
Abstract
In past decades, cancer research has focused on genetic alterations that are detected in malignant tissues and contribute to the initiation and progression of cancer. These changes include mutations, copy number variations, and translocations. However, it is becoming increasingly clear that epigenetic changes, including alternative splicing, play a major role in cancer development and progression. There are relatively few studies on the contribution of alternative splicing and the splicing factors that regulate this process to cancer development and progression. Recently, multiple studies have revealed altered splicing patterns in cancers and several splicing factors were found to contribute to tumor development. Studies using high-throughput genomic analysis have identified mutations in components of the core splicing machinery and in splicing factors in several cancers. In this review, we will highlight new findings on the role of alternative splicing and its regulators in cancer initiation and progression, in addition to novel approaches to correct oncogenic splicing.
Introduction
Approximately 95% of human genes encode more than one product.Citation1,2 This phenomenon is achieved by the process of alternative splicing, which is regulated by both cis and trans acting elements. The cis elements in the pre-mRNA sequence are recognized by a large family of factors called splicing factors, the trans-acting factors that recruit or repel the spliceosomal machinery to catalyze splicing at specific splice sites (for a detailed review seeCitation3,4).
In the past 15 years, multiple studies have revealed altered splicing patterns in cancersCitation5-10 and several splicing factors were found to contribute to tumor initiation.Citation11-15 It is therefore not surprising that recent studies using high-throughput genomic and exomic analysis of a variety of cancers have revealed mutations in components of the core splicing machinery and in splicing factors.Citation16-22 Although identification of mutations in splicing factors does not necessarily indicate a role for aberrant splicing in tumor initiation, these findings have given rise to a growing field of research on the role of alternative splicing factors in tumor initiation and progression, as well as in invasiveness, metastasis, and drug resistance. In this review we will focus mainly on the roles of splicing factors in these processes. Other levels of regulation, which will not be discussed here, include transcriptional regulation in splicing, the role of RNA polymerase II kinetics,Citation23-25 and histone modifications.Citation26,27
Genomic Evidence for the Role of Splicing Factors in Cancer
Mutations in splicing factors
In recent years many genome-wide studies have been performed to screen for genomic mutations in tumor samples.Citation28-31 Some of these screens have unveiled mutations in components of the splicing machinery. The first study to identify mutations in splicing factors was that of Yoshida et al.Citation16 In this study, 29 patients with myelodysplastic syndrome (MDS) were screened by whole exome sequencing and mutations in splicing components were identified in 16 out of 29 patients. These mutations were in the genes for U2AF1 (also known also as U2AF35), ZRSR2, SRSF2 (also known as SC35), SF3A1, SF3B1, and PRPF40B, although only one case was found for each of the last 3 genes. This initial study was expanded to include 582 patients with myeloid neoplasms, this time specifically searching for mutations in the aforementioned genes as well as in U2AF65, SF1, and SRSF1. Once again, mutations were found in genes for the splicing factors U2AF1, SRSF2, ZRSR2, SF3B1, SF3B1, PRPF40B, and U2AF65 with differential occurrence but mostly in U2AF35, SRSF2, ZRSR2, and SF3B1.Citation16 This discovery led to multiple follow-up studies that have been performed on a variety of hematological malignancies.Citation19,21,22,32-37 To date, mutations in splicing factors have been found mainly in hematological cancers, and to a lesser extent in solid tumors.Citation38 Data from these sequencing studies have been reviewed recently;Citation39 here, we will highlight the most commonly mutated genes and their relevance to tumor development.
SF3B1, a gene encoding one of the U2 snRNP components, was found to be mutated in 75% of patients with MDS,Citation16 as well as in patients with chronic lymphocytic leukemia (CLL)Citation35 and to a much lesser extent in solid tumors.Citation21 However, mutation in SF3B1 did not correlate with a negative prognosis in patients with MDS.Citation33,34 Analysis of SF3B1 mutation profiles in hematopoietic neoplasms and their biological and clinical importance have been recently reviewed.Citation37,40 SF3B1 was also found to be mutated in lung adenocarcinomaCitation18 and uveal melanoma.Citation19
U2AF1, the gene encoding the 3´ splice site binding protein U2AF35, was found to be mutated in 6% of patients with myeloid neoplasms,Citation16 and in patients with lung adenocarcinoma.Citation18
SRSF2, which encodes the SR protein SRSF2 (SC35), was found to be mutated in 28% of patients with chronic myeloid leukemia (CML).Citation16 Mutations were also detected in patients with MDS, acute myeloid leukemia (AML), myeloproliferative neoplasms, and juvenile myelomonocytic leukemia.Citation16,36 Another study by Lasho and colleaguesCitation34 found that 32 out of 187 (17%) patients with primary myelofibrosis (PMF) had a mutation in SRSF2. Most of these mutations were point mutations at position P95, although some patients had deletions (delP95-R102). Interestingly and importantly, survival of patients with mutations in SRSF2 was worse than that of patients with mutations in SF3B1.Citation34
Gene amplifications and copy number variation
Alterations in gene copy number of several splicing factors have also been detected in various cancers .
