
Abstract
Downregulation of G protein-coupled receptor kinase 2 (GRK2) in endothelial cells has recently been identified as a relevant event in the tumoral angiogenic switch. Based on the effects of altering GRK2 dosage in cell and animal models, this kinase appears to act as a hub in key signaling pathways involved in vascular stabilization and remodeling. Accordingly, decreased GRK2 expression in endothelial cells accelerates tumor growth in mice by impairing the pericytes ensheathing the vessels, thereby promoting hypoxia and macrophage infiltration. These results raise new questions regarding the mechanisms by which transformed cells trigger the decrease in GRK2 observed in human breast cancer vessels and how GRK2 modulates the interactions between different cell types that occur in the tumor microenvironment.
Abbreviations
| ALK | = | activin receptor-like kinase |
| Ang2 | = | angiopoietin 2 |
| AR | = | adrenergic receptor |
| BMP | = | bone morphogenetic protein |
| EC | = | endothelial cell |
| FGF | = | fibroblast growth factor |
| GM-CSF | = | granulocyte-macrophage colony-stimulating factor |
| HUVEC | = | human umbilical vein endothelial cell |
| IH | = | infantile hemangioma |
| NF-kB | = | nuclear factor kappa-light-chain-enhancer of activated B cells |
| PDGF | = | platelet-derived growth factor |
| PDGFR | = | PDGF receptor |
| S1P | = | sphingosine-1-phosphate |
| TGFβ1 | = | transforming growth factor beta 1 |
| VEGF | = | vascular endothelial growth factor |
| VEGFR | = | VEGF receptor |
| VSMC | = | vascular smooth muscle cell |
Vascular networks play an essential role in maintaining the homeostasis and growth of vertebrate organisms, and the somewhat complex process of angiogenesis is essential for vessel formation during embryonic development and physiological tissue remodeling.Citation1 Multiple cell types participate in physiological angiogenesis, during which numerous signaling pathways must be engaged and coordinated precisely through independent and interdependent factors. However, angiogenesis is induced aberrantly in association with inflammation or tumors. Indeed, the tumor microvasculature is usually very angiogenic and leaky, with enlarged and dilated vessels that are lined with immature walls as a result of the loss of pericytes.Citation2,3 These features cause deficient blood supply to the tumor, and the ensuing hypoxia triggers the secretion of diverse factors with proangiogenic and proinflammatory activities, further fostering abnormal vascularization and inflammation. In such a harsh microenvironment, transformed cells often became more aggressive, displaying increased proliferation, invasiveness, and drug resistance.Citation2 Through reciprocal interactions in the tumor-associated vasculature, tumor-infiltrated immune cells and transformed cells emerge as fundamental factors underlying cancer progression.
G protein-coupled receptor kinase 2 (GRK2) was recently shown to be a key element in the creation of a permissive microenvironment for tumor progression. In this review we discuss the evidence that links GRK2 and other GRKs to the regulation of different aspects of vessel formation in pathophysiological contexts. Specifically, we will focus on how GRK2 might affect the interaction between vascular cells and other non-tumor cells in the tumor microenvironment, how the latter might affect vascular GRK2 expression, and the potential relevance of vascular maturation regulated by GRK2.
GRK2 Regulates Physiological and Tumor Vascularization
G protein-coupled receptor kinases (GRKs) were initially identified as key players in the desensitization and internalization of multiple G protein-coupled receptors (GPCRs). In addition to such canonical roles, GRKs also promote G protein-independent transduction cascades downstream of GPCRs and initiate alternative signaling pathways by phosphorylating and/or interacting with partners other than GPCRs. Although the current GRK2 “interactome” includes several potential modulators of vascular homeostasis and remodeling, only recently has this kinase been shown to be necessary for vessel formation and stability. Angiogenesis involves several early morphogenetic steps, during which endothelial cells (ECs) polarize, migrate, establish cell–cell contacts, and form vessel lumens. These events are followed by a stabilization step that is driven by pericyte apposition.Citation1 Upon GRK2 downmodulation the behavior of primary microvascular ECs is altered, as characterized by an increase in motility and enhanced downstream signaling in response to key angiogenic stimuli such as vascular endothelial growth factor (VEGF), sphingosine-1-phosphate (S1P), or serum. Similarly, these cells lose the capability to organize into tubular structures and the balanced secretion of proinflammatory and proangiogenic factors is disrupted.
Endothelial GRK2 dosage modulates transforming growth factor beta 1 (TGFβ1)-mediated pathways.Citation6 TGFβ1 is fundamental for the activation and resolution of angiogenic events because diverse angiogenic stimuli control its production and secretion, and TGFβ itself upregulates other key angiogenic modulators.Citation7,8 The cellular responses triggered by TGFβ1 in ECs are complex and contribute positively or negatively to endothelial activation, in part due to the co-existence of 2 receptors that drive opposite effects, activin receptor-like kinase 1 (ALK1) and ALK5, and their functional cross-modulation.Citation8,9 In this scenario, GRK2 levels can alter the balance in TGFβ1 signaling through ALK5 and ALK1 receptorsCitation6 (see below). Consistent with this array of cell-autonomous endothelial defects, neovascularization is impaired in both global and endothelium-specific GRK2 knockout mice. In experimental models of angiogenesis, loss of GRK2 compromises the ability of ECs to differentiate and fuse into tubular structures and the adherence of pericytes required for the formation of structured vessels. In more physiological settings, such as embryonic or postnatal retinal neovascularization, mice with a global or endothelium-specific deficit of GRK2 develop vessels with altered morphometrics and reduced mural coating. In the mouse retina, GRK2 depletion causes delayed remodeling of the primary plexus, which has an excessive vascular density and abnormal endothelial-pericyte interactions that might impede effective pruningCitation6. Remarkably, a reduced level of GRK2 in the endothelium accelerates tumor growth in mice, in addition to increasing the size of intratumoral vessels, reducing pericyte coverage, and enhancing macrophage infiltration,Citation6 thereby strengthening many of the known hallmarks of the tumor microvasculature.
GRK2 at the Crossroads of Endothelial Activation and Vascular Maturation
The dynamic expression of GRK2 in the endothelium of the developing vasculature suggests that it fulfills a pivotal role in vascular maturation and maintenance, as does its ability to regulate key signaling pathways downstream of platelet-derived growth factor (PDGF), TGFβ1, and bone morphogenetic protein 9 (BMP9) receptors that are engaged in vascular stabilization and remodeling. Indeed, the immature vascular phenotype associated with GRK2 depletion further corroborates this role. The cellular dosage of GRK2 is important in determining how ECs respond to different physiological stimuli, such as VEGF, fibroblast growth factor (FGF), PDGF, or TGFβ, which orchestrate not only the functional specification of activated ECs, but also their maturation (). This dynamic interplay and the balance of the “tip” and “stalk” EC ratio are central to angiogenic activation. Such aspects are defined upon VEGF activation of ECs in pre-existing vessels through Notch-mediated cell fate determination.Citation10 Thus, VEGF upregulates the Notch ligand Delta-like 4 (Dll4), which mediates lateral inhibition of VEGF signaling in neighboring cells by downregulating VEGFR2 and upregulating VEGFR1, a decoy receptor for the VEGF ligand.Citation11 As a result, a graded response to VEGF is established in which the less common high responding cells become tip cells and sprout toward the source of VEGF, while most of the adjacent cells are instructed to become weaker responders and convert into stalk cells that follow the guiding tip cell and proliferate.Citation12 However, vessels in the tumor microenvironment are leaky and immature as a result of their exposure to high levels of VEGF-A derived from tumor cells, endothelial cells, and platelets. Such abnormal vascularization is related to the negative and positive effects of VEGF on PDGF-induced pericyte chemotaxis and differentiationCitation13 and sprouting activityCitation2, respectively. GRK2-deficient ECs do not produce more VEGF-A but they do display enhanced VEGF-dependent downstream signaling and chemotactic responses.Citation6 It is tempting to speculate that GRK2 downmodulation could simulate the effects of an excessive and chronic VEGF challenge in tumors that are poorly equipped with this angiogenic factor.
