasticity deficit stemmed from enhanced signaling through p75 neurotrophin receptors (p75NTRs) and the phosphatase and tensin homolog (PTEN), leading to antagonism of intracellular TrkBR cascades and LTP. This study suggests HD therapeutics should target p75NTR signaling, not TrkBR.
Abbreviations
| BDNF | = | brain-derived neurotrophic factor |
| dSPN | = | direct pathway spiny projection neuron |
| HD | = | Huntington's disease |
| iSPN | = | indirect pathway spiny projection neuron |
| LTP | = | long term potentiation |
| mHtt | = | mutant huntingtin |
| NGF | = | nerve growth factor |
| NMDA | = | N-methyl D-aspartate |
| NR2B | = | N-methyl D-aspartate receptor subtype 2B |
| NT3 | = | neurotrophin 3 |
| NT4 | = | neurotrophin 4 |
| p75NTR | = | p75 neurotrophin receptor |
| PI3K | = | phosphoinositide-3 kinase |
| PKB | = | protein kinase B |
| PTEN | = | phosphatase and tensin homolog deleted on chromosome 10 |
| TrkBR | = | tyrosine kinase receptor B |
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by a polyglutamine expansion in the coding region of the huntingtin gene.Citation1 Among the brain regions most conspicuously affected are the striatum and cerebral cortex.Citation2 In fact, progressive loss of enkephalin expression, a marker of indirect pathway striatal projection neurons (iSPNs), and ultimately striatal neuronal atrophy, are among the most well described HD pathologies.Citation3 According to the classical model of basal ganglia motor control,Citation4,5 impaired iSPN function should compromise the ability to suppress unwanted movements, consistent with the choreic motor symptoms associated with early to mid-stage HD.Citation2,6
In recent years, the impairment of iSPNs has been attributed to diminished trophic support of the striatum by the cerebral cortex.Citation7 Specifically, the delivery of cortically made brain-derived neurotrophic factor (BDNF) to the striatum has been posited to be responsible for iSPN atrophy. Despite the wide acceptance of this model, there are key aspects of it that have not been tested. In particular, it has never been shown that BDNF release by cortical axons and binding to postsynaptic TrkBRs is down-regulated in HD models at the point in time when motor symptoms begin to emerge. To address this need, we developed a novel synaptic plasticity protocol that requires engagement of TrkBR signaling cascades by BDNF. In wild-type iSPNs, LTP induction at axospinous corticostriatal synapses requires co-activation of TrkBRs, N-methyl-D-aspartate (NMDA) receptors, and A2a adenosine receptors. In conjunction with patch clamp recording and 2 photon laser scanning microscopy, 2 photon uncaging of glutamate or optogenetic activation of cortical terminals can be used to monitor LTP induction on a spine-by-spine basis. This type of LTP is only seen at spines that have cortical synapses, consistent with the cortex being the predominant source of striatal BDNF.Citation8 The ability to support this TrkBR-dependent synaptic plasticity was progressively lost in iSPNs in brain slices from BACHD and Q175 mouse modes of HD. By the time HD mice were 6 months of age – an age when the motor symptoms are very evident – LTP in iSPNs was completely lost but still normal in neighboring direct pathway SPNs (dSPNs).
Why was BDNF/TrkBR-dependent synaptic plasticity lost in HD mice? One explanation would be that BDNF production and delivery to iSPNs (but not dSPNs) is impaired. As difficult as this is to envision given the overlap in the cortical projections to these 2 cell types,Citation9,10 it is possible. However, using qPCR primers recognizing 6 regions spanning the BDNF gene, normalized to a panel of at least 6 reference genes, we found no evidence of altered cortical BDNF expression in 6 month-old HD mice; nor did we find altered TrkBR expression in either dSPNs or iSPNs in these models. Furthermore, there was no deficit in cortical or striatal BDNF protein at this age and activity-dependent phosphorylation of striatal TrkBRs was unperturbed. Thus, diminished BDNF production, delivery or activation of TrkBRs cannot be the culprit.
Why did our results ostensibly differ from those of previous reports?Citation7,11,12 One possibility is that the procedure used to calibrate estimates of mRNA abundance using quantitative PCR in previous studies yielded spurious results. Nearly all studies reporting changes in BDNF were normalized to a single variable reference gene, either GAPDH or β-actin; both transcripts can be affected by a variety of factors which are undesirable for a ‘housekeeping’ gene. We avoided this situation by using a weighted average of at least 6 mRNAs.Citation13 Another possibility is that the level of mutant huntingtin (mHtt) expression is critical to phenotype; there is no doubt for example that high levels of mHtt can disrupt axonal trafficking of BDNF.Citation14 The level of expression needed to have a measurable impact might not be reached in the heterozygous BACHD and Q175 models used in our work. Indeed, in homozygous Q175 mice, cortical expression of BDNF was reduced at 6 months of age. Lastly, the stage in the evolution of the disease might be critical to phenotype. Our work focused on ages when symptoms were becoming clear. But as the disease evolves, other regions and mechanisms might come into play. It is important to remember that the cerebral cortex and the basal ganglia are part of an interdependent neural network. Nevertheless, from the therapeutic standpoint, the earliest events in a pathophysiological cascade should be the best targets.