Table 1. Altered splicing factor levels in cancer tissues
SRSF1, the gene encoding for SR protein SRSF1 (SF2/ASF), was found to be overexpressed in lung, colon, and breast tumors, and amplified in breast cancer.Citation12,41
SRSF3, the gene encoding the SR protein SRSF3 (also known as SRp20), has been found to be amplified in lung cancer and cervical cancer.Citation15
SRSF6, encoding the SR protein SRSF6 (SRp55), has been found to be amplified in 12% of lung and breast tumors, and in 37% of colon tumors samples.Citation11 SRSF6 has also been shown to be amplified in skin cancer.Citation42
HNRNPA2B1, which encodes the hnRNP protein hnRNP A2/B1, was found to be amplified and overexpressed in glioblastoma tumors.Citation13
RBFOX1 (A2BP1), the gene encoding the splicing factor RBFOX1, was found to be deleted in 10% of glioblastoma cases and to act as a tumor suppressor in this cancer.Citation43
The accumulation of data showing that genes encoding splicing factors are mutated, amplified, or deleted in cancer has strengthened the link between splicing factors and cancer initiation. However, only limited evidence exists for a direct role of these mutations or copy number variations as driver mutations in cancer. In addition, post-transcriptional modifications, such as phosphorylation on the splicing factor itself, can affect the function or localization of the protein,Citation44,45 suggesting another layer of regulation. This will not be discussed here but examples are provided elsewhere.Citation46-49
Mechanisms of Transformation by Splicing Factors
Two major families of alternative splicing factors have been studied in depth: SR proteins and hnRNPs. Although it is generally thought that the SR proteins promote alternative splicing whereas hnRNPs inhibit it, there is accumulating evidence that both SR proteins and hnRNPs work through a combinatorial effect of positive and negative regulation to control alternative splicing. Additional RNA binding proteins such as SAM68,Citation50,51 members of the RBM family,Citation52-54 and HuR,Citation55-57 will not be discussed here.
The oncogenic activities of SR proteins
SR proteins are a family of 12 proteins containing 1 or 2 RNA-recognition motifs (RRMs) and a C-terminal RS domain (arginine–serine repeats).Citation58 SR proteins are required for constitutive pre-mRNA splicing as well as alternative splicing, and also have other non-splicing functions in the cell including mRNA nuclear export, nonsense-mediated mRNA decay (NMD),Citation59 translation,Citation60,61 genomic stability,Citation62 cell cycle progression,Citation63 and miRNA biogenesis.Citation64 In recent years several SR proteins were found to play a causative role in several types of cancer. In many cases, SR proteins regulate alternative splicing events leading to enhanced production of pro-oncogenic isoforms and reduced formation of tumor suppressive isoforms, although in other cases the oncogenic activity of SR proteins is independent of splicing regulation.
SRSF1
SRSF1 plays an important role in regulating many splicing events in the cell and is one of the most-studied splicing factors in the context of its biological and pathological effects (for a recent review see Citation65). SRSF1 is overexpressed in different cancer types and acts as a proto-oncogene. Overexpression of SRSF1 in immortalized rodent fibroblasts and human mammary epithelial cells results in oncogenic transformation as evidenced by proliferation, resistance to apoptosis, and ability to form tumors in mice.Citation12,41 Furthermore, SRSF1 overexpression in lung adenocarcinoma cells results in a more aggressive phenotype and confers resistance to anticancer drugs such as carboplatin and paclitaxel.Citation66
SRSF1 is known to have a variety of functions in addition to its role as an alternative splicing factor and its proto-oncogenic potential is most likely due to a combinatorial effect of these functions. Several mechanisms have been identified that contribute to its pro-oncogenic activity. SRSF1 has been shown to activate the mTORC1 signaling pathway by bypassing upstream components such as AKT.Citation67,68 In addition, blockade of mTORC1 can abolish the transformation effect of SRSF1 in mouse immortal fibroblasts. SRSF1 has also been shown to activate the Ras-MAPK pathway by increasing the expression of B-Raf, and the RRM1 domain of SRSF1 is necessary for this activation.Citation69
