
Abstract
S-phase checkpoints are triggered in tumor cells in response to DNA replication stress caused by the tumor microenvironment or oncogenes. A recent report from our laboratory showed that tumor cells and more normal epithelial cells have a very different response to replication stress. In this Author's View, the implications of this finding are discussed.
Abbreviations
| ATM | = | ataxia telangiectasia-mutated |
| ATR | = | ATM and Rad3 related |
| CHK1 | = | checkpoint kinase 1 |
| DSBs | = | DNA double-strand breaks |
| ROS | = | reactive oxygen species |
| RPA | = | replication protein A |
| ssDNA | = | single-stranded DNA |
Genome instability is now widely recognized as a critical driver of tumor development, having reached the status of one of the hallmarks of cancer. Events leading to genetic instability are initiated by DNA damage or DNA replication stress induced by exposure to exogenous or endogenous agents that modify DNA structure. However, there is also growing evidence that oncogenes cause DNA replication stress through alteration of DNA synthesis precursor poolsCitation1 or by inappropriately driving pre-tumor cells into S phase.Citation2 Nevertheless, cells can deal with such stresses through DNA repair mechanisms that recognize and correct DNA damage and DNA damage responses that trigger cell cycle arrest, resolve structures blocking replication, or commit cells to death if the damage is too great.Citation3
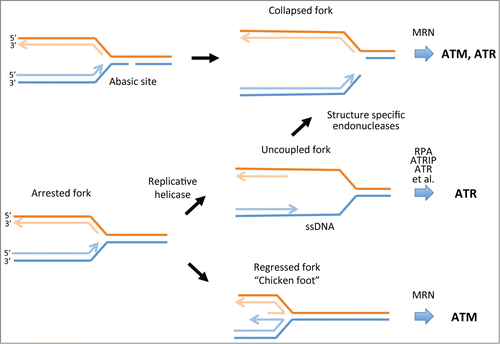
DNA damage response pathways have been most widely studied in tumor cells because of the importance of these pathways in ensuring tumor cell survival in hostile environments (e.g., hypoxia or nutrient deprivation). These studies provide evidence for a number of different responses to DNA replication stress induced by these conditionsCitation4 (). Prominent among replication fork responses is uncoupling of the replication and helicase complexes at forks when replication is disrupted. This leads to the formation of stretches of single-stranded DNA (ssDNA) that become coated by the replication protein A complex (RPA). In turn, RPA–ssDNA provides a substrate for recruitment of ataxia telangiectasia-mutated and RAD3-related (ATR) protein through the ATR interacting protein (ATRIP) and a host of other factors to activate the ATR-checkpoint kinase 1 (CHK1) signaling cascade.Citation5 Another potential response is fork collapse. This may occur through cleavage of ssDNA regions that are insufficiently protected by RPA by structure-specific endonucleasesCitation6 or by replication through abasic lesions in DNA that can be generated by reactive oxygen species (ROS) in tumor cells. Depending on the structure of the collapsed fork and its processing, such events trigger activation of ataxia telangiectasia-mutated (ATM) or ATR signaling cascades. An alternative fate is reversal of slowed or arrested replication forks to form so-called chicken foot structures that facilitate bypass of damaged DNA or difficult to replicate DNA. Again, depending on how such structures are processed, ATM and/or ATR signaling cascades can be activated. Most studies of tumor cells indicate that ATR-CHK1 signaling is primarily activated, suggesting that ssDNA is a key intermediate at arrested forks. This is supported by numerous studies demonstrating formation of RPA foci and ssDNA. Additionally, many studies have shown that ATR-CHK1 signaling (but not ATM) is a crucial determinant of replication fork and cell fate, as inhibitors of these checkpoint kinases trigger fork collapse and/or apoptosis.Citation6,7 In some tumor cell lines these fates require co-treatment with replication inhibitors whereas in others the checkpoint inhibitors are effective as single agents.
Figure 1. Fate of DNA replication forks in response to DNA damage or replication stress. Some forms of DNA damage such as abasic sites that may arise through the action of ROS can directly cause replication fork collapse. Such events trigger the activation of ATM or ATR, depending on the processing of the end by the MRE11/RAD50/NBS1 (MRN) complex or other DNA damage response proteins that may be recruited to the collapsed fork. Uncoupling of polymerase and helicase complexes at arrested forks can lead to the formation of stretches of ssDNA. These become coated with RPA, which then recruits ATRIP and ATR and a number of other proteins to activate the ATR-CHK1 protein kinase cascade. If ssDNA is insufficiently protected by RPA, structure-specific endonucleases such as MUS81 can cleave ssDNA to collapse the fork. Arrested forks may also undergo regression to produce the so-called chicken foot structure. This response potentially enables replication complexes to bypass DNA lesions or difficult to replicate regions. This structure also produces a substrate for the 3′–5′ exonuclease activity of MRE11 that can activate ATM. Regressed forks can also be resolved by a number of other pathways that generate DSBs or ssDNA to restart DNA synthesis. ATM, ataxia telangiectasia-mutated; ATR, ATM and Rad3 related; CHK1, checkpoint kinase 1; DSBs, DNA double-strand breaks; ROS, reactive oxygen species; RPA, replication protein A; ssDNA, single-stranded DNA.
