
Abstract
The PI3K/AKT/mTOR signaling is important for cell proliferation, survival, and metabolism. Hyperactivation of this pathway is one of the most common signaling abnormalities observed in cancer and a substantial effort has recently been made to develop molecules targeting this signaling cascade. However, it is becoming evident that PI3K inhibitors used as single agents do not elicit dramatic or durable responses. Given the numerous mechanisms mediating intrinsic and acquired resistance to these agents, hypothesis-based combinatorial strategies are probably needed to fully exploit their antitumor activity. In the first part of this review, we briefly dissect the PI3K/AKT/mTOR axis and list the most advanced compounds targeting different nodes of this cascade. The second part focuses on what we believe to be the most promising rationale-based therapeutic combinations with PI3K/AKT/mTOR inhibitors in solid tumors, with special emphasis on breast cancer.
Keywords:
Introduction
Recent genomic sequencing efforts have allowed investigators to decipher the molecular portraits of different types of cancer. This has led to the identification of several actionable gene alterations and paved the way toward personalized medicine.
Hyperactivation of the phosphoinositide-3-kinase (PI3K) signaling cascade, either by overexpression of upstream receptor tyrosine kinases or deregulation of several elements of the pathway, is one of the most frequent pathway alterations in cancer.
The PI3K pathway is highly conserved among species and has been proposed to play a key role in the regulation of multiple cellular events, including growth, proliferation, cell cycle progression, and survival. Direct pharmacological inhibition of the PI3K signaling is, therefore, an attractive clinical strategy and a number of PI3K pathway inhibitors are currently under clinical development. However, unlike other genomic alterations, there is no consensus regarding PIK3CA mutations as cancer drivers. As a consequence, inhibition of the PI3K pathway alone does not usually translate to dramatic antitumor activity. This could potentially be explained as follows:
The therapeutic window is narrow because normal cells also require PI3K signaling for survival. As a consequence, severe adverse effects (e.g., hyperglycemia) often manifest before full inhibition of the target in tumor cells.
Inhibition of the PI3K pathway leads to activation of compensatory pathways that can limit the sensitivity to these agents.Citation1-4
In this article, we review the main inhibitors of the PI3K/AKT/mTOR axis, focusing on those furthest along in the clinical pipeline, and propose hypothesis-based combinations that could potentially improve their antitumor activity.
PI3K: Structure and Biochemistry
PI3K enzymes are classified into 3 classes (Class I to III) according to their structural and biochemical properties. Because of their role in human cancer, in this review we will discuss only the Class I PI3Ks.
Class I PI3Ks are characterized by the presence of a catalytic subunit (p110) that forms a heterodimeric complex with the regulatory subunit (p85). The catalytic subunit is encoded by 1 of 4 genes, PIK3CA (p110α), PIK3CB (p110β), PIK3CD (p110δ), and PIK3CG (p110γ). All of these isoforms use phosphatidylinositol-(4,5)-biphosphate as a substrate. Whereas p110α and p110β are expressed in virtually all cell types, p110δ and p110γ are specifically enriched in leukocytes (reviewed inCitation5 andCitation6).
The catalytic subunit p110 contains a C-terminal kinase domain that is responsible for the lipid enzymatic activity, a helical domain with a yet unknown function, a C2 domain that has been suggested to bind the cellular membrane, a Ras-binding domain (RBD), and an N-terminal adaptor-binding domain (ABD) that is responsible for the interaction with the regulatory subunitCitation7 ().
Figure 1. Structure and biochemistry of PI3K. (A) The domains of PI3K catalytic (p110α) and regulatory (p85α) subunits are represented. The connecting arrow indicates the domains involved in the interaction between these 2 subunits. BD (Binding Domain), RBD (Ras-BD), SH3 (SRC Homology 3), PR (Proline-Rich), BH (BcR Homology), SH2 (SRC Homology 2), iSH2 (inter-SH2). (B) Phosphorylation of the phosphatidylinositol 4,5-bisphosphate inositol ring at position 3-OH (red). Arrows indicate the direction catalyzed by PI3K or the phosphatase PTEN.
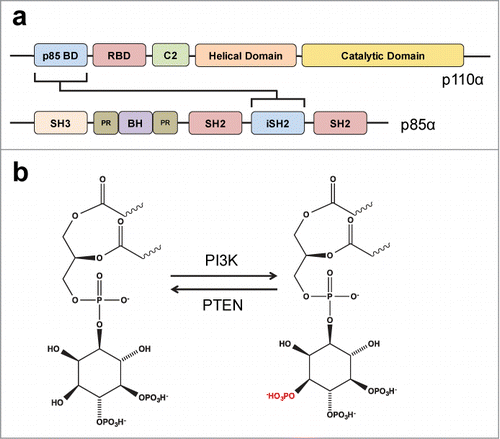
Class I enzymes are further divided into 2 groups, A and B, based on the regulatory subunit that they interact with. The Class IA isoforms, p110α, β, and δ, are associated with p85α or p85β subunits, whereas the Class IB isoform p110γ interacts with p101 or p87.Citation8
Alternative splicing of PIK3R1, the gene encoding p85, gives rise to 4 different isoforms with p85α being the most common. p85α contains 2 Src-homology 2 (SH2) domains that bind to phosphorylated tyrosine residues of receptor tyrosine kinases (RTKs) and an intermediate SH2 (iSH2) domain that interacts with and inhibits the p110 subunitCitation7 (). Upon RTK stimulation, SH2 domains recognize the phosphorylated sites and release the inhibitory effect of the iSH2 domain on the catalytic subunit. This allows the p110 subunit to exert its enzymatic activity using the abundant phosphatidylinositol-(4,5)-biphosphate (PIP2) present in the inner plasma membrane as a substrate. Biochemically, PI3K phosphorylates the hydroxyl (-OH) group at position 3 of the inositol ring in inositol phospholipids.Citation9 This reaction generates the second messenger phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) that triggers downstream signaling of the pathway. This reaction is reversed by both phosphatase and tensin homolog (PTEN, 3-phosphatase)Citation10 and SH2 domain-containing inositol 5’-phosphatase (SHIP, 5-phosphatase),Citation11 enzymes that dephosphorylate the inositol ring and convert PIP3 to PIP2 or phosphatidylinositol-(3,4)-biphosphate, respectively. Both PIP3 and phosphatidylinositol-(3,4)-biphosphate have been shown to regulate the function of multiple downstream effectors by recruiting them into the plasma membraneCitation12,Citation13 ().
Downstream of PI3K
AKT, a Ser/Thr kinase that belongs to the AGC family of the human kinome, is one of the most studied kinases and is considered a key output of the PI3K pathway because of its large number of substrates. There are 3 isoforms of AKT (1–3), encoded by the respective genes AKT1, AKT2, and AKT3.Citation14 Although it remained elusive for several years, there is now some consensus about the mechanism of activation of this kinase. Upon generation of the second messenger PIP3 in the membrane by PI3K, both phosphoinositide-dependent kinase-1 (PDK1, a constitutively active kinase) and AKT are recruited to the membrane through their pleckstrin homology domains (PHs), which recognize PIP3 with high affinity. The proximity of these kinases allows PDK1 to phosphorylate AKT at residue T308 of the activation loop (T-loop).Citation15,Citation16 Subsequently, AKT is phosphorylated at residue S473 of the hydrophobic motif by the rapamycin-insensitive mTOR complex 2 (mTORC2).Citation17 This phosphorylation is considered necessary to fully activate the kinase activity of AKT (); however, several reports suggest that phosphorylation at the T-loop may be sufficient to engage AKT activity on selected substrates.Citation18,Citation19
Figure 2. The PI3K/AKT pathway. Main transduction signals of the PI3K pathway. Blue phospholipids indicate PIP3 second messenger. Arrows indicate activation while bars represent inhibition. Red bars show pharmacologic targets of the pathway. Images were taken from Servier Medical Art.
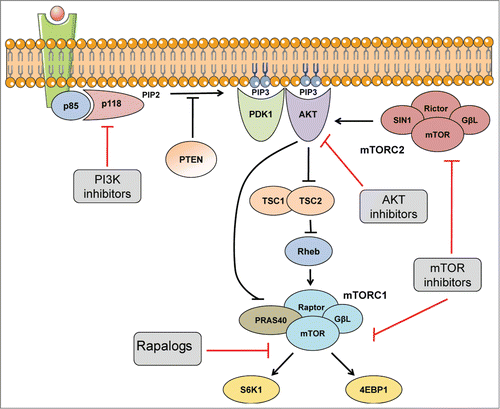
Activated AKT in turn phosphorylates several substrates involved in apoptosis and cell cycle regulation. For example, AKT is able to phosphorylate and inhibit Bad, a member of the Bcl-2 family,Citation20 and caspase 9,Citation21 2 main regulators of the mitochondrial apoptotic pathway. It also inhibits p21CIP,122 and p27KIP,Citation23 proteins that are directly involved in the inhibition of cell cycle progression. Moreover, AKT can also prevent the nuclear localization of the forkhead transcription factors FOXO1, 3, 4, and 6,Citation24 which are involved in the transcriptional regulation of several genes including the proapoptotic genes CD95L, BCL2L11 (BIM), BBC3 (PUMA), and genes encoding the cell cycle inhibitors CDKN2A (p21CIP) and CDKN2B (p27KIP). In addition to these effectors, AKT can phosphorylate PRAS40 and TSC2, 2 negative regulators of mTORC1 activity ()Citation25,Citation26 thus linking the PI3K/AKT pathway with the mTORC1 pathway.