Some of the splicing functions of SRSF1 have been shown to contribute directly to its pro-oncogenic activity. SRSF1 promotes skipping of exon 4 in the CCND1 transcript to generate the oncogenic cyclin D1b isoform in prostate cancer.Citation70 SRSF1 switches the splicing pattern of BIN1 in breast cancer to generate the BIN1+12a isoform that no longer binds MYC.Citation41 The BIN1 protein interacts with the product of the MYC proto-oncogene, suppressing its oncogenic activity. The inclusion of exon 12a thus abolishes the tumor suppressor activity of BIN1 by interfering with its MYC binding. SRSF1 also regulates alternative splicing of the proapoptotic gene BIM. In breast cancer, SRSF1 generates 2 new splicing isoforms of BIM: BIM γ1 and γ2. These isoforms lack exons 2 and 3, which encode the BH3 domain of BIM, and as a result act as antiapoptotic proteins.Citation41 SRSF1 also controls the splicing of caspase-9 in lung cancer.Citation71 Two splice variants, proapoptotic caspase-9a and antiapoptotic caspase-9b, are derived from the CASP9 gene; SRSF1 promotes the generation of caspase-9b. SRSF1 enhances the inclusion of exon 13b of the gene encoding Mnk2.Citation12 Recently, it was shown that SRSF1 altered the ratio of the Mnk2 isoforms in breast cancer cells, reducing production of the Mnk2a isoform and enhancing Mnk2b. The Mnk2a isoform acts as a tumor suppressor by activating the p38-MAPK stress pathway, whereas the Mnk2b isoform cannot activate the p38-MAPK pathway but activates eIF4E phosphorylation and is pro-oncogenic.Citation72 An additional study has found that favored production of the Mnk2b isoform through the action of SRSF1 in pancreatic ductal adenocarcinoma results in resistance to the drug gemcitabine,Citation73 further supporting the contribution of SRSF1 to the cancerous phenotype. Another splicing target of SRSF1 is the RPS6KB1 gene encoding the ribosomal protein S6K1. SRSF1 promotes expression of the short isoform (isoform-2) of S6K1. Whereas S6K1 isoform-1 acts as a tumor suppressor by blocking Ras-induced transformation, the short isoform-2 possesses oncogenic properties by activating mTORC1.Citation74 SRSF1 also regulates alternative splicing of the tyrosine kinase receptor MST1R (also known as RON) and enhances generation of the ΔRON isoform, which is constitutively active as a result of skipping of exon 11. This isoform was documented to enhance motility and invasion in several cell lines.Citation75
SRSF2
Although SRSF2 (known also as SC35) was found to be mutated in many hematopoietic cancer types, not much is known about its role as a tumor promoter or in tumor progression. Nevertheless, there is some experimental evidence supporting its role in cancer. SRSF2 was found to be overexpressed in a panel of neuroendocrine lung tumors. In these cases, SRSF2 contributed to the cancerous phenotype by causing cells to enter S phase. However, this effect is not achieved through splicing, but rather by cooperation with the transcription factor E2F1. SRSF2 is required for E2F1-mediated transcription of S-phase genes such as cyclin E and p45SKP2.Citation76
A direct role for the splicing function of SRSF2 in cancer has also been demonstrated. SRSF2 was found to interfere with alternative splicing of the KLF6 gene, a tumor suppressor. Expression of SRSF2 results in increased generation of the isoform containing exon1a. This exon has an early termination sequence that leads to the production of a protein that lacks the DNA binding domain and thus, unlike wild type KLF6, cannot act as a tumor suppressor.Citation77
SRSF2 has also been shown to have a tumor suppressor role. SRSF2 was found to cooperate with E2F1 to alter VEGF-A splicing. VEGF-A has several splice variants that are proangiogenic and are upregulated in human tumors. However, alternative splicing of exon 8 of VEGFA-A leads to different isoforms of the same length, but with 6 distinct C-terminal amino acids. These isoforms play an antiangiogenic role and are downregulated in some tumors. SRSF2 promotes a shift in the ratio of VEGF isoforms in favor of the antiangiogenic isoforms.Citation78 In addition, SRSF2 cooperates with E2F1 to control the splicing of c-flip, caspases-8 and -9, and Bcl-x, favoring the proapoptotic isoforms.Citation79
SRSF2 also plays a positive role in the response to cisplatin treatment. In this case, cisplatin stabilizes SRSF2 levels, leading to an increase in the caspase 8L isoform that promotes cell death via apoptosis.Citation80 More studies are needed to better understand the pro- and antioncogenic activities of SRSF2.
SRSF3 (SRp20)
The smallest member of the SR family, this protein contributes to cellular proliferation and apoptosis through its RNA processing functions. The role of SRSF3 in disease was reviewed recently;Citation81 in this review we will focus on its involvement in cancer.