In our recent paper,Citation8 we sought to determine how more normal cells respond to replication stress. Surprisingly little is known about the response of normal epithelial cells despite the epithelial origin of most tumors. Our evidence indicated that the ATR-CHK1 signaling pathway was not activated when human telomerase reverse transcriptase-immortalized normal human urothelial (hTERT-NHU) cells were treated with replication inhibitors. Critical downstream targets of ATR (CHK1-Ser345 and RPA2-Ser33) were not phosphorylated and the formation of RAD51 foci (which are normally generated by replication stress in an ATR-CHK1–dependent manner) was suppressed. In contrast, the ATM signaling cascade was activated within 2 hours of treatment. S-phase progression was slowed in these cells but there was no induction of apoptosis. Consistent with the failure to activate CHK1, CHK1 inhibitors had little effect on these cells in the presence or absence of the replication inhibitors. In contrast, a highly malignant bladder cancer cell line (EJ) showed rapid activation of the ATR-CHK1 signaling cascade and acute sensitivity to CHK1 inhibitors. The response of the hTERT-NHU cells is similar to that of human embryonic stem cells, which also fail to activate CHK1, induce RAD51 foci, or generate ssDNA following replication stress.Citation9 Also, CHK1 inhibitors have little effect on these cells. So how do ATM inhibitors affect hTERT-NHU cells exposed to replication stress? Unexpectedly, our evidence suggests that they trigger a G1 checkpoint instead of S-phase arrest. This response may be a consequence of the role of ATM in controlling ROS generation; when ATM is inhibited ROS accumulation could trigger a G1 checkpoint.Citation10 However, a ROS inhibitor did not relieve the G1 block and this is a question that requires further investigation. Interestingly, EJ bladder tumor cells do not arrest in G1 after treatment with the ATM inhibitor (and we have not found any reports in the literature of such an arrest in tumor cells), but show a weak accumulation in S phase following treatment with the ATM inhibitor alone and robust induction of apoptosis following treatment with a combination of ATM and replication inhibitors.
These findings lead us to speculate that replication forks in non-tumor cells may respond differently to fork arrest than those in tumor cells. The formation of ssDNA appears to be suppressed, suggesting that events such as the uncoupling of replication and helicase complexes that trigger ATR-CHK1 signaling may be more tightly controlled in hTERT-NHU. Whether this indicates that non-tumor cells have specific mechanisms to prevent polymerase-helicase uncoupling is an interesting question that deserves further study. And do these findings have any implications for therapy? They suggest that CHK1, and possibly ATM, inhibitors are particularly effective in preferentially killing at least some types of tumor cells. However, responses to these inhibitors are very diverse, underlining once again the need for biomarkers that will predict which tumor cells will respond.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
References
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genetic instability in early stages of cancer development. Cell 2011; 145:435-46; PMID:21529715; http://dx.doi.org/10.1016/j.cell.2011.03.044
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638-42; PMID:17136094; http://dx.doi.org/10.1038/nature05327
- Ciccia A, Elledge SJ. The DNA damage reponse: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/10.1016/j.molcel.2010.09.019
- Petermann E, Helleday T. Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 2010; 11:683-7; PMID:20842177; http://dx.doi.org/10.1038/nrm2974
- Cimprich KA, Cortez D. ATR: an esential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9:616-27; PMID:18594563; http://dx.doi.org/10.1038/nrm2450
- Toledo LI, Altmeyer M, Rask M-B, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013; 155:1088-103; PMID:24267891; http://dx.doi.org/10.1016/j.cell.2013.10.043
- Myers K, Gagou ME, Zuazua-Villar P, Rodriguez R, Meuth M. ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress. PLoS Genet 2009; 5:e1000324; PMID:19119425; http://dx.doi.org/10.1371/journal.pgen.1000324
- Wang W-T, Catto JW, Meuth M. Differential response of normal and malignant urothelial cells to CHK1 and ATM inhibitors. Oncogene 2014; PMID:25043304; http://dx.doi.org/10.1038/onc.2014.221; [Epub ahead of print
- Desmarais JA, Hoffmann MJ, Bingham G, Gagou ME, Meuth M, Andrews PW. Human embryonic stem cells fail to activate CHK1 and commit tom apoptosis in response to DNA replication inhibitors. Stem Cells 2012; 30:1385-93; PMID:22553144; http://dx.doi.org/10.1002/stem.1117
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004; 431:997-1002; PMID:15496926; http://dx.doi.org/10.1038/nature02989