The importance of PDK1 and AKT in mediating PI3K downstream signaling has been exploited as a suitable node for pharmacological inhibition. Although PDK1 inhibitors are being used in a preclinical setting (for an excellent review see referenceCitation27), many AKT inhibitors are currently under clinical development.
AKT inhibitors are highly specific and potent, and consequently on-target adverse effects such as severe hyperglycemia can limit their use. One of the first inhibitors reported to inhibit AKT is the phospholipid analog perifosine, which inhibits the PH domain of AKT.Citation28 Despite promising clinical activity in early studies, perifosine has failed to increase overall survival in metastatic CRC when administered in combination with capecitabine.Citation29 Other inhibitors of AKT are being investigated, including the allosteric inhibitor MK2206Citation30 and the catalytic inhibitors GDC-0068,Citation31 AZD5363,Citation32 and GSK690693.Citation33 These orally bioavailable agents are undergoing different phases of clinical development (Phase I-III) in multiple solid and hematological malignancies. Of note, most of these trials are being tested in combination with other agents such as CDK, RTK, ER, and MEK inhibitors.
mTOR
mTOR, a 289-kDa serine-threonine protein kinase, is at the core of the PI3K/AKT pathway and acts as a master integrator of multiple upstream signals.Citation34 mTOR senses and responds to environmental cues such as nutrient availability, stress, and mitogens to regulate protein synthesis through a highly orchestrated and complex mechanism.
mTOR was originally identified in the early 1990sCitation35 and was later shown to form 2 discrete complexes with distinct roles in the control of cell growth.Citation36
mTOR complex 1 (mTORC1), which contains mTOR, Deptor, Raptor, mLST8, and PRAS40, has been considered a master regulator of cell growth and metabolism that signals to 4E-binding protein (4EBP1) and 40S ribosomal protein S6 kinase (S6K), both of which are important in the physiological control of translation.Citation37 mTORC1 promotes protein synthesis by phosphorylating 4EBP1, which in turn prevents 4EBP1 from binding to the eukaryotic initiation factor 4E (eIF4E), enabling eIF4E to initiate cap-dependent translation. On the other hand, activation of S6K1 by mTORC1 leads to an increase in mRNA biogenesis and cap-dependent translation.Citation37 mTORC1 has also been shown to activate RNA PolI transcription and thus rRNA synthesis through a process involving the protein phosphatase 2A (PP2A) and the transcription initiation factor IA (TIFIA).Citation38
The second complex (mTORC2), which is rapamycin-insensitive,Citation36,Citation39 consists of at least 6 different proteins. The complex core is formed by mTOR, Deptor, and mLST8 but, instead of Raptor, it contains 3 other proteins: rapamycin-insensitive companion of mTOR (Rictor), mammalian stress-activated protein kinase interacting protein (mSIN1), and protein observed with Rictor-1 (Protor-1).Citation40 It is known that mTORC2 activity responds to growth factors, but how mTORC2 is regulated upstream and the exact molecular function of most of its interacting proteins remain elusive. Functionally, mTORC2 regulates organization of the actin cytoskeleton through the phosphorylation of protein kinase Cα and also activates AKT through phosphorylation at S473.Citation17,Citation41
Although other kinases such as DNA-PK and ATM have been suggested to phosphorylate AKT at S473,Citation42,Citation43 studies in Rictor, Sin1, and LST8 knockout mice have shown that an intact mTORC2 complex is required for maximal phosphorylation and activation of AKT in mouse embryonic fibroblasts.Citation19,Citation44
Genomic Alterations in the Pathway: PIK3CA and PTEN
PIK3CA, the gene encoding p110α, is the most commonly mutated gene among the components of the PI3K pathway.Citation45 Although there are 3 main mutation hotspots within the helical (E545 and E542) and kinase (H1047) domains,Citation46 other mutations can be found across the whole gene. The E542K and E545K mutations located in the helical domain induce an important electrostatic switch in these residues by reversing the ionic charge. This modifies the interaction of the N-terminal SH2 domain of the regulatory subunit p85 with the helical and kinase domain of p110α, comparable to binding at phospho-Y residues.Citation47,Citation48 In the case of the H1047R mutation within the kinase domain, it has been established that this alteration increases the kinase activity by inducing a new orientation of the C-terminal loop toward the plasma membrane, in which the active site has better access to its substrate PIP2.Citation49,Citation50
Other mutations include the R39 (R39C and R39H) and the R88Q mutations that occur in the ABD domain. These mutations are reported to disrupt the interaction between the ABD and kinase domains of p110α, thus augmenting its activity. In addition, 2 mutations within the C2 domain, N345L and E453Q, have been proposed to alter the interaction between this domain and the iSH2 domain of the regulatory subunit, again increasing the kinase activity of p110α.Citation47
Another well-studied alteration that can lead to hyperactivation of the pathway is loss of function of the tumor suppressor PTEN.Citation51 Low PTEN phosphatase activity results in increased levels of PIP3 and consequent activation of downstream PI3K effectors such as AKT and mTORC1 (). PTEN is a 55-kDa enzyme containing, among other structural motifs, a phosphatase domain that controls the catalytic activity of the enzyme and a C2 domain that is responsible for lipid binding.Citation10 Somatic mutations of PTEN are found throughout the entire gene, although there is a slightly higher frequency at the R130 residue. Somatic PTEN mutations are relatively frequent in endometrial carcinoma and glioblastoma, and PTEN copy number loss is common in prostate, breast, and ovary cancer and glioblastoma.Citation52,Citation53
PI3K inhibitors
There are currently approximately 200 clinical trials testing the activity, safety, and efficacy of different PI3K inhibitors (www.clinicaltrial.gov). The first inhibitors, isolated almost 2 decades ago, were not specific for PI3K and commonly inhibited other kinases, especially phosphatidylinositol 3-kinase-related kinases (PIKK) such as mTOR and DNA-PK, which contain structurally similar active sites.Citation54
Because PI3K uses ATP as a phosphate donor to phosphorylate the substrate PIP2, most inhibitors work as competitive ATP mimetics.Citation55 The first known inhibitor of PI3K, wortmannin, is a furanosteroid derivative isolated from the fungus Penicillium wortmanii, which acts as a potent and irreversible pan-PI3K inhibitor (IC50 ≈ 4.2 nM).Citation56 Unfortunately, this inhibitor also targets other members of the PIKK family and MAPK. Similarly, viridin, a natural steroid derived from Trichoderma viridae, has been shown to inhibit PI3K in a potent and irreversible manner (IC50 ≈5 nM).Citation57 Using the structure of the furanosteroid wortmannin as a pharmacophore, multiple drugs have been developed including PX-866,Citation58 which is currently in Phase I/II trials. Another natural product that was shown to inhibit PI3K is the flavonoid quercetin,Citation55 which was used as a model in the synthesis of LY294002. Although LY294002 has been used for many years as a tool in the field of PI3K research, it actually has relatively low potency toward PI3K (IC50 ≈ 1.4 μM) and co-inhibits mTOR and DNA-PK.Citation59 LY294002 has also been used as a model to create the β-specific inhibitors TGX-115, TGX-126, TGX-221, and TGX-286, which were initially designed as antithrombotic agents because of the involvement of PI3Kβ in hemostasis.Citation60 Another molecule, phenylquinazoline, inhibits PI3Kα with an IC50 of 1.3 μM, and is also a common pharmacophore used as a backbone to create novel PI3K inhibitors such as PI-103, a potent (IC50 ≈ 3.6 nM) but not very selective inhibitor of PI3Kα and β.Citation61,Citation62 The most clinically promising drug derived from a morpholino quinazoline is probably the thienopyrimidine GDC-0941, which is considered a pan PI3K inhibitor although it preferentially targets PI3Kα (IC50 ≈ 3 nM).Citation63 This molecule has shown good pharmacokinetic properties in animals and humans and is currently in Phase II trials for metastatic estrogen receptor (ER)-positive breast cancer and non-small cell lung cancer.
Another agent derived from the heterocyclic compound quinoxalin (benzopyrazine) is XL147,Citation64 which is being tested in combination with other therapies including chemotherapy and targeted agents such as erlotinib and trastuzumab.
NVP-BKM120 is a pan-PI3K inhibitor that is currently in clinical development in multiple combinations. It was originally derived from the 2-morpholino-6-aminopyridyl-pyrimidine scaffold, based on the structure of PI3Kγ.Citation65 This dimorpholino pyrimidine derivative shows approximate equipotency toward all of the Class IA isoforms of PI3K and is currently in multiple Phase II and III trials for breast, prostate, endometrial, glioblastoma, and lung cancer, among others.
Although these early inhibitors have shown some promise in the treatment of cancer, since the elucidation of the PI3K crystal structure in 2007Citation7 the design of PI3K inhibitors has improved, yielding compounds of higher potency and selectivity. As a result, isoform-specific PI3K inhibitors that could potentially increase efficacy while limiting off-target toxicity have been developed and are now entering the clinic.Citation8
The first reported PI3Kα specific inhibitor was the 2-aminothiazole-derivative NVP-BYL719 that inhibits the α isoform with an IC50 ≈ 5 nM and shows similar potency toward the mutant versions H1047R and E545K.Citation66 In fact, both preclinical and clinical studies have shown that PIK3CA mutations are associated with response to PI3Kα inhibition with agents like NVP-BYL719,Citation67 -Citation70 and this compound is currently being tested in the clinic for breast, head and neck, and gastrointestinal tumors. Similarly, INK1117 (also known as MLN-1117) has been reported as a specific PI3Kα inhibitor and is currently undergoing Phase I trials.Citation71 Recently, a new molecule, GDC-0032, has been reported to be a PI3Kβ sparing inhibitor. This agent is a highly potent and selective inhibitor of PI3Kα and PI3Kδ (IC50 ≈ 0.3 nM and 0.12 nM, respectively), but shows 3-fold greater potency toward the oncogenic mutants H1047R and E545K. GDC-0032 is currently in Phase I clinical trials and has shown promising results.Citation69 Finally, another notable drug is the PI3Kδ inhibitor CAL-101, a phenylquinazolin derivative that selectively inhibits the delta isoform of PI3K with an IC50 ≈ 70 nM, compared to the other isoforms (IC50 >1 μM).Citation72 Of note, this compound was the first PI3K inhibitor to be approved by the US Food and Drug Administration (FDA) and will be available for the treatment of chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), and follicular lymphoma (FL). In , we summarize the combinations with isoform-specific PI3K inhibitors that are currently under clinical investigation.