Figure 1. Endothelial-specific loss of GRK2 impairs maturation and vascular remodeling in adult and developmental angiogenesis. (A) Functional angiogenesis encompasses several sequential and interdependent steps that require proper control of GRK2 expression. The initial activation phase involves perivascular cell detachment from pre-existing vessels, and the specification of activated endothelial cells into tip and stalk populations as a result of the integration of VEGF, ALK1, and PDGF signals with the Notch pathway. During the resolution phase, ECs cease to migrate and proliferate and, instead, initiate tubulogenesis and membrane basement formation. Downmodulation of GRK2 deregulates the downstream signaling to such co-operating factors of endothelial specification, which might alter tip/stalk specification and/or the maintenance of activated ECs. Vessel maturation and remodeling also occur in the resolution phase, as mural cells are recruited for endothelial tube investment. Increased secretion of the mural cell attractant PDGF by GRK2-deficient endothelial cells could disturb the local gradients needed to drive adequate intercellular apposition of pericytes with ECs. (B) GRK2 regulates the TGFβ-dependent state of the endothelium via simultaneous modulation of the ALK5 and ALK1 pathways. GRK2 downmodulation not only decreases direct inhibition of Smad2/3 (in a kinase activity-dependent manner) but could also alleviate ALK1 signaling-mediated lateral inhibition of the ALK5 pathway. Stronger activation of ALK5 with concurrent attenuated ALK1 signaling might alter the balance between the activation and resolution phases of angiogenesis, thereby impairing sprouting and maturation. ALK, activin receptor-like kinase; EC, endothelial cell; PDGF, platelet-derived growth factor; TGFβ1, transforming growth factor beta 1; VEGF, vascular endothelial growth factor.
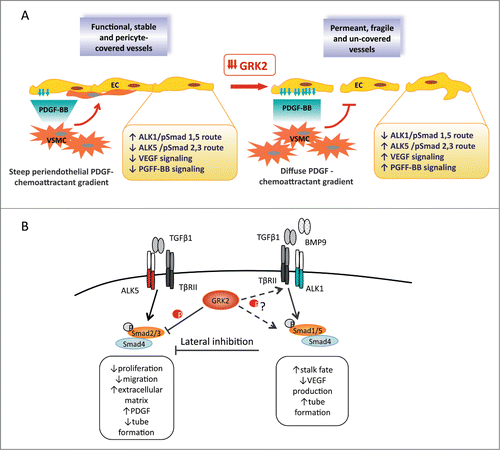
In addition to the potential impact on VEGF signaling, GRK2 also influences secretion of endothelial PDGF-BB, a factor that regulates several steps in vessel formation. During angiogenesis, PDGF-BB expression is restricted to immature capillary structures (angiogenic sprouts) and is concentrated in the tip ECs. Although the primary vascular target cells for PDGF-BB are pericytes and vascular smooth muscle cells (VSMCs), the endothelium also responds to this factor as FGF2-activated ECs can upregulate PDGFR receptors for autocrine PDGF-BB stimulation.Citation14 Such autocrine regulation, although not well characterized, has been related to increased sprouting and remodeling, proliferation, morphological reorganization, and anastomosis of ECs (for a review see ref. 15). Since PDGF-BB secretion is enhanced by GRK2 downmodulation in ECs,Citation6 this could exacerbate the “tip” cell character in ECs and augment the number of unstable and immature vessels. This scenario is consistent with the role of GRK2 as a desensitizer of PDGFR in VSMCsCitation5, whereby downregulation of PDGFR-β signaling is a prerequisite for the maturation of ECs.Citation15 However, the most relevant functions of PDGF-BB are as a potent recruiter of mural cells and in the promotion of pericyte/VSMC coverage of nascent vessels.Citation15,16 Secretion and periendothelial retention of PDGF-BB must be tightly regulated to form the proper chemotactic-like gradients that efficiently guide pericytes to nascent vessels and thereafter ensure their stable apposition.Citation15,17 The higher levels of PDGF-BB secreted by GRK2-deficient ECs might disturb such local gradients, contributing to the dissociation of pericytes from the vessel in both xenograft tumors and in the developing and normal adult tissues of mutant mice.Citation6
Abnormal PDGF-BB secretion by GRK2-deficient ECs could be secondary to improper tip/stalk specification, or may reflect GRK2-mediated impairment of signaling pathways engaged in its synthesis. Downmodulation of GRK2 stimulates the ALK5-dependent Smad2/3 phosphorylation branch of the TGFβ1 signaling pathway (), which is reported to promote PDGF-BB transcription.Citation18 The enhanced stimulation of ALK5-dependent Smad2/3 phosphorylation observed in GRK2+/- ECs could be explained by the previously reported direct inhibitory effect of GRK2 on Smad2/3 activity as a result of GRK2-mediated phosphorylation of these factors within the linker domain.Citation19 Conversely, a decrease in GRK2 markedly impairs the ALK1-Smad1/5 branch in response to either TGFβ1 or the ALK1-specific BMP9 ligand,Citation6 a member of the TGFβ family that influences vascular quiescence.Citation20,21 Therefore, GRK2 levels are able to modulate TGFβ1-mediated ALK1/ALK5 pathways in endothelial cells by both inhibiting the ALK5 route at the level of Smad2/3 and by positively contributing to the activation of the ALK1 route through an uncharacterized mechanism. Upon GRK2 downmodulation, both relief of the direct inhibition of Smad2/3 and decreased ALK1 signaling-mediated lateral inhibition of the ALK5 pathwayCitation22 would contribute to the observed unbalance in TGFβ1 signaling toward the ALK5 route. ALK1 signaling is involved in maturation events such as arteriovenous specification of developmental vasculature,Citation23 mural recruitment,Citation24 and adult vessel homeostasis.Citation25 Vascular malformations detected upon GRK2 downregulation might affect all of these ALK1-regulated processes. In addition, the reduction in GRK2 could influence the crosstalk between ALK1 and Notch that is involved in endothelial fate selection and the differentiation of periendothelial cells.Citation26 In particular, BMP9 activity on stalk cells stimulates Smad1/5 and its downstream gene expression, including Notch targets that prevent tip cell specification and the migration of ECs.Citation26 Thus, both ALK1 and Notch cascades cooperate in maintaining the stalk cell fate of EC cells,Citation26 which will ultimately undergo a process of tubulogenesis to form vessels.Citation10 Inhibition of the tumor upregulated Notch ligand Dll4 results in excessive tip cell formation and proliferation, formation of immature vessels, and tissue ischemia,Citation11,27 resembling the effects of GRK2 downmodulation. ALK1 activation also counteracts FGF-induced EC proliferation and VEGF-stimulated angiogenesis,Citation28 making it tempting to speculate that ECs with little GRK2 might be prone to adopt a tip phenotype as a result of weaker ALK1/Notch synergism and stronger ALK1/VEGF antagonism during EC commitment. However, GRK2-deficient ECs do not behave as “full” tip cells, as suggested by the impaired sprouting and the reduced branching and filopodia formation at early developmental stages of retinal vasculature. This is probably related to the fact that GRK2 also influences other angiogenic pathways. In this regard, GRK2 downmodulation could simultaneously strengthen the restrictive effect of the ALK5-Smad2/3 signaling branch on sprouting.Citation29
Overall, these results support the concept that GRK2 is a central regulator of endothelial activation and vessel maturation, representing a common signaling hub for different effector pathways operating in distinct steps of angiogenesis.
How Does the Tumor Milieu Instruct Endothelial Cells to Express Less GRK2?
Endothelial GRK2 protein expression is diminished in vessels of human breast cancer but not in benign lesions, which correlates with the increased vascular tortuosity, permeability, and pericyte depletion in tumors. Poor vascular remodeling might adversely skew progression toward malignancy and metastasis, and this may be actively determined by systemic hormonal and stress conditions, the stromal microenvironment of the tumor and, ultimately, by transformed cells.Citation30-32 In this regard, conditioned media from either melanoma or human breast cancer cell lines of luminal or basal-like lineages, but not that from normal mammary cells, significantly reduces the endothelial GRK2 dosage.Citation6 Moreover, GRK2 is downregulated in activated human microvascular ECs that are co-cultured with human colorectal cancer cells in the presence of blood components.Citation6 Interestingly, although these data suggest that aberrant vascular expression of GRK2 might be a common feature of cancer, this may not be the case for all types of cancer. Indeed, different tumor cells display a distinct ability to downmodulate endothelial GRK2, which is triggered by LS180 but not by HT-29 colon cancer cells, and is triggered more rapidly and potently by MCF7 than by Hs578T breast cancer cells. We speculate that such variability might have implications in the clinical outcome of different tumors.