SRSF3 was found to be overexpressed in ovarian cancer.Citation82 Knockdown of SRSF3 in ovarian cell lines resulted in inhibition of cell growth and induction of apoptosis through downregulation of Bcl-2.Citation83 SRSF3 overexpression was also detected in other cancer types including lung, breast, stomach, skin, bladder, colon, liver, thyroid, and kidney.Citation15 In vitro experimental systems indicate that SRSF3 is required in order for cells to enter the G2/M phase. SRSF3 expression is also required for tumor maintenance as knockdown of SRSF3 results in a decrease in tumor size in vivo.Citation15 SRSF3 was found to be translocated with BCL6 in follicular lymphoma.Citation84
A direct role for the splicing function of SRSF3 in cancer is seen in the response of cells to treatment with drugs such as doxorubicin. In ovarian cancer, SRSF3 expression correlates with splicing of MRP1, a member of the ATP binding cassette transporter family that is associated with multidrug resistance.Citation85 Some of these spliced isoforms can decrease the effect of treatment with drugs such as doxorubicin, resulting in increased drug resistance.Citation82
On the other hand, a hepatic-specific SRSF3 knockout mouse model developed tumors with aging. This phenomenon was correlated with aberrant splicing patterns of Igf2 and Insr, which led to a more mitogenic profile. In addition, decreased levels of SRSF3 were observed in patients with hepatocellular carcinoma (HCC), strengthening the role of SRSF3 as a tumor suppressor in liver cancer. This intriguing finding suggests distinct roles for the same factor in different tissues, adding more complexity to the already complex field of splicing factors and cancer.
SRSF6 (SRp55)
SRSF6 was found to be amplified in breast, lung, and colon cancer. Overexpression of SRSF6 was able to transform non-cancerous cell lines and cause tumor formation in mice.Citation11 Transgenic mice overexpressing SRSF6 show hyperplasia of the sensitized skin due to dysregulation of tissue homeostasis. This mechanism involves splicing of the extracellular matrix protein tenascin-C, resulting in increased expression of isoforms that are known to play a role in several cancers.Citation42
SRSF9
SRSF9 expression has been found to be elevated in multiple types of cancer such as glioblastoma, colon adenocarcinoma, squamous cell lung carcinoma, and malignant melanoma.Citation86 SRSF9 overexpression was able to transform NIH 3T3 cells to form colonies in soft agar in vitro and to form tumors in nude mice, whereas downregulation of SRSF9 in colon cancer cell lines reduced their colony formation ability.Citation86 The mechanism by which SRSF9 accomplishes this is not known.
The oncogenic activities of hnRNP splicing factors
The heterogeneous nuclear ribonucleoproteins (hnRNPs) are a large family of proteins containing more than 20 members with common structural domains.Citation87 hnRNPs have roles in various cellular processes such as RNA metabolism, DNA repair, telomere biogenesis, cell signaling, and regulation of gene expression at transcriptional, RNA processing, and translational levels. Emerging evidence suggests their involvement in tumor development and progression. More specifically, hnRNPs have been shown to function in proliferation, apoptosis, angiogenesis, and cell invasion.Citation88 In this review we will elaborate on recent studies implicating hnRNP proteins in cancer.
hnRNP A1
hnRNP A1 has been found to be deregulated in various types of cancers, including colon,Citation89 lung,Citation90 and liver,Citation91 usually leading to overexpression of hnRNP A1 mRNA and protein. Expression levels are correlated with increased proliferationCitation92,93 and tumor metabolism.Citation94,95 hnRNP A1 was found to be overexpressed in HCC.Citation91,96 Knockdown of hnRNP A1 in metastatic HCC cells caused a decrease in cell invasion, whereas upregulation of hnRNP A1 in poorly metastatic HCC cells led to a significant increase in their invasive ability. This effect correlated with CD44v6 expression.Citation91 Another study found that hnRNP A1 functions as a tumor promoter in model systems of overexpression in non-tumorigenic liver progenitor cell lines, in part due to increased proliferation.Citation96
hnRNP A1 is required for RON alternative splicing; expression of hnRNP A1 decreases the formation of the ΔRON isoform, which is known to drive epithelial-to-mesenchymal transition (EMT). As a result the cells undergo mesenchymal-to-epithelial transition (MET), which leads to the establishment of secondary tumors.Citation97 The fact that hnRNP A1 and SRSF1 act in opposite manners on RON alternative splicing supports the view that tight regulation of splicing factors is necessary for metastasis of cancer cells.
hnRNP A2/B1
Like hnRNP A1, hnRNP A2/B1 is also deregulated in various types of cancers. hnRNP A2 was found to be overexpressed in breast,Citation98 brain,Citation13 liver,Citation99 lung,Citation100,101 pancreas,Citation102 and GICitation99 cancers. Some controversy exists with regard to the role of hnRNP B1, an isoform of hnRNP A2, in cancer. These isoforms are identical except for 12 amino acids in the N-terminal region.Citation103 hnRNP B1 has also been reported to be overexpressed in some cancers, such as esophagusCitation104 and lung.Citation105-108 However, in contrast to hnRNP A2, forced overexpression of hnRNP B1 in normal mouse liver cells was unable to transform these cells.Citation96
Forced overexpression of hnRNP A2 was able to transform both normal mouse fibroblastCitation13 and normal mouse liver progenitor cells,Citation96 whereas knockdown of hnRNP A2 reduced the cancerous phenotype of brainCitation13 or liverCitation96 cancer cell lines injected into mice. In addition, knockdown of hnRNP A2 induced apoptosis in breast cancer cell linesCitation109 or reduced proliferation in Colo16 and HaCaT cells.Citation110
hnRNP A2 can activate the RAS-MAPK pathway in liver cancer cell lines. This is achieved by favoring formation of the wild type A-Raf isoform and decreasing formation of the dominant negative short A-Raf isoform that lacks the kinase domain.Citation96 In brain cancer, hnRNP A2 is required for development and maintenance of tumors and regulates splicing of the RON tyrosine kinase receptor. hnRNP A2 promotes the formation of the ΔRON isoform, which acts as a constitutively active receptor.Citation13
The hnRNP alternative splicing factors also play a role in the regulation of cancer metabolism. hnRNP A2, hnRNP A1, and polypyrimidine tract-binding protein (PTB, also known as hnRNP I) control splicing of PKM enzyme mRNA,Citation94,111 resulting in 2 mutually exclusive isoforms. The PKM1 isoform, which is generated by the inclusion of exon 9, promotes oxidative phosphorylation. Inclusion of exon 10 generates the PKM2 isoform that promotes aerobic glycolysis and the “Warburg effect." The PKM2 isoform is crucial for rapidly growing cells and is highly expressed during embryogenesis. However, cancer cells were also found to express this isoform.Citation112
Other members of the hnRNP protein family have also been studied in the context of cancer but not much is known about their function.