Table 1. Combinations with pan-PI3K inhibitors under clinical development
Table 2. Combinations with isoform-specific PI3K inhibitors under clinical development
mTORC1 inhibitors: rapalogs
Much of our knowledge about mTORC1 comes from studies that elucidated the mechanism of action of rapamycin, an allosteric inhibitor that directly binds to mTORC1 and inhibits downstream phosphorylation of its substrates. Specifically, rapamycin binds to the immunophilin FKB12 to generate a highly specific complex that binds to the FKBP12-rapamycin-binding (FKB) domain of mTOR.Citation37
In contrast to its effects on mTORC1, rapamycin cannot interact with mTORC2.Citation39 For this reason, mTORC1 is known as a rapamycin-sensitive complex and mTORC2 as a rapamycin-insensitive complex. This paradigm, however, may not be entirely accurate as it has been reported that prolonged treatment of U937 lymphoma cells and PC3 prostate cancer cells with rapamycin also inhibits mTORC2 activity.Citation73 This decrease in mTORC2 activity appears to be sufficient to inhibit AKT signaling.Citation34 Thus, rapamycin is considered to be a universal inhibitor of mTORC1 and seems to be a cell-type specific inhibitor of mTORC2 in a rapamycin time- and dose-dependent manner.
The physicochemical properties of rapamycin are not optimal for pharmacological development and as a consequence several analogs termed “rapalogs” have been developed. These analogs, such as temsirolimus, everolimus, and ridaforolimus, exhibit higher solubility and better pharmacokinetic properties than rapamycin.Citation75 Temsirolimus has also been used in combination with the aromatase inhibitor letrozole in patients with metastatic breast cancer in a Phase III study although the combination of drugs did not show a benefit over letrozole alone.Citation76
Everolimus achieved regulatory approval for use in pancreatic neuroendocrine tumors based on a Phase III trial in which it prolonged progression-free survival when compared to best supportive care.Citation77 Another Phase III study demonstrated superiority for everolimus over placebo in patients with metastatic renal cell carcinoma (mRCC) with progression after vascular endothelial growth factor receptor-tyrosine kinase inhibitor therapy, leading to approval for this indication.Citation78 Everolimus has also been approved by the FDA for use in combination with anti-estrogen therapy in hormone-receptor positive HER2-negative breast cancer.Citation79 At the present time, ridaforolimus, formerly known as deferolimus, has no approved indications. All 3 agents are currently under investigation across many clinical trials.
Despite a plethora of preclinical data on rapamycin and its analogs, these molecules have not shown universal antitumor activity in clinical trials. This may be in part due to an important limitation of the rapalogs: the paradoxical increase in the AKT activity resulting from feedback loops triggered by mTORC1 inhibition. S6K (one of the key substrates of mTOR) inhibits IRS1, the adaptor protein linking the IGF-1 receptor and PI3K. This effect leads to a reduction of input into the PI3K pathway coming from the stimulation of the insulin/IGF-1 receptors. The inhibition of mTORC1 releases the S6K-IRS1-PI3K feedback inhibitory loop and results in increased AKT activity.Citation1 Thus, additional targeting of other key members of the pathway may be required to overcome the effects of this feedback for maximal efficacy in certain cellular contexts.
mTOR kinase inhibitors
The finding that mTORC2 has a direct role in the activation of AKT, combined with the limited clinical activity of rapalogs in many tumors, has led to the development of ATP-competitive inhibitors of mTOR kinase that potently inhibit both mTORC1 and mTORC2 complexes. Interestingly, these compounds have been shown to inhibit mTORC1 more potently than the rapalogs.Citation80,Citation81 For example, the mTOR kinase inhibitor AZD8055 inhibits 4EBP1 phosphorylation more effectively than rapamycin and also effectively inhibits mTORC2 and AKT S473 phosphorylation.Citation82 However, the inhibition of AKT signaling seems to be transient, as inhibition by AZD8055 causes activation of RTKs, which in turn induce PI3K signaling and reactivate AKT activity and signaling.Citation82 Combined inhibition of mTOR kinase and RTKs fully abolishes AKT signaling resulting in tumor regression.Citation83
INK128 is another mTORC1/2 inhibitor for which in vitro and in vivo data have demonstrated successful inhibition of mTORC1 (S6K and 4EBP1) and mTORC2 (AKT at S473).Citation84 Interestingly, this agent has also shown marked activity in cell lines that are resistant to rapamycin and pan-PI3K inhibitors.Citation71 A Phase I trial testing the activity of this molecule is ongoing in patients with solid tumors. Other compounds that aim to inhibit both mTOR complexes and potentially have a more profound antitumor activity than rapalogs are AZD2014, CC-223, and OSI-027.
Combinations of PI3K Inhibitors with RTK Inhibitors
HER2-targeted therapies have produced clinical improvements in patient survival, both in the adjuvant and metastatic setting. However, despite initial responses the emergence of resistance occurs in the vast majority of patients with metastatic disease. Activation of the PI3K pathway, either by loss of PTEN or by the presence of PIK3CA mutations, is perhaps the most widely accepted mechanism of resistance to anti-HER2 therapy.Citation84-Citation88 Activating mutations in PIK3CA occur in approximately 25–30% of HER2-amplified breast cancers (TCGA) and may be important for response to HER2-targeted therapies. For example, introduction of a PIK3CA H1047R activating mutation in HER2-driven mammary tumors in MMTV/neu transgenic mice accelerates tumor onset and progression, and generates resistance to anti-HER2 therapy.Citation89 This evidence paved the way to investigate the efficacy of therapeutic strategies combining anti-HER2 agents with PI3K inhibitors. In preclinical models, this strategy proved to be synergistic in cells or tumors resistant to anti-HER2 therapy.Citation84,Citation86,Citation90–92 In a clinical setting, trastuzumab in combination with everolimus showed promising antitumor activity in heavily pretreated patients who had progressed to trastuzumab-based therapy.Citation93-Citation95 Further, in a recent study testing the effect of the PI3K inhibitor NVP-BKM120 in a similar population, researchers reported clinical responses in patients with tumors possessing an activated PI3K pathway.Citation96 Additional trials testing other PI3K inhibitors in combination with anti-HER2 agents are currently ongoing.
Interestingly, a recent study highlighted the need to design triple inhibition strategies to effectively delay and/or avoid tumor relapse, especially in patients with large tumor burdens.Citation97 Although toxicity remains a limiting factor when translating these combinatorial therapeutic strategies to patients, concomitant treatment of HER2-driven cancers with synergistic combinations targeting the PI3K/AKT pathway and additional driver events might render cancer cells vulnerable to efficient eradication and lead to curative regimes for patients.
Treatment with PI3K/AKT inhibitors leads to upregulation/activation of RTKs such as EGFR, HER3, and IGFR1 that, in turn, can limit the antitumor effects of these therapies by increasing PI3K signaling or triggering the activation of other compensatory pathways.Citation2,Citation4,82,98 This provides the rationale to test combinations of PI3K inhibitors with agents that block the activity of RTKs. Due to space limitations we are unable to list the large (and increasing) number of preclinical studies conducted in this regard. Likewise, many clinical trials testing the efficacy of PI3K inhibitors and anti-RTK agents (including EGFR, HER2, HER3, FGFR, IGFR, and CD20) have been launched. Details are available at www.clinicaltrial.gov and are summarized in and .
Combinations of PI3K Inhibitors with Endocrine Therapy
Up to 75% of breast tumors are hormone receptor (HR)-positive, expressing estrogen receptor (ER), progesterone receptor (PR), or both. These nuclear receptors are both targets and predictors of response to anti-estrogen therapy such as selective estrogen receptor modulators (SERMs), aromatase inhibitors (AIs), or selective estrogen receptor degraders (SERDs). Despite the fact that HR-positive breast tumors generally have a favorable prognosis and effective therapies exist, up to one-third of women diagnosed with this type of cancer will relapse after 5 years of adjuvant therapy with tamoxifenCitation99 and up to 20% of women treated with adjuvant AIs will undergo recurrence 10 years after the initial diagnosis.Citation100 Several mechanisms of resistance to hormone therapy have been proposedCitation101; however, a common theme emerging from many of these studies is the importance of the activation of growth factor receptors in endocrine resistance.