Influence of the proinflammatory milieu of transformed cells and the intratumor immune cell infiltrate
Despite evidence that GRK2 expression is downmodulated at the transcriptional level, there is little information available about the mechanisms that govern this process.Citation33,34 Analysis of the human GRK2 gene promoter in aortic smooth muscle cells revealed that transcription is enhanced by factors related to vasoconstriction downstream of the GPCR-Gq signaling pathways,Citation35 whereas proinflammatory mediators such as interleukin (IL)-1β, tumor necrosis factor (TNF)α, and interferon (INF)γ have the opposite effect. Interestingly, IL-1β and TNFα are often released by tumor cells in response to local hypoxia (). However, other tumor factors released in relation to the transforming phenotype could also be implicated in the paracrine regulation of GRK2.Citation36 Identification of the tumor-secreted factors that influence vascular GRK2 expression is an interesting goal for future studies.
Figure 2. Vascular expression of GRK2 in the tumor microenvironment. Several cellular components of the tumor stroma and of tumor cells themselves might be actively involved in the downmodulation of GRK2 in endothelial cells (ECs). Within the reactive tumor stroma, macrophages and transformed cells secrete a variety of proinflammatory chemokines and cytokines, and bioactive NO and ROS molecules that target ECs might also trigger GRK2 downmodulation through different mechanisms (inhibition of promoter activity or protein synthesis, stimulation of protein degradation, or catalytic blockade). Hypoxia associated with the tumor-induced pathological remodeling of vessels can also decrease GRK2 expression in ECs, thereby causing greater vessel disorganization that in turn creates further pockets of hypoxia and escalates immune infiltration. Systemic chronic inflammation that results from continuously challenging the vasculature with circulating and tissue inflammatory factors could also alter GRK2 expression at the endothelium. Likewise, behavioral stress is responsible for local intratumor release of norepinephrine, which might promote GRK2 protein decay in a βAR-subtype or cell context-specific manner. AR, adrenergic receptor; EC, endothelial cell; NO, nitric oxide; ROS, reactive oxygen species.
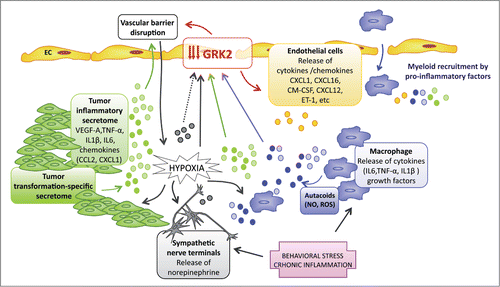
In addition, transformed cells interact in a paracrine manner with distinct non-neoplastic components of the tumor, mainly fibroblasts and immune cells, which leads to a “smoldering” inflammation state that encourages cancer cell proliferation and survival.Citation37,38 Secretion of diverse chemokines and VEGF-A by tumor cells attracts myeloid cells, which in turn release a cocktail of cytokines (including TNFα, IL-1β, and IL-6) that could directly reduce GRK2 transcription in ECs. Moreover, inflammatory mediators appear to affect post-transcriptional regulation of GRK2 protein and its activity. Thus, autacoids such as reactive oxygen species (ROS), nitric oxide (NO), and low-molecular-weight S-nitrosothiols (SNO) are actively released by infiltrating proinflammatory macrophages at earlier stages of tumor growth and can inhibit GRK2 translationCitation39 and catalytic activity by means of protein S-nitosylation.Citation40 In addition, cytokines whose levels are increased by the tumor, such as IL-6, promote proteasome-dependent degradation of GRK2 in blood peripheral mononuclear cells.Citation33, Citation41 Furthermore, proteasome-dependent GRK2 downmodulation was noted upon acute exposure of brain organotypic cultures to hypoxia/ischemiaCitation42 and in human umbilical vein endothelial cells (HUVECs; personal observations). Since GRK2 ubiquitination and proteasomal turnover is enhanced by several receptors (including the β2AR receptor, see below) through a mechanism involving GRK2 phosphorylation by c-Src or MAPK,Citation43,44 it is also possible that activation of these pathways in the EC tumor context may trigger kinase reduction.
Thus, it is tempting to suggest that the relationship between inflammation and vascular GRK2 might constitute a self-perpetuating pathological cycle that fosters tumor progression. The vessel dysfunction promoted by GRK2 downmodulation would trigger stronger hypoxia and myeloid infiltration, which in turn would drive further endothelial GRK2 depletion. In this scenario, the use of antitumoral antioxidant compounds to curtail this aberrant cycle and “normalize” the tumor vasculature could prove to be a useful approach.
Influence of tumor stress associated with activity of the sympathetic nervous system
Behavioral stress is another tumor-related event that could regulate vascular GRK2 expression. Cumulative evidence indicates that chronic neurosympathetic activity affects the activity of the immune system,Citation45 as well as the malignant growth and dissemination of tumor cells, through direct stimulation of β2-adrenergic receptors (ARs).Citation45,46 Several processes appear to underlie the influence of βARs on tumor progression, including myeloid recruitment or increased proinflammatory secretion, and angiogenesis triggered by adrenergic stimulation of tumor and immune cells.Citation47 It is tempting to suggest that exposure of tumor vessels to circulating adrenaline (released by the adrenal medulla) or, more significantly in a tumor context, to tissue ischemia-induced norepinephrine release from the sympathetic nerves,Citation47,48 might influence endothelial cells and the expression of GRK2. The interplay between βAR signaling and vascular GRK2 expression has mainly been studied in the context of cardiovascular and macrovasculature homeostasis.Citation49,50 The level of circulating catecholamines and βAR activity correlates positively with GRK2 protein and mRNA expression in different tissues and cell types.Citation51 These effects seem to be mediated by β1AR activation as β1AR (but not β2AR) transgenic mice display increased levels of GRK2.Citation52 In contrast, stimulation of β2AR triggers GRK2 protein degradation in a diversity of cell types.Citation43,44 Although it has not been directly tested, it is tempting to suggest that the sympathetic tumoral stress acting through β2AR could reduce GRK2 expression in endothelial cells (thus exacerbating pathological features of the tumor vasculature).
βAR activation appears to be involved in the stimulation of proliferation of both adult and circulating EC precursor cells and in functional neoangiogenesis by means of VEGF stimulation.Citation53,54 In contrast, defective βAR expression impairs the vascularization of damaged tissue,Citation54 suggesting that tight control of adrenergic activity is needed to integrate angiogenic responses in injury and repair. In keeping with this, β-blockers have been suggested to play a protective role in reducing the incidence of all cancer types through their influence in tumor angiogenesis by inhibiting β2AR receptor-mediated effects on both malignant cells and tumor vesselsCitation45,47; however, the extent of such protection varies among tumors, and moreover shows no relationship with clinical outcome in some cohorts of breast cancer patients.Citation55 These discrepancies might be related to different off-target β2AR affinities of the most commonly used selective β1-blockers, the distinct proportion and dosage of receptor subtypes in βAR-bearing tumoral and endothelial cells within a particular tumor, and/or the different relative influence of βAR-mediated effects on the behaviour of malignant cells versus the tumor microenvironment in tumor growth and progression. Remarkably, infantile hemangioma (IH), which results from abnormal proliferation of endothelial cells, has been shown to be effectively treated with non-selective β-blockers such as propanolol.Citation56 These benign vascular neoplasms are characterized by densely packed endothelial and pericyte cells that form immature vessels resembling those resulting from GRK2 downmodulation. Hemangioma-derived endothelial cells (HemECs) are rounded in shape and display increased growth and migratory properties, whereas proliferative pericytes show a diminished ability to adhere to and stabilize blood vessels.Citation56 Moreover, HemECs appear to be more sensitive to paracrine/external stimulation by VEGF-ACitation56, and the proliferating phase of IH is paralleled by excessive VEGF production and increased VEGFR-2 activity, which are partially dependent on β2AR signaling.Citation57 Based on these features it is tempting to suggest that HemECs may express reduced GRK2 levels and that the effects of propanolol on vessel stabilization may be partially mediated by upregulation of this kinase. However, in the context of other tumor pathologies, the effects of βAR challenge (and β-blocker therapy) on endothelial GRK2 might be not straightforward, depending on the particular targeted cell type (tumoral versus stromal components), cell microenvironment conditions (hypoxic, inflamed, or tumoral), the specific adrenergic ligand (epinephrine versus norepinephrine), or the relative abundance of the βAR subtypes, which may distinctly affect the signaling pathways related to protein kinase degradation.