hnRNP C
hnRNP C has been shown to increase translation of c-myc mRNA via use of an internal ribosome entry site (IRES).Citation113
hnRNP H
This splicing factor was found to be overexpressed in glioblastoma.Citation14 Moreover, hnRNP H was shown to act as an oncogene in this context by controlling the splicing of 2 important genes; hnRNP H controls the splicing of RON to form the ΔRON isoform which is constitutively active, and the splicing of IG20/MADD,Citation14 an adaptor protein that is involved in apoptosis through caspase-8 activity. hnRNP H promotes formation of the MADD product, which mediates cell survival, over the IG20 product, which triggers apoptosis.Citation14 hnRNP H is also overexpressed in head and neck carcinomas. One possible role for hnRNP H in cancer is to block apoptosis, as knockdown of hnRNP H increases cell death via caspase-3 activity.Citation114
One mechanism of transformation by hnRNP H is by regulating the alternative splicing of ARAF, reducing the formation of a short isoform of ARAF that lacks the Ras binding domain and acts as an inhibitor of the Ras-MAPK pathway.Citation115
hnRNP I (PTB1)
This family member was shown to bind the pyrimidine-rich region in introns. PTB1 and its variant nPTB are important regulators of neuronal development and their ratio changes during development.Citation116,117
hnRNP I was found to be overexpressed in ovarian,Citation82,118 brainCitation119 and breast cancers.Citation120 In brain cancer it was found to promote the skipping of an exon in FGF1, leading to the production of a high-affinity receptor.
In ovarian cancer, changes in splicing of MRP1, a member of the ATP binding cassette transporter family that is associated with multidrug resistance, correlated with expression of hnRNP I and SRSF3, suggesting a role for hnRNP I in cancer drug resistance.Citation82
hnRNP M
The hnRNP M gene has been found to be overexpressed in colon carcinoma.Citation121 In addition, hnRNP M was found to promote EMT in breast cancer through alternative splicing of CD44; hnRNP M binds to GU-rich elements in the transcript to favor skipping of exon 8. It has been shown that knockdown of hnRNP M can block EMT in vitro and in vivo. Indeed, hnRNP M levels correlated with a more aggressive breast cancer phenotype and its overexpression promoted metastasis of breast cancer cells into the lungs in an in vivo mouse model.Citation122 Importantly, hnRNP M can compete with another splicing regulator, ESRP1 (epithelial splicing regulatory protein 1), which also binds to GU-rich elements and promotes exon inclusionCitation123 (see below).