Although BDNF expression and delivery to the striatum were not altered in HD mice, TrkBR signaling was, as determined by phosphorylation of its downstream target, protein kinase B (PKB, also known as Akt). Surprisingly however, signaling elements downstream of TrkBRs appeared to be intact. Rather, the inability of TrkBR activation to induce LTP was due to attenuated signaling through an immediate player in the TrkBR cascade: phosphoinositide-3 kinase (PI3K). This deficit was traced back to p75NTR. While overall p75NTR mRNA and protein expression did not appear to be altered, expression of its downstream target PTEN was increased. Thus, p75NTR signaling was amplified in iSPNs from HD mice (). Indeed, the capacity to support LTP was completely restored in BACHD iSPNs by inhibiting p75NTR and PTEN activity.
Figure 1. Diagram of TrkBR and p75NTR signaling in iSPNs in normal and HD conditions.
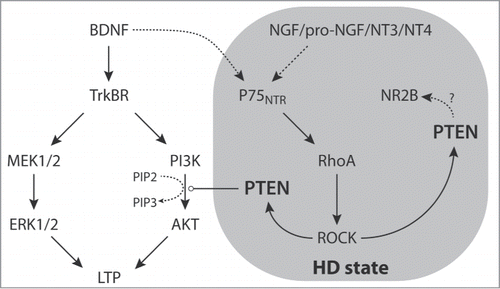
p75NTRs can be activated by a number of ligands, including nerve growth factor (NGF), pro-NGF, neurotrophin 3 (NT3), neurotrophin 4 (NT4), and even BDNF itself, albeit less effectively than TrkBRs.Citation15,16 This raises the possibility that, in HD, BDNF is stepping on both the accelerator (TrkBRs) and the brake (p75NTRs) at the same time. This is consistent with other work showing that the balance between TrkBR and p75NTR signaling is altered in HD and that this imbalance can lead to synapse-specific adaptations.Citation17 If this is the case, compounds that are agonists at TrkBRs, but not p75NTRs, could be effective therapeutics.Citation18,19 The other possibility is that p75NTR signaling is being driven in HD by one of the other agonists at this receptor.
Regardless of the activation mechanism of p75NTRs, their signaling is amplified by increased PTEN expression in symptomatic HD iSPNs. It is interesting to speculate about other potential consequences elevated PTEN activity may have. A well-described synaptic pathology in HD mouse models is increased insertion of N-methyl D- aspartate receptor subtype 2B (NR2B)-containing extrasynaptic NMDA receptors.Citation20 Besides shunting PI3K signaling by dephosphorylating PtdIns(3,4,5)P3 into PtdIns(4,5)P2, PTEN physically interacts with NR2B containing NMDARs, enhancing extrasynaptic NMDAR function.Citation21 If such an interaction is at play in iSPNs, PTEN may represent another means of enhancing extrasynaptic NMDARs in HD, and the neurotoxic cascades extrasynaptic NR2B receptors are associated with.
Though pathological p75NTR signaling in HD iSPNs seems to be a consequence of elevated PTEN expression, it is a poor therapeutic target because of its role as a tumor suppressor.Citation22 Interestingly, a recent epidemiological study showed that the incidence of cancer is lower in HD patients,Citation23 a phenomenon that is consistent with elevated PTEN expression. p75NTR is a much better therapeutic target as it is developmentally downregulated in most parts of the brain, suggesting it is dispensable. Although we don't know of a selective p75NTR antagonist, conditional knockouts of p75NTR in adult mice are feasible, making their role, and their viability as a therapeutic target, testable.