GRK2 and Tumor Angiogenesis: Beyond Endothelial Regulation
Although our results highlight the vascular bed as the main functional target, other cellular components of the tumor stroma could contribute to GRK2-dependent malignant phenotypes (). In this regard, infiltration of immune cells into the tumor microenvironment is a critical event in cancer progression.Citation32,37,58 Interestingly, tumor progression is enhanced in global systemic GRK2 hemizygous mice, a model in which other cell types besides endothelial cells also have diminished GRK2. Notably, despite showing similar endothelial expression of GRK2, tumor growth is greater in Tie2Cre-GRK2fl/− than in Tie2Cre-GRK2fl/fl mice (), suggesting that differences in growth may also rely on non-endothelial host components, such as macrophages. Indeed, these cells are major inflammatory components associated with malignant tumors and although their conventional role is to combat pathogens and to assist in adaptive immune responses, in most cases they enhance tumor cell proliferation and invasion.Citation32,58,59 Circulating monocytes are actively recruited into damaged tissues where they specifically differentiate into 2 major states with different activities depending on local cues: the classically activated (M1)-polarized macrophages that are cytotoxic to microbes or malignant cells and stimulate the lymphocyte immune responses or the alternatively activated (M2)-polarized macrophages involved in immunosuppression and wound healing/tissue repair. In the tumor milieu, monocytes lean toward a differentiation state that shares similarities with the M2-polarized macrophages, thereby displaying potent immunosuppressive, proangiogenic, and protumoral properties.Citation59,60
Table 1. Tumor growth in different mouse models with global or endothelial-specific alterations in GRK2 levels
Figure 3. Proposed direct and vascular-mediated effects of GRK2 in angiogenesis induced by tumor-associated macrophages. GRK2-deficient endothelial cells (ECs) display an enhanced ability to trigger macrophage migration and an altered secretory profile of CXCL12, CXCL1, ET-1, and other macrophage regulatory factors that favor attraction, rolling, and extravasation of macrophages. Monocyte and macrophage chemotaxis is also dependent on GRK2 levels, and GRK2 downmodulation might influence the differentiation of myeloid cells in the tumor stroma. Some pro-M2 factors such as IL33 can reduce GRK2 expression, which would alleviate GRK2-mediated CCL2 receptor desensitization and the inhibition of the alternative NFkB pathway, thereby favoring M2 polarization. An increase in the presence of tumor-associated macrophages and of macrophage polarization to M2-like profiles supports malignant proliferation, survival, angiogenesis, and stroma remodeling.
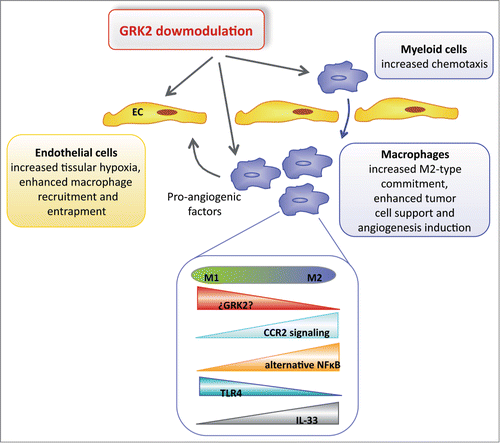
Effect of endothelial GRK2 dosage on tumor macrophage recruitment
Specific signals recruit monocytes and guide them into the hypoxic areas of the tumor; indeed, the highest density of tumor-associated macrophages (TAMs) is found in such hypoxic foci in breast and other tumors.Citation61,62 These foci are created by the concurrence of a dysfunctional microvasculature, which limits the oxygen supply, and the presence of nearby tumor cells with high rates of oxygen consumption. In these hypoxic areas, TAMs no longer fulfill inflammatory/cytotoxic roles but, rather, are committed to adopt a protumoral M2-like behavior. Both monocyte recruitment and hypoxia-induced commitment of macrophages appear to be fostered not only by endothelial GRK2 downmodulation but also by the GRK2 monocyte dosage. Several CC chemokines that act through GPCRs and are produced by tumor cells, fibroblasts, and ECs specifically attract and activate mononuclear cells, with CCL2 or CCL5 being those most widely implicated in human breast and melanoma cancer. However, other chemoattractants may also be involved, such as VEGF, the endothelins ET1 or ET2, and those produced by activated macrophages such as CCL4, colony-stimulating factor-1, or CXCL8. Reduced GRK2 expression alters the endothelial production of several of these factors and might help recruit monocytes to tumors, as indicated by the stronger presence of macrophages in cancer lesions that develop in endothelial GRK2 specific-knockout mice and by the enhanced monocyte chemotaxis produced by the EC-derived secretome from these mice.Citation6 Thus, higher levels of potent macrophage chemoattractants, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), placental growth factor 2 (PIGF2), Factor III, or CXCL16, are produced by GRK2+/− mouse lung ECs,Citation6 while there is also an increase in the factors that facilitate monocyte adhesion and extravasation through endothelial layers, such as CXCL12 and ET-1.Citation63 In addition, secretion of the CXCR2 ligand KC (CXCL1) in the conditioned media of these cells is strongly upregulated, which is involved in monocyte chemotaxisCitation64 and arrest on the endothelium as well as flow and extravasation.Citation65 Within the tumor mass, macrophages seem to follow a gradient of hypoxia-induced chemoattractants such as VEGF, CXCL8, or ET2, which are mainly produced by tumor cells.Citation62 In summary, endothelium-specific downmodulation of GRK2 could favor the recruitment of macrophages to discrete regions of the tumor by direct (EC secretome) and indirect (leaky vessels leading to hypoxia) mechanisms, thereby fueling tumor growth.
GRK2 and chemotactic responses of myeloid cells
Monocytes/macrophages from systemic GRK2 hemizygous mice may display more potent chemotactic responses toward tumor-derived homing signals, which might contribute to the more effective growth of tumors in GRK2+/- and Tie2Cre-GRK2fl/− mice than in Tie2Cre-GRK2 fl/+ and Tie2Cre-GRK2fl/fl mice (). GRK2 is known to play an important role in the motility of immune cells and is expressed strongly in different types of immune cells, representing an important regulator of the T lymphocyte, macrophage, and neutrophil responses during inflammation.Citation66 Consistent with the “classic” role of GRK2 in GPCR desensitization, reduction of its expression significantly enhances the chemotactic response of lymphocytes and neutrophils toward CCR5, CCR2, CXCR2, and chemotactic receptors for substance P, S1P, or formyl-peptides.Citation66,67 Several reports have reflected the inability of neutrophils with increased GRK2 levels to migrate toward CXCL2 or CXCL8 chemoattractants,Citation68 while mobilization of macrophages from global GRK2 hemizygous mice to CCL5 is enhanced.Citation69 Interestingly, lipopolysaccharide activation of TLR4 favors CCL2-induced migration of macrophages by disengaging GRK2-dependent CCR2 receptor desensitization.Citation70 Overall, it is tempting to suggest that the concurrent downmodulation of GRK2 in vascular endothelial cells and circulating monocytes might foster further tumor growth ().
Interestingly, ECs and macrophages share common precursor progenitors of myeloid origin during embryonic development, suggesting that Tie2-driven recombinase activation and GRK2 depletion would affect both cell types at some point in their differentiation. Despite the fact that expression of GRK2 in macrophages from Tie2-Cre/GRK2fl/fl mice is no different from that in wild-type animals,Citation71 supporting the importance of the endothelial component in the differential growth of tumors observed in this animal model, it is still possible that in a pathological setting, minor myeloid Tie2-positive lineages would be selectively mobilized to participate in tumor growth. In fact, a subset of circulating Tie2-expressing monocytes (TEMs) has been shown to represent a relevant proportion of the tumor immune cellular infiltrate distinct from TAMs.Citation32,72 TEMs are a rare cell population in normal tissues but they actively home to the tumor stroma in response to Ang2, a Tie2-ligand produced by activated ECs and angiogenic vessels. Selective depletion of TEMs inhibits tumor angiogenesis and causes tumor regression despite the presence of TAMs, suggesting that TEMs are critical for the tumor angiogenic switch. It has been proposed that, in addition to Ang2, TEMs might use tumor-secreted non-inflammatory mediators other than those used by TAMs to be guided into avascular, non-inflamed tumor regions. It is tempting to suggest that low levels of GRK2 could facilitate the chemotaxis of these cells toward some relevant cues, as occurs in the migration of macrophages toward inflammatory mediators. Changes in GRK2 expression levels by TEMs (or TAMs) might modulate the tumor homing of these cellular populations and the outcome of tumor progression, an interesting issue to address in future studies.