Given that PI3K is the most frequently altered pathway in ER+ breast tumors, deregulation of the PI3K pathway through activation of upstream receptor tyrosine kinases or mutations in the downstream effectors as a potential mediator of endocrine resistance has been one of the main research areas. For example, it has been described that upregulation of ERBB2 (which signals mainly through the PI3K pathway) mediates resistance to anti-estrogen therapyCitation102,Citation103 thus establishing the rationale for the combination of anti-estrogen and anti-HER2 therapy.Citation104 However, constitutive activation of PI3K does not seem to confer this resistant phenotype.Citation105-Citation107 Despite the lack of a correlation between response to anti-estrogen therapy and PI3K alterations, there is preclinical evidence pointing to the benefit of a combination of inhibitors of ER and the PI3K pathway.Citation108,Citation109 In fact, we have reported that PI3K pathway inhibition upregulates ER transcriptional activity and increases cell survival dependency on ER, which translates into a synergism between PI3K inhibitors and ER degraders with increased tumor control.Citation110 Furthermore, the Bolero-2 trial highlighted that inhibiting both mTOR and ER signaling with the combination of everolimus and exemestane has a superior effect in terms of progression-free survival.Citation79 Also, early-phase clinical trials introducing novel drugs that directly target PI3K have demonstrated that combination strategies have an excellent response rate in selected patients.Citation111,Citation112
The PI3K and androgen signaling axes are also critical drivers of the pathogenesis of prostate cancer and the 2 most commonly activated pathways in this disease. Mutation or deletion of PTEN and amplification of PIK3CA are 2 of the most common genetic aberrations observed in prostate cancer genomics studies,Citation113,Citation114 and virtually all untreated primary prostate cancers initially respond to androgen deprivation therapy. Further, it has been demonstrated that these 2 signaling cascades reciprocally regulate one another, such that inhibition of one pathway induces activation of the other.Citation115 Thus, there is great interest in targeting these pathways simultaneously in prostate cancer. PI3K pathway inhibitors and androgen-targeted therapies have shown promising activity in preclinical studies,Citation115,Citation116 and several early-phase clinical trials are ongoing.
Dual PI3K/mTOR blockade
An interest in targeting multiple important components in the PI3K/AKT/mTOR pathway has encouraged the development of dual inhibitors that might suppress the growth, proliferation, and survival of cancer cells. The activity of these compounds differs from that of rapalogs or dual mTOR inhibitors, as they are able to block both mTOR complexes in addition to the PI3K isoforms in an ATP-competitive manner. Several mTOR/PI3K dual inhibitors, such as SF1126, NVP-BEZ235, XL765, GDC-0980, PF-04691502, PKI-587, GSK2126458, and PWT3359, have shown activity in preclinical models and are in early-stage clinical trials. For example, NVP-BEZ235 has been reported to inhibit tumor growth in preclinical models of prostate, breast, pancreatic, and renal cancers, multiple myeloma, and sarcomas.Citation117 Other studies have shown that breast cancer cell lines with HER2 amplification and/or PIK3CA mutations are highly sensitive to NVP-BEZ235.Citation90 These and other findings have allowed NVP-BEZ235 to enter Phase I/II clinical trials in patients with advanced solid tumors, including breast cancer.Citation117 XL765 is a PI3K/mTOR inhibitor that showed pathway inhibition and a reduction in cell proliferation in a Phase I dose-escalation study of patients with solid tumors.Citation118 Another dual PI3K/mTOR inhibitor, GDC-0980, has demonstrated broad preclinical activity in breast, ovarian, lung, and prostate cancer models.Citation119 It has also been shown to be active against tumor cells bearing mutations in PI3K, PTEN, or KRASCitation120 and is currently in Phase I clinical development.
Recent work has demonstrated that mTORC1 inhibition is required for sensitivity to p110α inhibitors in PIK3CA-mutant breast cancer.Citation67 This study demonstrated that breast cancer cell lines and patient tumors that are resistant to the PI3K inhibitor NVP-BYL719 have active mTORC1 signaling. Thus, sustained mTORC1 activation may limit the effects of NVP-BYL719 on tumor growth. This hypothesis was confirmed by the demonstration that adding everolimus to NVP-BYL719 resulted in reversal of resistance both in vitro and in vivo.Citation67
Additionally, work from our laboratory showed that progressive loss of PTEN leads to clinical resistance to the PI3Kα inhibitor NVP-BYL719. Specifically, loss of PTEN in NVP-BYL719-sensitive cell lines and in patients that progress to this agent leads to resistance to PI3Kα inhibition. Since PTEN-deficient preclinical models mostly rely on the p110β subunit of the PI3K holoenzyme, concomitant inhibition of p110α and p110β isoforms of PI3K resensitizes the cells to NVP-BYL719.Citation121
Combination of PI3K and MEK inhibitors
The RAS/RAF/MEK/ERK pathway is frequently dysregulated in human cancer and alterations in this pathway have been found to lead to tumorigenesis and resistance to several therapies. The PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways are known to interact at multiple levels. Examples of this crosstalk include direct activation of PI3K by RAS via its interaction with the catalytic subunit of PI3KCitation122 and phosphorylation of TSC2 by ERK that suppresses TSC2 function and promotes activation of mTORC1.Citation123 Moreover, the 90-kDa ribosomal S6 kinase (RSK), which lies downstream of the RAS-ERK pathway, was found to phosphorylate TSC2 at Ser1798 and inactivate its tumor suppressor function leading to mTORC1 signaling and increased translation.Citation124 RSK was also reported to phosphorylate Raptor, providing another link between RSK and the mTOR pathway.Citation125
The interaction between the PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways may explain the modest single-agent activity of agents targeting one of these pathways. Thus, to achieve optimal anticancer effects, dual blockade of these pathways may be necessary. Indeed, studies of dual pharmacological inhibition of these pathways have shown that combination treatment increases antiproliferative activity. For example, the dual pan-PI3K and mTOR inhibitor NVP-BEZ235 in combination with a MEK inhibitor show successful synergy in shrinking KRAS-mutant lung cancers or EGFR-mutant lung cancers.Citation126,Citation127 Furthermore, targeting of PI3K/mTOR in combination with MEK inhibitors was shown to be necessary to effectively inhibit growth of NRAS-mutant melanoma cells in vitro and in vivo.Citation128 Several Phase I and II clinical trials examining PI3K inhibitors plus MEK inhibitors in patients with advanced solid tumors are currently undergoing or completeCitation129 (). Examples include studies of a MEK inhibitor (MEK162) plus a PI3K inhibitor (BKM120 or BYL719) in adult patients with advanced solid tumors and studies of a MEK inhibitor (GSK1120212) in combination with a AKT inhibitor (GSK2110183) in patients with solid tumors and multiple myeloma (www.clinicaltrials.gov) ().
Combination of PI3K inhibitors with DNA damaging agents
Aberrations in DNA damage repair (DDR) and cell cycle checkpoint proteins are present in nearly all cancers, subverting standard cell cycle arrest mechanisms and allowing genomic aberrations to persist unrepaired. Furthermore, DNA damaging therapies such as chemotherapy and radiotherapy are cornerstones of cancer therapy and are administered to the vast majority of cancer patients at some point during their disease course. The DNA damage signaling and PI3K signaling networks intersect at multiple nodes,Citation130 and several preclinicalCitation131 -Citation134 and clinicalCitation135 studies have supported the notion that DNA damage activates PI3K/AKT pathway signaling. Further, activation of PI3K signaling has been implicated in the promotion of chemo- and radioresistance.Citation136 -Citation138 Therefore, it has been hypothesized that combining therapies targeting the PI3K-AKT signaling axis with standard DNA damaging therapies in PI3K-driven tumors may induce synergistic cancer cell killing.
Perhaps the most widely studied PI3K signaling protein involved in the DDR is AKT. Numerous studies have demonstrated that AKT is phosphorylated on S473 within minutes of DNA damage.Citation42,Citation139,140 Furthermore, AKT S473, but not AKT T308, accumulates at foci of DNA damage.Citation43,Citation139 However, the exact mechanism of AKT activation following DNA damage and its specific role in the DNA repair cascade is controversial and may vary depending on the cell's genetic and epigenetic profile. Several studies have demonstrated that the phosphorylation of AKT on S473 is dependent on DNA-PK.Citation43,Citation140,141 On the other hand, other studies have suggested that induction of AKT S473 following radiation is independent of both DNA-PK and PI3K signaling, but instead is dependent on the activation of ATM.Citation139 Another possible mechanism of AKT activation is provided by multiple studies demonstrating that radiation can induce activation of all 4 ErbB receptors, and therefore downstream PI3K signaling, in a ligand-independent manner.Citation142,Citation143
Given the evidence that AKT is activated by DNA damage, several studies have investigated the specific role of AKT in the DDR. In one study, AKT1 was found to form a complex with DNA-PKcs following DNA damage, resulting in enhanced activation and autophosphorylation of DNA-PKcs at S2056, thus allowing DNA-PKcs to dissociate from sites of DNA damage and other critical downstream DNA damage repair proteins to access the DNA damage sites.Citation144 Additionally, AKT phosphorylates multiple downstream targets, and several of these have been shown to participate in the DDR. For example, one study demonstrated that following DNA damage AKT2 mediates phosphorylation and inhibition of GSK-3β, an enzyme important for phosphorylating and activating the p53 inhibitor MDM2.Citation140 Further, AKT2 knockdown (but not PI3K knockdown) markedly reduced p53 accumulation in response to radiation.Citation140 AKT-mediated inhibition of GSK-3β has been also shown to upregulate expression of MRE11, a component of that MRN complex that is one of the earliest proteins recruited to sites of DNA double-strand breaks.Citation145
However, the relationship between AKT and DNA damage signaling is complex, and many other targets that are inhibited by AKT actually enhance DNA damage checkpoints and repair. For example, FOXO3A has been shown to directly associate with ATM, p53, and Chk2, enhancing the activation of these proteins,Citation146,Citation147 and to regulate both the G1Citation148 and G2/MCitation149 DNA damage checkpoints. Thus, FOXO3A inactivation through AKT phosphorylation might be expected to impair the DDR. Further, AKT has also been shown to inhibit Chk1Citation150 and Wee1Citation151 through inhibitory phosphorylations on S280 and S642, respectively, thus promoting progression through the cell cycle. It is unclear, therefore, whether hyperactivation of AKT signaling can abrogate the ability of cells to arrest at the G2/M checkpoint and repair DNA damage prior to mitosis.