References
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington“s disease chromosomes. Cell 1993; 72:971-83; PMID:8458085; http://dx.doi.org/10.1016/0092-8674(93)90585-E
- Raymond LA, Andre VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function. Neuroscience 2011; 198:252-73; PMID:21907762; http://dx.doi.org/10.1016/j.neuroscience.2011.08.052
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 1988; 85:5733-7; PMID:2456581; http://dx.doi.org/10.1073/pnas.85.15.5733
- Alexander GE, Crutcher MD. Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 1990; 13:266-71; PMID:1695401; http://dx.doi.org/10.1016/0166-2236(90)90107-L
- Gerfen CR, Surmeier DJ. Modulation of Striatal Projection Systems by Dopamine. Annu Rev Neurosci 2011; 34:441-66; PMID:21469956; http://dx.doi.org/10.1146/annurev-neuro-061010-113641
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev 2010; 90:905-81; PMID:20664076; http://dx.doi.org/10.1152/physrev.00041.2009
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol 2007; 81:294-330; PMID:17379385; http://dx.doi.org/10.1016/j.pneurobio.2007.01.003
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997; 389:856-60; PMID:9349818; http://dx.doi.org/10.1038/39885
- Kress GJ, Yamawaki N, Wokosin DL, Wickersham IR, Shepherd GMG, Surmeier DJ. Convergent cortical innervation of striatal projection neurons. Nat Neurosci 2013; 16:665-7; PMID:23666180; http://dx.doi.org/10.1038/nn.3397
- Wall NR, La Parra De M, Callaway EM, Kreitzer AC. Differential Innervation of Direct- and Indirect-Pathway Striatal Projection Neurons. Neuron 2013; PMID:23810541
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 2001; 293:493-8; PMID:11408619; http://dx.doi.org/10.1126/science.1059581
- Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu X-H, Tao J, Yamazaki I, Li S-H, Sun YE, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci 2008; 28:6182-95; PMID:18550760; http://dx.doi.org/10.1523/JNEUROSCI.0857-08.2008
- Pfister C, Tatabiga MS, Roser F. Selection of suitable reference genes for quantitative real-time polymerase chain reaction in human meningiomas and arachnoidea. BMC Res Notes 2011; 4:275; PMID:21806841; http://dx.doi.org/10.1186/1756-0500-4-275
- Gauthier LR, Charrin BC, Borrell-Pagès M, Dompierre JP, Rangone H, Cordelières FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004; 118:127-38; PMID:15242649; http://dx.doi.org/10.1016/j.cell.2004.06.018
- Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci 1995; 18:223-53; PMID:7605062; http://dx.doi.org/10.1146/annurev.ne.18.030195.001255
- Dechant G, Barde Y-A. The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci 2002; 5:1131-6; PMID:12404007; http://dx.doi.org/10.1038/nn1102-1131
- Brito V, Puigdellívol M, Giralt A, del Toro D, Alberch J, Ginés S. Imbalance of p75(NTR)/TrkB protein expression in Huntington's disease: implication for neuroprotective therapies. Cell Death Dis 2013; 4:e595; PMID:23598407; http://dx.doi.org/10.1038/cddis.2013.116
- Todd D, Gowers I, Dowler SJ, Wall MD, McAllister G, Fischer DF, Dijkstra S, Fratantoni SA, van de Bospoort R, Veenman-Koepke J, et al. A Monoclonal Antibody TrkB Receptor Agonist as a Potential Therapeutic for Huntington's Disease. PLoS ONE 2014; 9:e87923; PMID:24503862; http://dx.doi.org/10.1371/journal.pone.0087923
- Simmons DA, Belichenko NP, Yang T, Condon C, Monbureau M, Shamloo M, Jing D, Massa SM, Longo FM. A Small Molecule TrkB Ligand Reduces Motor Impairment and Neuropathology in R6/2 and BACHD Mouse Models of Huntington's Disease. J of Neurosci 2013; 33:18712-27; PMID:24285878; http://dx.doi.org/10.1523/JNEUROSCI.1310-13.2013
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RWY, Vasuta OC, Graham RK, Hayden MR, et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron 2010; 65:178-90; PMID:20152125; http://dx.doi.org/10.1016/j.neuron.2010.01.008
- Ning K, Pei L, Liao M, Liu B, Zhang Y, Jiang W, Mielke JG, Li L, Chen Y, El-Hayek YH, et al. Dual neuroprotective signaling mediated by downregulating two distinct phosphatase activities of PTEN. J Neurosci 2004; 24:4052-60; PMID:15102920; http://dx.doi.org/10.1523/JNEUROSCI.5449-03.2004
- Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function: the long and the short of it. Trends Biochem Sci 2014; 39:183-90; PMID: 24656806; http://dx.doi.org/10.1016/j.tibs.2014.02.006
- MD DJJ, MD PKS, MD PJS. Cancer incidence in patients with polyglutamine diseases: a population-based study in Sweden. Lancet Oncol 2012; 13:642-8; PMID:22503213; http://dx.doi.org/10.1016/S1470-2045(12)70132-8