A role for GRK2 in macrophage polarization?
Finally, decreased expression of GRK2 in the tumoral vascular bed and/or the myeloid infiltrate might also influence the phenotypic TAM commitment. Although M2 marker-based characterization of TAMs has not been addressed in tumors grown in Ti2Cre-GRK2fl/fl mice, the higher levels of adrenomedullin in the tumor stroma suggests there is an increase in M2-like differentiated TAMs in these mice.Citation6 M2 macrophages play a fundamental role in several aspects of tumor progression through their relationship with vascular cells in stimulating angiogenesis, with other immune cells in suppressing adaptive immunity, with fibroblasts to induce extracellular matrix remodeling, and with transformed cells to promote invasion and metastasis.Citation58 Different molecular pathways have been implicated in directing macrophage differentiation. Cytokines such as GM-CSF, IFN-γ, and IL-1, and toll-like receptor (TLR) activation can polarize monocytes toward the M1 macrophage subtype with a “proinflammatory” cytokine profile, whereas challenge with M-CSF, CCL2, and interleukins IL-4, IL-10, and IL-13 produces an “anti-inflammatory” cytokine profile similar to that of M2 macrophages.Citation60 Interestingly, the effectiveness of some of these pathways can be modified by GRK2 expression. CCR2-receptor responses to the M2-polarizing chemokine CCL2 are desensitized by GRK2 in different cell types.Citation73,74 Moreover, the NFkB pathway that is central to macrophage activation and differentiation is also modulated by GRK2.Citation75,76 Interestingly, the classic NFκB pathway is activated in macrophages during early stages of tumor initiation but this is not the case in advanced tumors, in which M2-like TAMs have high levels of p50 homodimers and the anti-inflammatory IL-10, an situation that we hypothesize could be favored in tumors growing in systemic GRK2 hemizygous mice. Furthermore, the M1-polarizing activation of TLR4 receptors promotes GRK2 upregulation in macrophagesCitation77 and neutrophils,Citation78 which also undergo a sort of M1 polarization,Citation79 whereas the M2-differentiating factor IL-33 prevents such upregulation in myelomonocytic cells.Citation80 Indeed, the re-routing of the polarization of adipose tissue-resident macrophages from a M2 to M1 phenotype in obesity is associated with upregulated GRK2 expression in many different cell types.Citation81 Therefore, it is tempting to suggest that reduced monocyte expression of GRK2 could favor M2-like polarization in the tumor milieu.
Role of Other GRKs in Tumor Angiogenesis: Brothers in Arms with GRK2?
There is cumulative evidence that changes in GRKs are relevant events in tissue-specific tumor progression, as their key roles in the regulation of many cell processes and signaling pathways can be conditionally harnessed by transformed cells depending on the tumor GPCR profile, oncogenic pathways, and environmental insults. The impact of different GRKs on tumor angiogenesis might be related to the modulation of GPCR activity, either by triggering receptor desensitization or by mediating G-protein independent receptor signaling.Citation4 Numerous chemokines act as direct pro-angiogenic factors, whereas other GPCR agonists (catecholamines, S1P, or thrombin) stimulate production of master angiogenic drivers such as HIF1α or VEGF.Citation82 The few studies addressing the role of GRKs in tumor angiogenesis have focused on the consequences of overall changes in GRK expression either in the transforming cell itself or in the host tumor tissue, but not specific alterations in the vascular component of the tumor. As such, endothelial GRK2 is the only isoform that has been directly implicated in the control of tumor angiogenesis.
In terms of other GRKs, downregulation of GRK3 has been correlated with increased proliferation of glioblastoma cells, both in a cell-autonomous manner and through a paracrine, endothelial-mediated effect.Citation83 Conversely, increased GRK3 expression has been reported in prostate tumors cells that require this kinase for survival and proliferation of the transformed cells, whereas its overexpression fosters primary tumor growth and metastasis through the induction of angiogenesis.Citation84 However, whether vascular GRK3 expression is altered or required to support angiogenesis in different tumors has not been addressed. Alternatively, expression of GRK5 (but not of GRK2) in endothelial cells attenuates the production and secretion of several proangiogenic cytokines by inhibiting NFkB transcriptional activity,Citation85 which provokes impaired in vitro tube formation and in vivo angiogenesis in models of wound healing and chronic ischemia. These results suggest that vascular GRK5 levels might influence tumor angiogenesis. Interestingly, intratumor administration of a GRK5-based inhibitor of NFkB activity impairs the formation of tumor vascularization.Citation86 However, the tumor component (tumor cells or vascular cells) involved in the antitumoral properties of GRK5-derived constructs and the cellular dosage of GRK5 in such tumor components remain unknown.
Furthermore, xenograft tumors implanted in systemic GRK6 knockout mice grow faster and more aggressively and with greater intratumor microvascular densities, which correlates with increased immune cell infiltration of myeloid origin and higher levels of stromal MMP9 and MMP2 metalloproteases.Citation87 GRK6 deficiency blunted CXCR2 desensitization, suggesting that enhanced tumor growth might be a consequence of improved chemotactic motility, tumor homing, and protumoral neutrophil activity in these animals. However, GRK6 deficiency may also affect the proangiogenic function of the vascular cells and, indeed, GRK6 expression has been reported to be confined to vascular structures in some tumor tissues.Citation83 Moreover, vascular ECs are one of the CXCR2-positive cellular components in the tumor stroma, and endothelial activation of CXCR2 augments the rate of angiogenesis.Citation88 Therefore, it is possible that downregulation of vascular GRK2 in tumors might coexist with alterations in the profile of other GRKs, which would together shape a protumoral vasculature. Interestingly, a compensatory increase in expression of the closely related GRK3 isoform was not detected in mouse lung endothelial cells from global GRK2 hemizygous mice, whereas expression of GRK6 was modestly decreased. However, the pathophysiological function of GRK2 in vessel formation might be unique as there is no functional compensation for its loss by other GRKs, as suggested by the vascular malformations of embryo and adult Tie2Cre-GRK2fl/fl mice. Overall, it is tempting to suggest that altered levels of GRKs other than GRK2 may direct the scale of the activation phase of angiogenesis and the number of vessels in tumors, whereas the expression of GRK2 may shape the phenotypic characteristics of these vessels, in line with the notion that tumor size correlates better with the quality of the vessels than with the vascular density.Citation89,90
Concluding Remarks and Perspectives
A decrease in endothelial GRK2 has a pivotal effect by contributing critically to the impaired pericyte–endothelial cell–cell interactions that characterize the immature tumor vasculature, thereby favoring vessel instability, increased permeability, and enhanced access of different cell types to the tumor in order to promote disease progression. Remarkably, vascular GRK2 is specifically downregulated in intratumoral vessels, whereas vessels in the normal tissue surrounding the tumor or in benign lesions retain kinase expression. Tumor-driven downregulation of endothelial GRK2 harnesses the role of GRK2 in vessel formation and stability in order to generate a microenvironment permissive for cancer progression. This challenging notion may have important pathophysiological implications and should prompt further studies to assess its feasibility as a novel marker of pathological vasculature and as a potential target for therapeutic strategies aimed at vessel normalization in cancer. The fact that GRK2 represents a common signaling hub for many receptors involved in endothelial activation and maturation suggests that its modulation may simultaneously rewire signaling from the VEFG, PDGF-BB, and TGFβ1 pathways.