hnRNP K
This hnRNP family member was found to be overexpressed in lungCitation124 and liverCitation125 cancers. Moreover, elevation of hnRNP K protein levels in head and neck or oral squamous cell carcinomas can be used as a biomarker for poor prognosis.Citation126
Role of Splicing Factors in EMT and Metastasis
Epithelial-to-mesenchymal transition (EMT) is a key step in tumor progression and metastasis.Citation127 Several studies have shown that this process is regulated at the transcriptional level and several transcription factors that control and execute the EMT program have been identified.Citation128,129 However, it is now becoming clear that regulation at the post-transcriptional level also occurs in EMT, specifically at the level of alternative splicing.Citation130 Several alternative splicing events have been characterized and the roles of the different splice variants in the process of EMT have been studied.Citation123,131-133 RNA deep sequencing methods were used to determine the EMT-driven alternative splicing program in breast cancer.Citation134 In addition to changes in the splicing pattern, the trans-acting proteins responsible for this pattern were identified. One of the best-studied splicing factors in this context is RBFox2. Expression of RBFox2 was shown to be higher during EMT and this factor is responsible for executing splicing of Cttn, Pard3, and DNM2, all of which are known to play a role in EMT.Citation135 Deletion of RBfox2, however, did not prevent EMT, suggesting redundancy in the regulation of the alternative splicing signature. As mentioned above, SRSF1 regulates splicing of the proto-oncogene tyrosine kinase receptor RON, which is involved in several physiological processes such as migration and invasion. Elevated expression of the constitutively active ΔRON splice variant results in enhanced EMT.Citation75 In recent years, additional splicing factors have also been found to regulate the splicing of RON. hnRNP H and hnRNP A2 were found to increase the production of the ΔRon variant in glioblastoma, resulting in a more aggressive phenotype.Citation13,14 In contrast, hnRNP A1 was found to compete with SRSF1 for binding to the same site on RON and prevent skipping of exon 11. In this way, hnRNP A1 contributes to the reverse process of MET.Citation97 MET is thought to be involved in later stages of metastasis, when cells begin to form secondary foci in metastatic organs. In some cases a splicing switch in EMT is controlled by the ESRP proteins, important mediators of the epithelial tissue identity that regulate many epithelial-specific alternative splicing events. The splicing activity of these factors is inhibited during EMT, and they might be considered tumor suppressors.Citation123,136,137 ESRP1 and ESRP2 control the splicing of CD44, FGFR2, CTNND1, and ENAH.Citation138,Citation123
Role of Alternative Splicing in Drug Resistance
One of the greatest problems in cancer treatment is recurrence of the disease and resistance of the tumors to anticancer drugs. In some cases this is due to acquired resistance to chemotherapeutic drugs that initially worked well. Drug resistance has been ascribed to several mechanisms, alternative splicing being one of them. A classic example is prolonged exposure of cancer cells to the BRAF inhibitor vemurafenib, which results in the loss of sensitivity to the drug.Citation139 In approximately 30% of vemurafenib-resistant melanoma tumors the identified mechanism is alternative splicing of BRAF;Citation140,141 skipping of exons 3–8 in the resistant tumors results in increased production of a BRAF isoform that lacks the Ras binding domain and can enhance constitutive dimerization, resulting in activation of the MAPK pathway in a Ras-independent manner. The trans-acting factor responsible for this alternative splicing has yet to be identified and how prolonged exposure to the drug results in this alternative splicing remains unknown.Citation142 Another striking example of the role of alternative splicing in drug resistance is alternative splicing of the androgen receptor (AR) gene in prostate cancer.Citation143 As this cancer is androgen/AR-dependent a first line of treatment is androgen depletion therapy, and indeed a response can be achieved by this method. However, resistance to this treatment may occur and one mechanism by which this is achieved involved alternative splicing of the AR gene that generates a product lacking the ligand binding domain.Citation144
Modulating Alternative Splicing as a Novel Therapeutic Approach
Given that aberrant splicing is one of the characteristic features of cancer it is obvious that the development of therapeutic approaches based on splicing modulation would be the focus of intense research.Citation145 The aim of these approaches is to either alter or inhibit the splicing machinery to convert it to the non-cancerous state, or to degrade the cancer-specific splicing isoforms that contribute to tumor development, maintenance, or progression. Several approaches will be discussed and are outlined in .
Figure 1. Splicing-based therapeutic approaches. The diagram shows a cassette exon splicing event. Three exons (rectangles) and 2 introns (black lines) are shown. The black box in the middle exon represents an exonic splicing enhancer (ESE) sequence that is recognized by the enhancer splicing factor (black SF). The orange box in the middle exon represents an exonic splicing silencer (ESS) sequence that is recognized by the splicing factor silencer (orange SF). SF3B binds to the branch point sequence (BPS) in the intron. (A) No treatment. In this scenario 2 mRNAs can be generated: one in which the middle exon is included in the mRNA (mRNA A) and a second in which the middle exon is skipped (mRNA B). The width of the black arrows indicates that formation of mRNA A is favored. (B) SF3B inhibitor (SSA) prevents binding of SF3B to the BPS, resulting in inhibition of splicing. In this case the intronic sequences are retained in the mRNA and may contain a premature stop codon. This mRNA will be recognized by the NMD machinery and degraded. Alternatively, the premature stop codon will not lead to degradation of the mRNA, but rather to the production of a truncated protein. Another outcome is that the transcript will be retained in the nucleus. SF3B inhibitors act as general inhibitors of splicing and do not target a specific mRNA. (C) Inhibition of specific splicing factors. Small molecules that block activity (e.g., phosphorylation) of a specific SF (in this example a SF that binds to the ESE) will prevent its binding to the cis elements in the pre-mRNA and result in reduced inclusion of the exon. Other silencer SFs will still bind to the pre-mRNA to favor skipping of this exon. As a result, more mRNA B will be generated. (D) ASOs/siRNAs directed against the SF mRNA will lead to degradation of the SF mRNA, and in this case to decreased binding to the ESE resulting in decreased inclusion of the middle exon. Other SFs can still bind to the pre-mRNA and favor skipping and production of mRNA B. (E) Specific ASOs designed to prevent binding of the SF to the cis elements in the mRNA (ESE) will lead to decreased inclusion of the middle exon. Other SFs can still bind to the pre-mRNA and favor production of mRNA B. (F) Specific ASOs designed to prevent binding of the splicing machinery result in skipping of the exon and production of more mRNA B.