Despite these complexities, most preclinical evidence suggests that the net effect of AKT loss is increased sensitivity to DNA damaging agents. For example, AKT−/− mice exhibit decreased survival following total body irradiation compared with their AKT-proficient littermates.Citation152 Similarly, AKT−/− thymocytes are more susceptible to irradiation-induced apoptosis than wild-type thymocytes.Citation152 Several studies have also shown that AKT1 knockdown sensitizes cancer cells to radiotherapy.Citation134,Citation139,145,153
Given the extensive evidence that PI3K signaling is induced following DNA damage, for many years there has been interest in using PI3K inhibitors to sensitize to DNA damaging therapies. In fact, multiple studies have demonstrated that pharmacologic inhibitors of the PI3K pathway are capable of sensitizing cancer cells to DNA damaging therapies in vitro and in vivo.Citation83,Citation134,144,Citation154–156 However, the vast majority of these studies relied heavily on PI3K pathway inhibitors like LY294002, wortmannin, PI103, and especially NVP-BEZ235, which have significant activity against DNA-PK and lower activity against ATM and ATR.Citation157,Citation158 In fact, in a study from Kevan Shokat's group, inhibitors of PI3K were found to frequently inhibit DNA-PK, and p110α was found to cluster with DNA-PK in target space by structural activity relationship analysis despite sharing limiting sequence homology.Citation158 Additionally, 5 of the 6 chemotypes that inhibited p110α with an IC50 lower than 5 μM also potently inhibited DNA-PK in this study. Given that DNA-PK plays a central role in the DDR, particularly in non-homologous end joining, caution should be used when drawing conclusions from studies that rely predominantly on these inhibitors to demonstrate enhanced efficacy from PI3K pathway inhibition and DNA damaging agents compared to either alone. Studies using more selective p110α inhibitors with limited activity against DNA-PK, such as NVP-BYL719 and GDC-0032, will help define the relationship between PI3K inhibition and sensitivity to DNA damaging agents.
Two recent studies have demonstrated a direct role for PI3K in homologous recombination via regulation of BRCA1 expression,Citation159 thereby inhibiting recruitment of Rad51 to foci of DNA damageCitation160 in patients with triple-negative breast cancer. These findings have led to a Phase I clinical trial combining the pan-PI3K inhibitor NVP-BKM120 and the PARP inhibitor olaparib in patients with triple-negative breast cancer or high-grade serous ovarian cancer.Citation161 However, in addition to the role of p110α in the DNA damage response, activation of p110β by DNA damage seems to play a critical role in recognition of DNA double-strand breaks and recruitment of NBS1 to sites of DNA damage.Citation162 Loss of p110β leads to impaired recruitment of downstream DNA damage repair mediators such as ATM, RAD17, γ-H2AX, and 53BP.Citation162
In contrast to AKT, several reports have shown that mTORC1 signaling is actually downregulated as a result of DNA damage.Citation163-Citation165 Oxidative stress was shown to downregulate mTORC1 signaling by activating ATM, leading to a phosphorylation chain resulting in sequential activation of LKB1, AMPK, and finally TSC2.Citation165 Given that DNA damage is also a major regulator of ATM signaling, the mechanism of DNA damage-induced downregulation of mTORC1 signaling may be similar. This suggests that both AKT and ATM signaling may converge on TSC2, and that mTORC1 output may depend on the relative contribution of both signals.
In summary, the relationship between DNA damage repair and PI3K signaling is complex and bidirectional, with each network influencing the other in multiple ways. However, the bulk of evidence supports the notion that DNA damage enhances AKT activation, thereby leading to activation of a signaling network that generally promotes survival following DNA damaging therapies like radiation and chemotherapy. Although the use of selective PI3K inhibitors to enhance the efficacy of DNA damaging agents represents a promising approach for some cancers, it is imperative to define molecular predictors for which patients are most likely to benefit from this approach in order to maximize the chance that this approach will be successful in the clinic.
Concluding Remarks
Although the precise biology and the mechanisms of oncogenicity of the PI3K/AKT pathway are still under investigation, this signaling cascade is an attractive target for cancer therapy because of the high frequency of aberrations seen across a wide spectrum of cancers. Nevertheless, PI3K inhibitors have limited efficacy when used as a single agent, in part due to the activation of compensatory pathways. Thus, combinatorial strategies seem to be the best approach in order to obtain the maximal therapeutic advantage from these agents.
In this review, we have discussed what we believe are some of the most promising strategies for combinatorial therapy with PI3K inhibitors. However, it should be mentioned that this field is continuously evolving with the development and testing of new combinations that we have not specifically discussed. Such approaches include cosuppression of cyclin-dependent kinases,Citation166,Citation167 Smoothened,Citation168 BCL-2,Citation169 histone deacetylase,Citation170 Janus kinase,Citation171 heat shock protein 90,Citation172 and many others.
Ultimately, given the intra- and inter-tumoral molecular heterogeneity underlying cancer, a single specific combinatorial strategy is unlikely to be uniformly effective for all patients. Thus, understanding which tumors are most prone to respond to specific combinations of PI3K inhibitors with other targeted agents will be critical to their successful implementation in the clinic.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Due to space limitation, the authors apologize for their inability to cite everyone who has contributed to this field of inquiry.
Funding
JC holds a fellowship from the Terri Brodeur Foundation; ME is an International Sephardic Education Foundation postdoctoral fellow; AB holds a Translational Research Fellowship from the Spanish Society of Medical Oncology (SEOM).
References
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500-8; PMID:16452206; http://dx.doi.org/10.1158/0008-5472.CAN-05-2925
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011; 19:58-71; PMID:21215704; http://dx.doi.org/10.1016/j.ccr.2010.10.031
- Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011; 30:2547-57; PMID:21278786; http://dx.doi.org/10.1038/onc.2010.626
- Tao JJ, Castel P, Radosevic-Robin N, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal 2014; 7:ra29
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9:550-62; PMID:19629070; http://dx.doi.org/10.1038/nrc2664
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002; 2:489-501; PMID:12094235; http://dx.doi.org/10.1038/nrc839
- Huang CH, Mandelker D, Schmidt-Kittler O, Samuels Y, Velculescu VE, Kinzler KW, Vogelstein B, Gabelli SB, Amzel LM. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science 2007; 318:1744-8; PMID:18079394; http://dx.doi.org/10.1126/science.1150799
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 2010; 11:329-41; PMID:20379207; http://dx.doi.org/10.1038/nrm2882
- Whitman M, Downes CP, Keeler M, Keller T, Cantley L. Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 1988; 332:644-6; PMID:2833705; http://dx.doi.org/10.1038/332644a0
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999; 99:323-34; PMID:10555148; http://dx.doi.org/10.1016/S0092-8674(00)81663-3
- Lioubin MN, Algate PA, Tsai S, Carlberg K, Aebersold A, Rohrschneider LR. p150Ship, a signal transduction molecule with inositol polyphosphate-5-phosphatase activity. Genes Dev 1996; 10:1084-95; PMID:8654924; http://dx.doi.org/10.1101/gad.10.9.1084
- Klippel A, Kavanaugh WM, Pot D, Williams LT. A specific product of phosphatidylinositol 3-kinase directly activates the protein kinase Akt through its pleckstrin homology domain. Mol Cell Biol 1997; 17:338-44; PMID:8972214
- Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 1997; 387:673-6; PMID:9192891; http://dx.doi.org/10.1038/42648
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261-74; PMID:17604717; http://dx.doi.org/10.1016/j.cell.2007.06.009
- Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 1997; 7:776-89; PMID:9368760; http://dx.doi.org/10.1016/S0960-9822(06)00336-8
- Alessi DR. Discovery of PDK1, one of the missing links in insulin signal transduction. Colworth Medal Lecture. Biochem Soc Trans 2001; 29:1-14; PMID:11356119; http://dx.doi.org/10.1042/BST0290001