Antiangiogenic treatments, such as those based on anti-VEGF strategies, commonly fail because of compensation by parallel pathways or resistance at the receptor level,Citation91 which could be overcome by targeting signaling hubs. However, several questions arise related to the use of endothelial GRK2 downmodulation to combat tumor angiogenesis. For example: Will preventing endothelial GRK2 downregulation ameliorate tumor growth or aggressiveness, and does GRK2 upregulation have the opposite effect in vascular remodeling? What are the relevant GRK2-interacting proteins involved in vessel maturation? Likewise, the precise contribution of GRK2 kinase activity to different aspects of vascular remodeling remains to be determined. While it is attractive to anticipate that vessel normalization by preventing the reduction in GRK2 might divert a tumor toward a dormant state, it is also tempting to suggest that a potential synergistic effect of GRK2 depletion and antiangiogenic therapies based on ALK1 blockageCitation92 could successfully trigger complete vessel regression. Conversely, it is unclear whether steady-state levels of endothelial GRK2 are related to metastasis. Cancer progression is characterized by the recruitment of macrophages to tumor tissues, a step fostered by vascular GRK2 downmodulation, and to premetastatic niches, where they condition the secondary localization of tumor cells. This latter step could also be facilitated in the context of reduced GRK2 expression.
Finally, epidemiological and clinical studies have shown that various chronic inflammatory diseases predispose patients to the risk of cancer, as noted for colorectal cancer in patients with Crohn's disease or ulcerative colitis, or pancreatic cancer in patients with chronic pancreatitis, among others.Citation93 In this regard, pathological angiogenesis in major inflammatory bowel conditions (ulcerative colitis and Crohn's disease) and in other inflammatory processes, such as rheumatoid arthritis or psoriasis, is potentially a key factor in perpetuating disease.Citation94 Remarkably, it is well documented that GRK2 levels are decreased in human patients suffering from inflammatory diseases (multiple sclerosis, rheumatoid arthritis, sepsis),Citation5,66 suggesting that suppressed GRK2 levels might be important for long-term immaturity of the vascular system and cancer risk.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Our laboratory is funded by grants from Ministerio de Educación y Ciencia (SAF2011–23800), Fundación Ramón Areces, The Cardiovascular Network (RECAVA) of Ministerio Sanidad y Consumo-Instituto Carlos III (RD12/0042/0012), Comunidad de Madrid Indisnet Network (S2011/BMD-2332) to F.M, and Fundación Eugenio Rodriguez Pascual, Fundación Ramón Areces, ISS-PIE, and Instituto de Salud Carlos III (PI11/00859) to P.P.
References
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011; 473:298-307; PMID:21593862; http://dx.doi.org/10.1038/nature10144
- Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 2011; 17:1359-70; PMID:22064426; http://dx.doi.org/10.1038/nm.2537
- Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 2011; 146:873-87; PMID:21925313; http://dx.doi.org/10.1016/j.cell.2011.08.039
- Watari K, Nakaya M, Kurose H. Multiple functions of G protein-coupled receptor kinases. J Mol Signal 2014; 9:1; PMID:24597858; http://dx.doi.org/10.1186/1750-2187-9-1
- Penela P, Murga C, Ribas C, Lafarga V, Mayor F, Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol 2010; 160:821-32; PMID:20590581; http://dx.doi.org/10.1111/j.1476-5381.2010.00727.x
- Rivas V, Carmona R, Munoz-Chapuli R, Mendiola M, Nogues L, Reglero C, Miguel-Martin M, Garcia-Escudero R, Dorn GW, 2nd, Hardisson D, et al. Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2. J Clin Invest 2013; 123:4714-30; PMID:24135140; http://dx.doi.org/10.1172/JCI67333
- van Meeteren LA, Goumans MJ, ten Dijke P. TGF-beta receptor signaling pathways in angiogenesis; emerging targets for anti-angiogenesis therapy. Curr Pharm Biotechnol 2011; 12:2108-20; PMID:21619534; http://dx.doi.org/10.2174/138920111798808338
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell 2003; 12:817-28; PMID:14580334; http://dx.doi.org/10.1016/S1097-2765(03)00386-1
- Orlova VV, Liu Z, Goumans MJ, ten Dijke P. Controlling angiogenesis by two unique TGF-beta type I receptor signaling pathways. Histol Histopathol 2011; 26:1219-30; PMID:21751154
- Tung JJ, Tattersall IW, Kitajewski J. Tips, stalks, tubes: notch-mediated cell fate determination and mechanisms of tubulogenesis during angiogenesis. Cold Spring Harb Perspect Med 2012; 2:a006601; PMID:22355796; http://dx.doi.org/10.1101/cshperspect.a006601
- Bridges E, Oon CE, Harris A. Notch regulation of tumor angiogenesis. Future Oncol 2011; 7:569-88; PMID:21463145; http://dx.doi.org/10.2217/fon.11.20
- Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell 2009; 16:196-208; PMID:19217422; http://dx.doi.org/10.1016/j.devcel.2009.01.015
- Greenberg JI, Shields DJ, Barillas SG, Acevedo LM, Murphy E, Huang J, Scheppke L, Stockmann C, Johnson RS, Angle N, et al. A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature 2008; 456:809-13; PMID:18997771; http://dx.doi.org/10.1038/nature07424
- Nissen LJ, Cao R, Hedlund EM, Wang Z, Zhao X, Wetterskog D, Funa K, Brakenhielm E, Cao Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest 2007; 117:2766-77; PMID:17909625; http://dx.doi.org/10.1172/JCI32479
- Hellberg C, Ostman A, Heldin CH. PDGF and vessel maturation. Recent Results Cancer Res 2010; 180:103-14; PMID:20033380; http://dx.doi.org/10.1007/978-3-540-78281-0_7
- Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res 2003; 314:15-23; PMID:12883993; http://dx.doi.org/10.1007/s00441-003-0745-x
- Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 2003; 17:1835-40; PMID:12897053; http://dx.doi.org/10.1101/gad.266803
- Taylor LM, Khachigian LM. Induction of platelet-derived growth factor B-chain expression by transforming growth factor-beta involves transactivation by Smads. J Biol Chem 2000; 275:16709-16; PMID:10828062; http://dx.doi.org/10.1074/jbc.275.22.16709
- Ho J, Cocolakis E, Dumas VM, Posner BI, Laporte SA, Lebrun JJ. The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J 2005;24:3247-3258; PMID:16121194; http://dx.doi.org/10.1038/sj.emboj.7600794
- David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 2007; 109:1953-61; PMID:17068149; http://dx.doi.org/10.1182/blood-2006-07-034124
- David L, Mallet C, Keramidas M, Lamande N, Gasc JM, Dupuis-Girod S, Plauchu H, Feige JJ, Bailly S. Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ Res 2008; 102:914-22; PMID:18309101; http://dx.doi.org/10.1161/CIRCRESAHA.107.165530
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell 2003; 12:817-828; PMID:14580334; http://dx.doi.org/10.1016/S1097-2765(03)00386-1
- Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 2000; 26:328-31; PMID:11062473; http://dx.doi.org/10.1038/81634
- Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A 2000; 97:2626-31; PMID:10716993; http://dx.doi.org/10.1073/pnas.97.6.2626
- Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, et al. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood 2008; 111:633-42; PMID:17911384; http://dx.doi.org/10.1182/blood-2007-08-107359