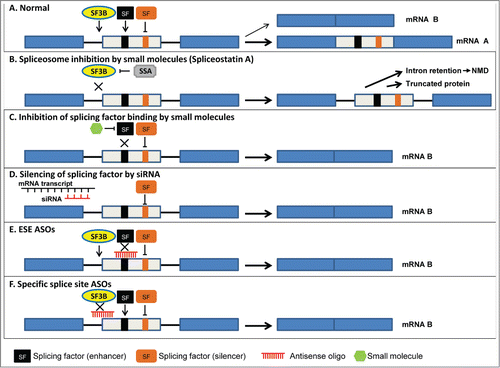
General inhibition of splicing by small molecules
Recent studies have attempted to directly target the spliceosome machinery as a global anticancer treatment.Citation146 Treatment of cells with amiloride, a diuretic agent that affects sodium transport and fluid homeostasis, causes alterations in alternative splicing. Amiloride changed the alternative splicing pattern of several oncogenic genes such as BCL-X, HIPK3, and RON/MISTR1,Citation147 most likely due to hypophosphorylation of the splicing factor SRSF1. Another small molecule used to interfere with splicing is spliceostatin A (SSA). SSA is a derivative of FR901464, a fermentation product of Pseudomonas bacteria. It has been shown that treatment with SSA prolongs the life of tumor-bearing mice.Citation148 SSA binds to splicing factor SF3b Citation149 and thus inhibits splicing in vitro and in cultured cells.Citation149-151 Another closely related molecule, sudemycin E, also has been shown to alter splicing.Citation152 Cells treated with sudemycin E were arrested in G2 and showed increased cell death. It has been suggested that this is due to changes in alternative splicing as well as changes in chromatin condensation.
Inhibition of splicing factor activity by small molecules
Although general inhibition of splicing by small molecules may be effective in reversing aberrant splicing in cancer, non-specific inhibition can lead to cellular toxicity. Therefore, there is a need for small molecules that inhibit only specific alternative splicing events. SR proteins are known to undergo both phosphorylation and dephosphorylation events that control the precise stage of splicing assemblyCitation153 and the splicing reaction.Citation154 Several kinases have been identified that phosphorylate SR proteins, including SRPK,Citation155,156 HhPRP4,Citation157 topoisomerase I,Citation158 Clk,Citation159 and DIRK1A (for a more detailed review see Citation44). Specific inhibition was achieved by inhibition of the kinase activity of topoisomerase I; as a result, phosphorylation of SRSF1 and other SR proteins is reduced and spliceosome assembly does not occur.Citation160 In addition, small molecules have been identified that can inhibit the phosphorylation of specific SR proteins.Citation161
As in the case of topoisomerase I inhibition, reduction of phosphorylation of SR proteins by Clk inhibition has been shown to interfere with the alternative splicing pattern of several target genes.Citation162 Inhibitors of other SR kinases, the SRPK proteins, were shown to reduce phosphorylation of SRSF4 and as a result interfere with viral replication.Citation163 This more specific approach may open up new windows for splicing-based therapeutics. However, more studies need to be performed to ascertain whether these small molecules can be effective, but not toxic.