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098-101; PMID:15718470; http://dx.doi.org/10.1126/science.1106148
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006; 127:125-37; PMID:16962653; http://dx.doi.org/10.1016/j.cell.2006.08.033
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 2006; 11:859-71; PMID:17141160; http://dx.doi.org/10.1016/j.devcel.2006.10.007
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91:231-41; PMID:9346240; http://dx.doi.org/10.1016/S0092-8674(00)80405-5
- Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998; 282:1318-21; PMID:9812896; http://dx.doi.org/10.1126/science.282.5392.1318
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 2001; 3:245-52; PMID:11231573; http://dx.doi.org/10.1038/35060032
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 2002; 8:1145-52; PMID:12244301; http://dx.doi.org/10.1038/nm759
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96:857-68; PMID:10102273; http://dx.doi.org/10.1016/S0092-8674(00)80595-4
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9:316-23; PMID:17277771; http://dx.doi.org/10.1038/ncb1547
- Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 2002; 10:151-62; PMID:12150915; http://dx.doi.org/10.1016/S1097-2765(02)00568-3
- Medina JR. Selective 3-phosphoinositide-dependent kinase 1 (PDK1) inhibitors: dissecting the function and pharmacology of PDK1. J Med Chem 2013; 56:2726-37; PMID:23448267; http://dx.doi.org/10.1021/jm4000227
- Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther 2003; 2:1093-103; PMID:14617782
- Bendell JC, Ervin TJ, Senzer NN, et al. Results of the X-PECT study: A phase III randomized double-blind, placebo-controlled study of perifosine plus capecitabine (P-CAP) versus placebo plus capecitabine (CAP) in patients (pts) with refractory metastatic colorectal cancer (mCRC). J Clin Oncol 2012; 30; abstract 3501
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010; 9:1956-67; PMID:20571069; http://dx.doi.org/10.1158/1535-7163.MCT-09-1012
- Lin J, Sampath D, Nannini MA, Lee BB, Degtyarev M, Oeh J, Savage H, Guan Z, Hong R, Kassees R, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res 2013; 19:1760-72; PMID:23287563; http://dx.doi.org/10.1158/1078-0432.CCR-12-3072
- Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, Li J, Gao B, Ji Q, Maynard J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 2012; 11:873-87; PMID:22294718; http://dx.doi.org/10.1158/1535-7163.MCT-11-0824-T
- Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ, McConnell RT, Gilmer TM, Zhang SY, Robell K, et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res 2008; 68:2366-74; PMID:18381444; http://dx.doi.org/10.1158/0008-5472.CAN-07-5783
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006; 6:729-34; PMID:16915295; http://dx.doi.org/10.1038/nrc1974
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991; 253:905-9; PMID:1715094; http://dx.doi.org/10.1126/science.1715094
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell 2002; 10:457-68; PMID:12408816; http://dx.doi.org/10.1016/S1097-2765(02)00636-6
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009; 10:307-18; PMID:19339977; http://dx.doi.org/10.1038/nrm2672
- Mayer C, Zhao J, Yuan X, Grummt I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev 2004; 18:423-34; PMID:15004009; http://dx.doi.org/10.1101/gad.285504
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6:1122-8; PMID:15467718; http://dx.doi.org/10.1038/ncb1183
- Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci 2009; 122:3589-94; PMID:19812304; http://dx.doi.org/10.1242/jcs.051011
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 2004; 14:1296-302; PMID:15268862; http://dx.doi.org/10.1016/j.cub.2004.06.054
- Viniegra JG, Martinez N, Modirassari P, Hernández Losa J, Parada Cobo C, Sánchez-Arévalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Ramón y Cajal S, Rojas JM, et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem 2005; 280:4029-36; PMID:15546863; http://dx.doi.org/10.1074/jbc.M410344200
- Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 2008; 30:203-13; PMID:18439899; http://dx.doi.org/10.1016/j.molcel.2008.02.024
- Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell 2006; 11:583-9; PMID:16962829; http://dx.doi.org/10.1016/j.devcel.2006.08.013
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304:554; PMID:15016963; http://dx.doi.org/10.1126/science.1096502
- Vogt PK, Kang S, Elsliger MA, Gymnopoulos M. Cancer-specific mutations in phosphatidylinositol 3-kinase. Trends Biochem Sci 2007; 32:342-9; PMID:17561399; http://dx.doi.org/10.1016/j.tibs.2007.05.005
- Gabelli SB, Huang CH, Mandelker D, Schmidt-Kittler O, Vogelstein B, Amzel LM. Structural effects of oncogenic PI3Kalpha mutations. Curr Top Microbiol Immunol 2010; 347:43-53; PMID:20593314
- Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, Williams RL. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science 2007; 317:239-42; PMID:17626883; http://dx.doi.org/10.1126/science.1135394
- Mandelker D, Gabelli SB, Schmidt-Kittler O, Zhu J, Cheong I, Huang CH, Kinzler KW, Vogelstein B, Amzel LM. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A 2009; 106:16996-7001; PMID:19805105; http://dx.doi.org/10.1073/pnas.0908444106
- Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A 2008; 105:2652-7; PMID:18268322; http://dx.doi.org/10.1073/pnas.0712169105
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275:1943-7; PMID:9072974; http://dx.doi.org/10.1126/science.275.5308.1943
- Myers MP, Tonks NK. PTEN: sometimes taking it off can be better than putting it on. Am J Hum Genet 1997; 61:1234-8; PMID:9399917; http://dx.doi.org/10.1086/301659
- Furnari FB, Lin H, Huang HS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A 1997; 94:12479-84; PMID:9356475; http://dx.doi.org/10.1073/pnas.94.23.12479
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014; 13:140-56; PMID:24481312; http://dx.doi.org/10.1038/nrd4204
- Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 2000; 6:909-19; PMID:11090628; http://dx.doi.org/10.1016/S1097-2765(05)00089-4
- Norman BH, Shih C, Toth JE, Ray JE, Dodge JA, Johnson DW, Rutherford PG, Schultz RM, Worzalla JF, Vlahos CJ. Studies on the mechanism of phosphatidylinositol 3-kinase inhibition by wortmannin and related analogs. J Med Chem 1996; 39:1106-11; PMID:8676346; http://dx.doi.org/10.1021/jm950619p
- Wipf P, Minion DJ, Halter RJ, Berggren MI, Ho CB, Chiang GG, Kirkpatrick L, Abraham R, Powis G. Synthesis and biological evaluation of synthetic viridins derived from C(20)-heteroalkylation of the steroidal PI-3-kinase inhibitor wortmannin. Org Biomol Chem 2004; 2:1911-20; PMID:15227545; http://dx.doi.org/10.1039/b405431h
- Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 2004; 3:763-72; PMID:15252137
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 1994; 269:5241-8; PMID:8106507
- Knight ZA, Chiang GG, Alaimo PJ, Kenski DM, Ho CB, Coan K, Abraham RT, Shokat KM. Isoform-specific phosphoinositide 3-kinase inhibitors from an arylmorpholine scaffold. Bioorg Med Chem 2004; 12:4749-59; PMID:15358300; http://dx.doi.org/10.1016/j.bmc.2004.06.022
- Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Okada M, Ohta M, Tsukamoto S, Parker P, Workman P, et al. Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem 2006; 14:6847-58; PMID:16837202; http://dx.doi.org/10.1016/j.bmc.2006.06.046
- Hayakawa M, Kaizawa H, Kawaguchi K, Ishikawa N, Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S, et al. Synthesis and biological evaluation of imidazo[1,2-a]pyridine derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem 2007; 15:403-12; PMID:17049248
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 2008; 51:5522-32; PMID:18754654; http://dx.doi.org/10.1021/jm800295d
- Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Braña I, Pandya SS, Scheffold C, Laird AD, Nguyen LT, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2014; 20:233-45; PMID:24166903; http://dx.doi.org/10.1158/1078-0432.CCR-13-1777
- Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther 2012; 11:317-28,; PMID:22188813; http://dx.doi.org/10.1158/1535-7163.MCT-11-0474
- Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett 2013; 23:3741-8; PMID:23726034; http://dx.doi.org/10.1016/j.bmcl.2013.05.007
- Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH, et al. mTORC1 Inhibition Is Required for Sensitivity to PI3K p110alpha Inhibitors in PIK3CA-Mutant Breast Cancer. Sci Transl Med 2013; 5:196ra99; PMID:23903756; http://dx.doi.org/10.1126/scitranslmed.3005747
- Juric D, Rodon J, Gonzalez-Angulo AM, et al. Abstract CT-01: BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study. Cancer Res 2012; 72: CT-01-