- Larrivee B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M, Eichmann A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell 2012; 22:489-500; PMID:22421041; http://dx.doi.org/10.1016/j.devcel.2012.02.005
- Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006; 444:1032-7; PMID:17183313; http://dx.doi.org/10.1038/nature05355
- Scharpfenecker M, van Dinther M, Liu Z, van Bezooijen RL, Zhao Q, Pukac L, Lowik CW, ten Dijke P. BMP-9 signals via ALK1 and inhibits bFGF-induced endothelial cell proliferation and VEGF-stimulated angiogenesis. J Cell Sci 2007; 120:964-72; PMID:17311849; http://dx.doi.org/10.1242/jcs.002949
- Mallet C, Vittet D, Feige JJ, Bailly S. TGFbeta1 induces vasculogenesis and inhibits angiogenic sprouting in an embryonic stem cell differentiation model: respective contribution of ALK1 and ALK5. Stem Cells 2006; 24:2420-7; PMID:17071858; http://dx.doi.org/10.1634/stemcells.2005-0494
- Hall K, Ran S. Regulation of tumor angiogenesis by the local environment. Front Biosci (Landmark Ed) 2010; 15:195-212; PMID:20036815; http://dx.doi.org/10.2741/3615
- Chakroborty D, Sarkar C, Basu B, Dasgupta PS, Basu S. Catecholamines regulate tumor angiogenesis. Cancer Res 2009; 69:3727-30; PMID:19383906; http://dx.doi.org/10.1158/0008-5472.CAN-08-4289
- De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol 2007; 28:519-24; PMID:17981504; http://dx.doi.org/10.1016/j.it.2007.09.004
- Penela P, Ribas C, Mayor F, Jr. Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell Signal 2003; 15:973-81; PMID:14499340; http://dx.doi.org/10.1016/S0898-6568(03)00099-8
- Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor F, Jr. Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res 2006; 69:46-56; PMID:16288730
- Ramos-Ruiz R, Penela P, Penn RB, Mayor F, Jr. Analysis of the human G protein-coupled receptor kinase 2 (GRK2) gene promoter: regulation by signal transduction systems in aortic smooth muscle cells. Circulation 2000; 101:2083-9; PMID:10790351; http://dx.doi.org/10.1161/01.CIR.101.17.2083
- Karagiannis GS, Pavlou MP, Diamandis EP. Cancer secretomics reveal pathophysiological pathways in cancer molecular oncology. Mol Oncol 2010; 4:496-510; PMID:20934395; http://dx.doi.org/10.1016/j.molonc.2010.09.001
- Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J Immunol Res 2014; 2014:149185; PMID:24901008; http://dx.doi.org/10.1155/2014/149185
- Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 2013; 13:759-71; PMID:24154716; http://dx.doi.org/10.1038/nrc3611
- Cobelens PM, Kavelaars A, Heijnen CJ, Ribas C, Mayor F, Jr., Penela P. Hydrogen peroxide impairs GRK2 translation via a calpain-dependent and cdk1-mediated pathway. Cell Signal 2007; 19:269-77; PMID:16963227; http://dx.doi.org/10.1016/j.cellsig.2006.06.009
- Whalen EJ, Foster MW, Matsumoto A, Ozawa K, Violin JD, Que LG, Nelson CD, Benhar M, Keys JR, Rockman HA, et al. Regulation of β-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell 2007; 129:511-22; PMID:17482545; http://dx.doi.org/10.1016/j.cell.2007.02.046
- Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J 1999; 13:715-25; PMID:10094932
- Lombardi MS, Vroon A, Sodaar P, van Muiswinkel FL, Heijnen CJ, Kavelaars A. Down-regulation of GRK2 after oxygen and glucose deprivation in rat hippocampal slices: role of the PI3-kinase pathway. J Neurochem 2007; 102:731-40; PMID:17437535; http://dx.doi.org/10.1111/j.1471-4159.2007.04576.x
- Penela P, Elorza A, Sarnago S, Mayor F, Jr. Beta-arrestin- and c-Src-dependent degradation of G-protein-coupled receptor kinase 2. EMBO J 2001; 20:5129-38; PMID:11566877; http://dx.doi.org/10.1093/emboj/20.18.5129
- Elorza A, Penela P, Sarnago S, Mayor F, Jr. MAPK-dependent degradation of G protein-coupled receptor kinase 2. J Biol Chem 2003; 278:29164-73; PMID:12738776; http://dx.doi.org/10.1074/jbc.M304314200
- Antoni MH, Lutgendorf SK, Cole SW, Dhabhar FS, Sephton SE, McDonald PG, Stefanek M, Sood AK. The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nat Rev Cancer 2006; 6:240-8; PMID:16498446; http://dx.doi.org/10.1038/nrc1820
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med 2006; 12:939-44; PMID:16862152; http://dx.doi.org/10.1038/nm1447
- Cole SW, Sood AK. Molecular pathways: β-adrenergic signaling in cancer. Clin Cancer Res 2012; 18:1201-6; PMID:22186256; http://dx.doi.org/10.1158/1078-0432.CCR-11-0641
- Hubens LE, Verloop WL, Joles JA, Blankestijn PJ, Voskuil M. Ischemia and reactive oxygen species in sympathetic hyperactivity states: a vicious cycle that can be interrupted by renal denervation? Curr Hypertens Rep 2013; 15:313-20; PMID:23754326; http://dx.doi.org/10.1007/s11906-013-0367-y
- Harris DM, Cohn HI, Pesant S, Eckhart AD. GPCR signalling in hypertension: role of GRKs. Clin Sci (Lond) 2008; 115:79-89; PMID:18593382; http://dx.doi.org/10.1042/CS20070442
- Dorn GW, 2nd. GRK mythology: G-protein receptor kinases in cardiovascular disease. J Mol Med (Berl) 2009; 87:455-63; PMID:19229505; http://dx.doi.org/10.1007/s00109-009-0450-7
- Rengo G, Lymperopoulos A, Leosco D, Koch WJ. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol 2011; 50:785-92; PMID:20800067; http://dx.doi.org/10.1016/j.yjmcc.2010.08.014
- Mialet-Perez J, Rathz DA, Petrashevskaya NN, Hahn HS, Wagoner LE, Schwartz A, Dorn GW, Liggett SB. Beta1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med 2003; 9:1300-1305; PMID:14502278; http://dx.doi.org/10.1038/nm930
- Iaccarino G, Ciccarelli M, Sorriento D, Galasso G, Campanile A, Santulli G, Cipolletta E, Cerullo V, Cimini V, Altobelli GG, et al. Ischemic neoangiogenesis enhanced by beta2-adrenergic receptor overexpression: a novel role for the endothelial adrenergic system. Circ Res 2005; 97:1182-9; PMID:16239589; http://dx.doi.org/10.1161/01.RES.0000191541.06788.bb
- Galasso G, De Rosa R, Ciccarelli M, Sorriento D, Del Giudice C, Strisciuglio T, De Biase C, Luciano R, Piccolo R, Pierri A, et al. beta2-Adrenergic receptor stimulation improves endothelial progenitor cell-mediated ischemic neoangiogenesis. Circ Res 2013; 112:1026-34; PMID:23418295; http://dx.doi.org/10.1161/CIRCRESAHA.111.300152
- Ji Y, Chen S, Xiao X, Zheng S, Li K. b-blockers: a novel class of antitumor agents. OncoTargets Ther 2012; 5:391-401; PMID:23226026; http://dx.doi.org/10.2147/OTT.S38403
- Ji Y, Chen S, Li K, Li L, Xu C, Xiang B. Signaling pathways in the development of infantile hemangioma J. Hematol Oncol 2014; 7:13-26; http://dx.doi.org/10.1186/1756-8722-7-13
- Ji Y, Chen S, Li K, Xiao X, Zheng S Xu T. The role of β-adrenergic receptor signaling in the proliferation of hemangioma-derived endothelial cells. Cell Division 2013; 8:1-12; PMID:23286511; http://dx.doi.org/10.1186/1747-1028-8-1
- Cook J, Hagemann T. Tumour-associated macrophages and cancer. Curr Opin Pharmacol 2013; 13:595-601; PMID:23773801; http://dx.doi.org/10.1016/j.coph.2013.05.017
- Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl) 2013; 91:411-29; PMID:23515621; http://dx.doi.org/10.1007/s00109-013-1021-5
- Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol 2008; 180:2011-7; PMID:18250403; http://dx.doi.org/10.4049/jimmunol.180.4.2011
- Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, Van Ginderachter JA. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol 2011; 55:861-7; PMID:22161841; http://dx.doi.org/10.1387/ijdb.113371dl
- Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood 2004; 104:2224-34; PMID:15231578; http://dx.doi.org/10.1182/blood-2004-03-1109
- Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol 2006; 80:1183-96; PMID:16997855; http://dx.doi.org/10.1189/jlb.0905495
- Traves SL, Smith SJ, Barnes PJ, Donnelly LE. Specific CXC but not CC chemokines cause elevated monocyte migration in COPD: a role for CXCR2. J Leukoc Biol 2004; 76:441-50; PMID:15155777; http://dx.doi.org/10.1189/jlb.1003495
- Smith DF, Galkina E, Ley K, Huo Y. GRO family chemokines are specialized for monocyte arrest from flow. Am J Physiol Heart Circ Physiol 2005; 289:H1976-84; PMID:15937099; http://dx.doi.org/10.1152/ajpheart.00153.2005
- Vroon A, Heijnen CJ, Kavelaars A. GRKs and arrestins: regulators of migration and inflammation. J Leukoc Biol 2006; 80:1214-21; PMID:16943386; http://dx.doi.org/10.1189/jlb.0606373
- Penela P, Nogues L, Mayor F, Jr. Role of G protein-coupled receptor kinases in cell migration. Curr Opin Cell Biol 2014; 27:10-7; http://dx.doi.org/10.1016/j.ceb.2013.10.005
- Arraes SM, Freitas MS, da Silva SV, de Paula Neto HA, Alves-Filho JC, Auxiliadora Martins M, Basile-Filho A, Tavares-Murta BM, Barja-Fidalgo C, Cunha FQ. Impaired neutrophil chemotaxis in sepsis associates with GRK expression and inhibition of actin assembly and tyrosine phosphorylation. Blood 2006; 108:2906-13; PMID:16849637; http://dx.doi.org/10.1182/blood-2006-05-024638
- Otten JJ, de Jager SC, Kavelaars A, Seijkens T, Bot I, Wijnands E, Beckers L, Westra MM, Bot M, Busch M, et al. Hematopoietic G-protein-coupled receptor kinase 2 deficiency decreases atherosclerotic lesion formation in LDL receptor-knockout mice. FASEB J 2013; 27:265-76; PMID:23047899; http://dx.doi.org/10.1096/fj.12-205351
- Liu Z, Jiang Y, Li Y, Wang J, Fan L, Scott MJ, Xiao G, Li S, Billiar TR, Wilson MA, et al. TLR4 Signaling augments monocyte chemotaxis by regulating G protein-coupled receptor kinase 2 translocation. J Immunol 2013; 191:857-64; http://dx.doi.org/10.4049/jimmunol.1300790
- Ciccarelli M, Sorriento D, Franco A, Fusco A, Del Giudice C, Annunziata R, Cipolletta E, Monti MG, Dorn GW, 2nd, Trimarco B, et al. Endothelial G protein-coupled receptor kinase 2 regulates vascular homeostasis through the control of free radical oxygen species. Arterioscler Thromb Vasc Biol 2013; 33:2415-24; PMID:23950144; http://dx.doi.org/10.1161/ATVBAHA.113.302262
- De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, Naldini L. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 2005; 8:211-26; PMID:16169466; http://dx.doi.org/10.1016/j.ccr.2005.08.002
- Aragay AM, Mellado M, Frade JM, Martin AM, Jimenez-Sainz MC, Martinez AC, Mayor F, Jr. Monocyte chemoattractant protein-1-induced CCR2B receptor desensitization mediated by the G protein-coupled receptor kinase 2. Proc Natl Acad Sci U S A 1998; 95:2985-90; PMID:9501202; http://dx.doi.org/10.1073/pnas.95.6.2985
- Kleibeuker W, Jurado-Pueyo M, Murga C, Eijkelkamp N, Mayor F, Jr., Heijnen CJ, Kavelaars A. Physiological changes in GRK2 regulate CCL2-induced signaling to ERK1/2 and Akt but not to MEK1/2 and calcium. J Neurochem 2008; 104:979-92; PMID:17971124; http://dx.doi.org/10.1111/j.1471-4159.2007.05023.x
- Patial S, Saini Y, Parvataneni S, Appledorn DM, Dorn GW, 2nd, Lapres JJ, Amalfitano A, Senagore P, Parameswaran N. Myeloid-specific GPCR kinase-2 negatively regulates NF-kappaB1p105-ERK pathway and limits endotoxemic shock in mice. J Cell Physiol 2011; 226:627-37; PMID:20717897; http://dx.doi.org/10.1002/jcp.22384
- Patial S, Luo J, Porter KJ, Benovic JL, Parameswaran N. G-protein-coupled-receptor kinases mediate TNFalpha-induced NFkappaB signalling via direct interaction with and phosphorylation of IkappaBalpha. Biochem J 2010; 425:169-78; http://dx.doi.org/10.1042/BJ20090908
- Loniewski K, Shi Y, Pestka J, Parameswaran N. Toll-like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol Immunol 2008; 45:2312-22; PMID:18180038; http://dx.doi.org/10.1016/j.molimm.2007.11.012
- Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A 2009; 106:4018-23; PMID:19234125; http://dx.doi.org/10.1073/pnas.0900196106
- Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: "N1" versus "N2" TAN. Cancer Cell 2009; 16:183-94; PMID:19732719; http://dx.doi.org/10.1016/j.ccr.2009.06.017
- Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA, Jr., Auxiliadora-Martins M, Basile-Filho A, McKenzie AN, Xu D, Cunha FQ, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med 2010; 16:708-12; PMID:20473304; http://dx.doi.org/10.1038/nm.2156
- Garcia-Guerra L, Nieto-Vazquez I, Vila-Bedmar R, Jurado-Pueyo M, Zalba G, Diez J, Murga C, Fernandez-Veledo S, Mayor F, Jr., Lorenzo M. G protein-coupled receptor kinase 2 plays a relevant role in insulin resistance and obesity. Diabetes 2010; 59:2407-17; PMID:20627936; http://dx.doi.org/10.2337/db10-0771
- Richard DE, Vouret-Craviari V, Pouyssegur J. Angiogenesis and G-protein-coupled receptors: signals that bridge the gap. Oncogene 2001; 20:1556-62; PMID:11313902; http://dx.doi.org/10.1038/sj.onc.1204193
- Woerner BM, Luo J, Brown KR, Jackson E, Dahiya SM, Mischel P, Benovic JL, Piwnica-Worms D, Rubin JB. Suppression of G-protein-coupled receptor kinase 3 expression is a feature of classical GBM that is required for maximal growth. Mol Cancer Res 2012; 10:156-66; PMID:22086906; http://dx.doi.org/10.1158/1541-7786.MCR-11-0411
- Li W, Ai N, Wang S, Bhattacharya N, Vrbanac V, Collins M, Signoretti S, Hu Y, Boyce FM, Gravdal K, et al. GRK3 is essential for metastatic cells and promotes prostate tumor progression. Proc Natl Acad Sci U S A 2014; 111:1521-6.
- Sorriento D, Ciccarelli M, Santulli G, Campanile A, Altobelli GG, Cimini V, Galasso G, Astone D, Piscione F, Pastore L, et al. The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proc Natl Acad Sci U S A 2008; 105:17818-23; PMID:19008357; http://dx.doi.org/10.1073/pnas.0804446105
- Sorriento D, Campanile A, Santulli G, Leggiero E, Pastore L, Trimarco B, Iaccarino G. A new synthetic protein, TAT-RH, inhibits tumor growth through the regulation of NFkappaB activity. Mol Cancer 2009; 8:97; PMID:19900276; http://dx.doi.org/10.1186/1476-4598-8-97
- Raghuwanshi SK, Smith N, Rivers EJ, Thomas AJ, Sutton N, Hu Y, Mukhopadhyay S, Chen XL, Leung T, Richardson RM. G protein-coupled receptor kinase 6 deficiency promotes angiogenesis, tumor progression, and metastasis. J Immunol 2013; 190:5329-36; PMID:23589623; http://dx.doi.org/10.4049/jimmunol.1202058
- Miyake M, Goodison S, Urquidi V, Gomes Giacoia E, Rosser CJ. Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1/2 and EGF pathways. Lab Invest 2013; 93:768-78; PMID:23732813; http://dx.doi.org/10.1038/labinvest.2013.71
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 2011; 91:1071-121; PMID:21742796; http://dx.doi.org/10.1152/physrev.00038.2010
- Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011; 10:417-27; PMID:21629292; http://dx.doi.org/10.1038/nrd3455
- Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med 2012; 2:a006593; PMID:23209176
- Cunha SI, Pietras K. ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 2011; 117:6999-7006; PMID:21467543; http://dx.doi.org/10.1182/blood-2011-01-330142
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860-7; PMID:12490959; http://dx.doi.org/10.1038/nature01322
- Pousa ID, Mate J, Gisbert JP. Angiogenesis in inflammatory bowel disease. Eur J Clin Invest 2008; 38:73-81; PMID:18226040; http://dx.doi.org/10.1111/j.1365-2362.2007.01914.x