- Juric D, Krop I, Ramanathan RK, et al. Abstract LB-64: GDC-0032, a beta isoform-sparing PI3K inhibitor: Results of a first-in-human phase Ia dose escalation study. Cancer Res 2013; 73:LB-64-; http://dx.doi.org/10.1158/1538-7445.AM2013-3363
- Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther 2014; 13:1117-29; PMID:24608574; doi:10.1158/1535-7163
- Jessen K, Wang S, Kessler L, et al. INK128 is a potent and selective TORC1/2 inhibitor with broad oral antitumor activity. Mol Cancer Ther 2009; 8: Supplement 1
- Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011; 117:591-4; PMID:20959606; http://dx.doi.org/10.1182/blood-2010-03-275305
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22:159-68; PMID:16603397; http://dx.doi.org/10.1016/j.molcel.2006.03.029
- Hudes GR. mTOR as a target for therapy of renal cancer. Clin Adv Hematol Oncol 2007; 5:772-4; PMID:17998894
- Hess G. Temsirolimus for the treatment of mantle cell lymphoma. Expert Rev Hematol 2009; 2:631-40; PMID:21082954; http://dx.doi.org/10.1586/ehm.09.57
- Seto B. Rapamycin and mTOR: a serendipitous discovery and implications for breast cancer. Clin Transl Med 2012; 1:29; PMID:23369283; http://dx.doi.org/10.1186/2001-1326-1-29
- Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011; 364:514-23; PMID:21306238; http://dx.doi.org/10.1056/NEJMoa1009290
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma : final results and analysis of prognostic factors. Cancer 2010; 116:4256-65; PMID:20549832; http://dx.doi.org/10.1002/cncr.25219
- Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 2012; 366:520-9; PMID:22149876; http://dx.doi.org/10.1056/NEJMoa1109653
- Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009; 5:725-6; PMID:19395872; http://dx.doi.org/10.4161/auto.5.5.8504
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 2009; 284:8023-32; PMID:19150980; http://dx.doi.org/10.1074/jbc.M900301200
- Rodrik-Outmezguine VS, Chandarlapaty S, Pagano NC, Poulikakos PI, Scaltriti M, Moskatel E, Baselga J, Guichard S, Rosen N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov 2011; 1:248-59; PMID:22140653; http://dx.doi.org/10.1158/2159-8290.CD-11-0085
- Kao GD, Jiang Z, Fernandes AM, Gupta AK, Maity A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J Biol Chem 2007; 282:21206-12; PMID:17513297; http://dx.doi.org/10.1074/jbc.M703042200
- Garcia-Garcia C, Ibrahim YH, Serra V, Calvo MT, Guzmán M, Grueso J, Aura C, Pérez J, Jessen K, Liu Y, et al. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin Cancer Res 2012; 18:2603-12; PMID:22407832; http://dx.doi.org/10.1158/1078-0432.CCR-11-2750
- Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, Wang B, Shao Z, Wang Z, Hu X. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer 2011; 11:248; PMID:21676217; http://dx.doi.org/10.1186/1471-2407-11-248
- Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 2008; 68:9221-30; PMID:19010894; http://dx.doi.org/10.1158/0008-5472.CAN-08-1740
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007; 12:395-402; PMID:17936563; http://dx.doi.org/10.1016/j.ccr.2007.08.030
- Nagata Y, Lan KH, Zhou XY, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004; 6:117-127; PMID:15324695; http://dx.doi.org/10.1016/j.ccr.2004.06.022
- Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sánchez V, Sutton CR, Cheng H, Perou CM, Zhao JJ, et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci U S A 2013; 110:14372-7; PMID:23940356; http://dx.doi.org/10.1073/pnas.1303204110
- Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008; 68:8022-30; PMID:18829560; http://dx.doi.org/10.1158/0008-5472.CAN-08-1385
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009; 15:429-40; PMID:19411071; http://dx.doi.org/10.1016/j.ccr.2009.03.020
- Miller TW, Forbes JT, Shah C, Wyatt SK, Manning HC, Olivares MG, Sanchez V, Dugger TC, de Matos Granja N, Narasanna A, et al. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clin Cancer Res 2009; 15:7266-76; PMID:19934303; http://dx.doi.org/10.1158/1078-0432.CCR-09-1665
- Andre F, Campone M, O'Regan R, Manlius C, Massacesi C, Sahmoud T, Mukhopadhyay P, Soria JC, Naughton M, Hurvitz SA. Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol 2010; 28:5110-5; PMID:20975068; http://dx.doi.org/10.1200/JCO.2009.27.8549
- Jerusalem G, Fasolo A, Dieras V, Cardoso F, Bergh J, Vittori L, Zhang Y, Massacesi C, Sahmoud T, Gianni L. Phase I trial of oral mTOR inhibitor everolimus in combination with trastuzumab and vinorelbine in pre-treated patients with HER2-overexpressing metastatic breast cancer. Breast Cancer Res Treat 2011; 125:447-55; PMID:21107682; http://dx.doi.org/10.1007/s10549-010-1260-x
- Morrow PH, Wulf GM, Booser DJ, et al. Phase I/II trial of everolimus (RAD001) and trastuzumab in patients with trastuzumab-resistant, HER2-overexpressing breast cancer. J Clin Oncol 2010; 28: abstr 1014
- Saura C, Bendell J, Jerusalem G, Su S, Ru Q, De Buck S, Mills D, Ruquet S, Bosch A, Urruticoechea A, et al. Phase Ib study of Buparlisib plus Trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on Trastuzumab-based therapy. Clin Cancer Res 2014; 20:1935-45; PMID:24470511; http://dx.doi.org/10.1158/1078-0432.CCR-13-1070
- Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2013; 2:e00747
- Garrett JT, Sutton CR, Kurupi R, Bialucha CU, Ettenberg SA, Collins SD, Sheng Q, Wallweber J, Defazio-Eli L, Arteaga CL. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110alpha inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res 2013; 73:6013-23; PMID:23918797; http://dx.doi.org/10.1158/0008-5472.CAN-13-1191
- Early Breast Cancer Trialists' Collaborative G: Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet 2005; 365:1687-717; PMID:15894097; http://dx.doi.org/10.1016/S0140-6736(05)66544-0
- Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M, Forbes JF; ATAC/LATTE investigators. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol 2010; 11:1135-41; PMID:21087898; http://dx.doi.org/10.1016/S1470-2045(10)70257-6
- Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 2009; 9:631-43; PMID:19701242; http://dx.doi.org/10.1038/nrc2713
- Massarweh S, Osborne CK, Creighton CJ, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res 2008; 68:826-33; PMID:18245484; http://dx.doi.org/10.1158/0008-5472.CAN-07-2707
- Dowsett M, Harper-Wynne C, Boeddinghaus I, Salter J, Hills M, Dixon M, Ebbs S, Gui G, Sacks N, Smith I. HER-2 amplification impedes the antiproliferative effects of hormone therapy in estrogen receptor-positive primary breast cancer. Cancer Res 2001; 61:8452-8; PMID:11731427
- Kaufman B, Mackey JR, Clemens MR, et al. Trastuzumab plus anastrozole versus anastrozole alone for the treatment of postmenopausal women with human epidermal growth factor receptor 2-positive, hormone receptor-positive metastatic breast cancer: results from the randomized phase III TAnDEM study. J Clin Oncol 2009; 27:5529-37; PMID:19786670; http://dx.doi.org/10.1200/JCO.2008.20.6847
- Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, Gonzalez-Angulo AM, Pusztai L, Symmans WF, Bardelli A, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc Natl Acad Sci U S A 2010; 107:10208-13; PMID:20479250; http://dx.doi.org/10.1073/pnas.0907011107
- Beelen K, Opdam M, Severson TM, et al. PIK3CA mutations, phosphatase and tensin homolog, human epidermal growth factor receptor 2 and insulin-like growth factor 1 receptor and adjuvant tamoxifen resistance in postmenopausal breast cancer patients. Breast Cancer Res 2014; 16: R13
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012; 486:353-60; PMID:22722193
- Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, Jiang A, Smith RA, Maira SM, et al. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov 2011; 1:338-51; PMID:22049316; http://dx.doi.org/10.1158/2159-8290.CD-11-0101
- Fox EM, Kuba MG, Miller TW, et al. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res 2013; 15:R55
- Bosch A. EH, Tao J., Castel P., Ibrahim Y, Bergamaschi A., Prat A., Scaltriti M., Baselga J. PI3K pathway inhibitors sensitize to anti-estrogen therapy as a result of increased estrogen receptor levels and signaling. Eur J Cancer 2013; 49(suppl 3):abstract 38
- Mayer IA, Abramson VG, Isakoff SJ, Forero A, Balko JM, Kuba MG, Sanders ME, Yap JT, Van den Abbeele AD, Li Y, et al. Stand Up to Cancer Phase Ib Study of Pan-Phosphoinositide-3-Kinase Inhibitor Buparlisib With Letrozole in Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer. J Clin Oncol 2014; 32(12):1202-9; PMID:24663045; http://dx.doi.org/10.1200/JCO.2013.54.0518
- Juric D SC, Cervantes A, Kurkjian C, Patel MR, Sachdev J, Mayer I, Krop IE, Oliveira M, Sanabria S, Cheeti S, et al. Ph1b study of the PI3K inhibitor GDC-0032 in combination with fulvestrant in patients with hormone receptor-positive advanced breast cancer. J Clin Oncol 2013; 31:abstract PD1-3.
- Barbieri CE, Baca SC, Lawrence MS, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012; 44:685-9; PMID:22610119; http://dx.doi.org/10.1038/ng.2279
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012; 487:239-43; PMID:22722839; http://dx.doi.org/10.1038/nature11125
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011; 19:575-86; PMID:21575859; http://dx.doi.org/10.1016/j.ccr.2011.04.008
- Thomas C, Lamoureux F, Crafter C, Davies BR, Beraldi E, Fazli L, Kim S, Thaper D, Gleave ME, Zoubeidi A. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther 2013; 12:2342-55; PMID:23966621; http://dx.doi.org/10.1158/1535-7163.MCT-13-0032
- Zhang YJ, Duan Y, Zheng XF. Targeting the mTOR kinase domain: the second generation of mTOR inhibitors. Drug Discov Today 2011; 16:325-31; PMID:21333749; http://dx.doi.org/10.1016/j.drudis.2011.02.008
- LoRusso P, Markman B, Tabernero J, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765, a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced solid tumors. J Clini Oncol 2009; 27:3502
- Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang J, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther 2011; 10:2426-36; PMID:21998291; http://dx.doi.org/10.1158/1535-7163.MCT-11-0446
- Wagner AJ, Burris III HA, de Bono JS, et al. Pharmacokinetics and pharmacodynamic biomarkers for the dual PI3K/mTOR inhibitor GDC-0980: Initial Phase I evaluation. Mol Cancer Therapeut 2009; 8: Supplement 1
- Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H, Ebbesen SH, Ainscough BJ, Ramu A, Iyer G, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)K inhibitor. Nature 2014 [epub; preprint]; doi:10.1038/nature13948; http://www.ncbi.nlm.nih.gov/pubmed/25409150
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994; 370:527-32; PMID:8052307; http://dx.doi.org/10.1038/370527a0
- Ma L, Teruya-Feldstein J, Bonner P, Bernardi R, Franz DN, Witte D, Cordon-Cardo C, Pandolfi PP. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res 2007; 67:7106-12; PMID:17671177; http://dx.doi.org/10.1158/0008-5472.CAN-06-4798
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 2004; 101:13489-94; PMID:15342917; http://dx.doi.org/10.1073/pnas.0405659101
- Carriere A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, Roux PP. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr Biol 2008; 18:1269-77; PMID:18722121; http://dx.doi.org/10.1016/j.cub.2008.07.078
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008; 14:1351-6; PMID:19029981; http://dx.doi.org/10.1038/nm.1890
- Faber AC, Li D, Song Y, Yeap BY, Bronson RT, Lifshits E, Chen Z, Maira SM, García-Echeverría C, Wong KK, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A 2009; 106:19503-8; PMID:19850869; http://dx.doi.org/10.1073/pnas.0905056106
- Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T, et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci U S A 2013; 110:4015-20; PMID:23431193; http://dx.doi.org/10.1073/pnas.1216013110
- Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol 2013; 71:1395-409; PMID:23443307; http://dx.doi.org/10.1007/s00280-013-2121-1
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160-6; PMID:17525332; http://dx.doi.org/10.1126/science.1140321
- Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res 2001; 61:3986-97; PMID:11358816
- Grana TM, Rusyn EV, Zhou H, et al. Ras mediates radioresistance through both phosphatidylinositol 3-kinase-dependent and Raf-dependent but mitogen-activated protein kinase/extracellular signal-regulated kinase kinase-independent signaling pathways. Cancer Res 2002; 62:4142-50; PMID:12124353
- Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev 2008; 22:436-48; PMID:18281460; http://dx.doi.org/10.1101/gad.1627008
- Kim IA, Bae SS, Fernandes A, Wu J, Muschel RJ, McKenna WG, Birnbaum MJ, Bernhard EJ. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res 2005; 65:7902-10; PMID:16140961
- Gupta AK, McKenna WG, Weber CN, Feldman MD, Goldsmith JD, Mick R, Machtay M, Rosenthal DI, Bakanauskas VJ, Cerniglia GJ, et al. Local recurrence in head and neck cancer: relationship to radiation resistance and signal transduction. Clin Cancer Res 2002; 8:885-92; PMID:11895923
- Mukherjee B, Choy H, Nirodi C, Burma S. Targeting nonhomologous end-joining through epidermal growth factor receptor inhibition: rationale and strategies for radiosensitization. Semin Radiat Oncol 2010; 20:250-7; PMID:20832017; http://dx.doi.org/10.1016/j.semradonc.2010.05.002
- Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer 2011; 11:239-53; PMID:21430696; http://dx.doi.org/10.1038/nrc3007
- West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat 2002; 5:234-48; PMID:12531180; http://dx.doi.org/10.1016/S1368-7646(02)00120-6
- Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 2011; 10:2218-32; PMID:21623170; http://dx.doi.org/10.4161/cc.10.13.16305
- Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci U S A 2008; 105:7785-90; PMID:18505846; http://dx.doi.org/10.1073/pnas.0703423105
- Park J, Feng J, Li Y, Hammarsten O, Brazil DP, Hemmings BA. DNA-dependent protein kinase-mediated phosphorylation of protein kinase B requires a specific recognition sequence in the C-terminal hydrophobic motif. J Biol Chem 2009; 284:6169-74; PMID:19144640; http://dx.doi.org/10.1074/jbc.C800210200
- Schmidt-Ullrich RK, Valerie KC, Chan W, et al. Altered expression of epidermal growth factor receptor and estrogen receptor in MCF-7 cells after single and repeated radiation exposures. Int J Radiat Oncol Biol Phys 1994; 29:813-9; PMID:8040028; http://dx.doi.org/10.1016/0360-3016(94)90570-3
- Contessa JN, Hampton J, Lammering G, Mikkelsen RB, Dent P, Valerie K, Schmidt-Ullrich RK. Ionizing radiation activates Erb-B receptor dependent Akt and p70 S6 kinase signaling in carcinoma cells. Oncogene 2002; 21:4032-41; PMID:12037685; http://dx.doi.org/10.1038/sj.onc.1205500
- Toulany M, Lee KJ, Fattah KR, Lin YF, Fehrenbacher B, Schaller M, Chen BP, Chen DJ, Rodemann HP. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Res 2012; 10:945-57; PMID:22596249; http://dx.doi.org/10.1158/1541-7786.MCR-11-0592
- Deng R, Tang J, Ma JG, Chen SP, Xia LP, Zhou WJ, Li DD, Feng GK, Zeng YX, Zhu XF. PKB/Akt promotes DSB repair in cancer cells through upregulating Mre11 expression following ionizing radiation. Oncogene 2011; 30:944-55; PMID:20956948; http://dx.doi.org/10.1038/onc.2010.467
- Tsai WB, Chung YM, Takahashi Y, Xu Z, Hu MC. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol 2008; 10:460-7; PMID:18344987; http://dx.doi.org/10.1038/ncb1709
- Chung YM, Park SH, Tsai WB, Wang SY, Ikeda MA, Berek JS, Chen DJ, Hu MC. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat Commun 2012; 3:1000; PMID:22893124; http://dx.doi.org/10.1038/ncomms2008
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000; 404:782-7; PMID:10783894; http://dx.doi.org/10.1038/35008115
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002; 296:530-4; PMID:11964479; http://dx.doi.org/10.1126/science.1068712
- Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, Mansukhani M, Murty VV, Gaciong Z, Meek SE, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005; 7:193-204; PMID:15710331; http://dx.doi.org/10.1016/j.ccr.2005.01.009
- Katayama K, Fujita N, Tsuruo T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol Cell Biol 2005; 25:5725-37; PMID:15964826; http://dx.doi.org/10.1128/MCB.25.13.5725-5737.2005
- Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev 2001; 15:2203-8; PMID:11544177; http://dx.doi.org/10.1101/gad.913901
- Jiang Z, Pore N, Cerniglia GJ, et al. Phosphatase and tensin homologue deficiency in glioblastoma confers resistance to radiation and temozolomide that is reversed by the protease inhibitor nelfinavir. Cancer Res 2007; 67:4467-73; PMID:17483362; http://dx.doi.org/10.1158/0008-5472.CAN-06-3398
- Fokas E, Im JH, Hill S, Yameen S, Stratford M, Beech J, Hackl W, Maira SM, Bernhard EJ, McKenna WG, et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res 2012; 72:239-48; PMID:22108822; http://dx.doi.org/10.1158/0008-5472.CAN-11-2263
- Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, Amici A, Boothman DA, Scaglioni PP. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res 2009; 69:7644-52; PMID:19789349; http://dx.doi.org/10.1158/0008-5472.CAN-09-0823
- Prevo R, Deutsch E, Sampson O, Diplexcito J, Cengel K, Harper J, O'Neill P, McKenna WG, Patel S, Bernhard EJ. Class I PI3 kinase inhibition by the pyridinylfuranopyrimidine inhibitor PI-103 enhances tumor radiosensitivity. Cancer Res 2008; 68:5915-23; PMID:18632646; http://dx.doi.org/10.1158/0008-5472.CAN-08-0757
- Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, Fernandez-Capetillo O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol 2011; 18:721-7; PMID:21552262; http://dx.doi.org/10.1038/nsmb.2076
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006; 125:733-47; PMID:16647110; http://dx.doi.org/10.1016/j.cell.2006.03.035
- Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2012; 2:1036-47; PMID:22915752; http://dx.doi.org/10.1158/2159-8290.CD-11-0348
- Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2012; 2:1048-63; PMID:22915751; http://dx.doi.org/10.1158/2159-8290.CD-11-0336
- Matulonis U, Wulf GM, Birrer MJ, et al. Phase I study of oral BKM120 and oral olaparib for high-grade serous ovarian cancer (HGSC) or triple-negative breast cancer (TNBC). J Clin Oncol 2014; 32:5s; http://dx.doi.org/10.1200/JCO.2013.49.4757
- Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear phosphoinositide 3-kinase beta controls double-strand break DNA repair. Proc Natl Acad Sci U S A 2010; 107:7491-6; PMID:20368419; http://dx.doi.org/10.1073/pnas.0914242107
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 2005; 102:8204-9; PMID:15928081; http://dx.doi.org/10.1073/pnas.0502857102
- Tee AR, Proud CG. DNA-damaging agents cause inactivation of translational regulators linked to mTOR signalling. Oncogene 2000; 19:3021-31; PMID:10871854; http://dx.doi.org/10.1038/sj.onc.1203622
- Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A 2010; 107:4153-8; PMID:20160076; http://dx.doi.org/10.1073/pnas.0913860107
- Yu C, Rahmani M, Dai Y, Conrad D, Krystal G, Dent P, Grant S. The lethal effects of pharmacological cyclin-dependent kinase inhibitors in human leukemia cells proceed through a phosphatidylinositol 3-kinase/Akt-dependent process. Cancer Res 2003; 63:1822-33; PMID:12702569
- Vora SR, Juric D, Kim N, et al. CDK 4/6 Inhibitors Sensitize PIK3CA Mutant Breast Cancer to PI3K Inhibitors. Cancer Cell 2014; 26:136-49; PMID:25002028; http://dx.doi.org/10.1016/j.ccr.2014.05.020
- Gruber Filbin M, Dabral SK, Pazyra-Murphy MF, Ramkissoon S, Kung AL, Pak E, Chung J, Theisen MA, Sun Y, Franchetti Y, et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: new therapeutic opportunities. Nat Med 2013; 19:1518-23; PMID:24076665; http://dx.doi.org/10.1038/nm.3328
- Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, Brugge JS. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012; 21:227-39; PMID:22340595; http://dx.doi.org/10.1016/j.ccr.2011.12.024
- Ellis L, Ku SY, Ramakrishnan S, Lasorsa E, Azabdaftari G, Godoy A, Pili R. Combinatorial antitumor effect of HDAC and the PI3K-Akt-mTOR pathway inhibition in a Pten defecient model of prostate cancer. Oncotarget 2013; 4:2225-36; PMID:24163230
- Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Müller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 2012; 22:796-811; PMID:23238015; http://dx.doi.org/10.1016/j.ccr.2012.10.023
- Chen MH, Chiang KC, Cheng CT, Huang SC, Chen YY, Chen TW, Yeh TS, Jan YY, Wang HM, Weng JJ, et al. Antitumor activity of the combination of an HSP90 inhibitor and a PI3K/mTOR dual inhibitor against cholangiocarcinoma. Oncotarget 2014; 5:2372-89; PMID:24796583