Targeted modulation of splicing by antisense oligonucleotides
In contrast to global alterations in splicing, a more subtle approach has been established to target only specific splicing events. One method in development is to use complementary sequences to the splice site to redirect the splicing machinery from the specific splice site.Citation164 This approach is based on the fact that trans-acting factors recognize and bind to specific cis elements on the mRNA to execute splicing. Antisense oligonucleotides (ASO) are synthetic nucleic acids (15–25 bases) that can bind cis elements on the mRNA through base pairing.Citation145,165 ASOs can be designed to either block factor binding to a cis element, preventing splicing from occurring at that specific site and resulting in enhanced inclusion of a specific exon, or to target splicing enhancers or silencers to either block or promote splicing. This approach has been used with some success, mainly in the treatment of neurodegenerative diseases such as Duchenne muscular dystrophyCitation166 and spinal muscular atrophy.Citation167,168
Targeting the cis elements by ASOs has major advantages over targeting the splicing factors directly. When protein expression of a specific factor is inhibited all of its biological functions are affected, including splicing, transport, or translation. Introduction of ASOs into the cells only affects the targeted mRNA and other cell functions remain unaffected. However, there are some disadvantages to this approach. One disadvantage is that the ASOs have to be absorbed by the target tissue/cells at a very high efficiency. This difficulty has been overcome in the treatment of neurodegenerative disease with ASOs.Citation169 Cancer treatment based on splicing modulation has also been attempted. To date, several targets have been studied, such as RON,Citation170 PKM,Citation171 Mdm,Citation172 mcl-1,Citation173 FGFR1,Citation174 and BCL-X.Citation175 For a further review on ASO-based treatment please see.Citation145
Summary and Future Scope
Splicing is a complex process requiring a combinatorial network of cis-acting elements and trans-acting factors. Elucidating the roles of the trans-acting splicing factors in cancer development and progression is challenging; these factors are involved in almost every layer of cellular function, they are ubiquitously expressed in all tissues with differential roles in different tissues,Citation87,176,177 and they regulate many steps of RNA processing including splicing, mRNA transport, mRNA stability, nonsense-mediated decay, and miRNA biogenesis.Citation60,62-64,178,179 In the past few years, it has become clear that splicing factors are major drivers of cancer initiation and progression. The involvement of splicing factors in multiple steps of RNA processing and the redundancy of activities among some of these factors makes it difficult to decipher the precise constellation of factors necessary for a specific splicing decision and for cancer development. Another hurdle in understanding the role of splicing factors in normal physiological conditions is the lack of good animal model systems. Mice that lack the alternative splicing factors SRSF1, SRSF2, or SRSF3 are non-viableCitation180-182 and therefore there is a need for more sophisticated mouse model systems such as inducible and/or tissue specific knockouts.Citation183 There is also a need for transgenic mice model systems that overexpress splicing factors. One recent example is the inducible transgenic SRSF6 mouse model, in which overexpression of this oncogenic SR protein was shown to induce skin hyperplasia.Citation42 In vitro modulation, such as overexpression or knockdown, is more common and has been used extensively, but has limitations. For example, the expression of some splicing factors is tightly regulated, with some factors auto-regulating their own expression. New techniques, such as cross-linking immunoprecipitation (CLIP) and modifications of CLIP, have been established to identify mRNA targets of splicing factors and characterize their cis-acting sequences.Citation184-192 Results of these studies will surely be applied to cancer research. The recent identification of recurrent mutations in spliceosomal componentsCitation16-22 reinforces the recognition of splicing factors as important drivers of cancer development and progression and as promising targets for the development of anticancer drugs. In this regard, newly identified alternative splicing events that contribute to cancer initiation and/or progression present promising targets for splicing modulation using modified antisense RNA oligos as described above. Resources such as The Cancer Genome Atlas (TCGA), which contains RNA-seq data from hundreds of tumors and corresponding normal tissues, will contribute to the identification of new alternative splicing events that drive tumor formation and maintenance. Modulation of these splicing events by antisense oligonucleotides or small molecules presents a new approach for cancer therapy (). Moreover, over the past few years increasing lines of evidence have suggested that splicing factors can act as driving oncogenes in several types of cancer, and their inhibition might be a new avenue in cancer therapy (). Although less well established, certain splicing factors can probably act as tumor suppressors through mechanisms that are yet to be elucidated. The field of alternative splicing is entering an exciting new era in which its implication in human diseases, and specifically in cancer research and treatment, is becoming increasingly important.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors wish to thank the Karni lab for helpful discussions.
Funding
This work was supported by the Israeli Science Foundation (ISF Grant no. 1290/12 to R.K.), Israel-US Bi-national Science Foundation (BSF Grant no. 2009026) and the Len & Susan Mark Initiative for Ovarian and Uterine/MMMT Cancers from the Israel Cancer Research Fund (ICRF).
References
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008; 40:1413-5; PMID:18978789; http://dx.doi.org/10.1038/ng.259
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470-6; PMID:18978772; http://dx.doi.org/10.1038/nature07509
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 2009; 10:741-54; PMID:19773805
- Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136, 701-18; PMID:19239890; http://dx.doi.org/10.1016/j.cell.2009.02.009
- Xu Q, Lee C. Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res 2003; 31:5635-43; PMID:14500827; http://dx.doi.org/10.1093/nar/gkg786
- Hui L, Zhang X, Wu X, Lin Z, Wang Q, Li Y, Hu G. Identification of alternatively spliced mRNA variants related to cancers by genome-wide ESTs alignment. Oncogene 2004; 23:3013-23; PMID:15048092; http://dx.doi.org/10.1038/sj.onc.1207362
- Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, et al. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol 2009; 16:670-6; PMID:19448617; http://dx.doi.org/10.1038/nsmb.1608
- Thorsen K, Sørensen KD, Brems-Eskildsen AS, Modin C, Gaustadnes M, Hein AM, Kruhøffer M, Laurberg S, Borre M, Wang K, et al. Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Mol Cell Proteomics 2008; 7:1214-24; PMID:18353764; http://dx.doi.org/10.1074/mcp.M700590-MCP200
- Guo X, Chen QR, Song YK, Wei JS, Khan J. Exon array analysis reveals neuroblastoma tumors have distinct alternative splicing patterns according to stage and MYCN amplification status. BMC Med Genomics 2011; 4:35; PMID:21501490; http://dx.doi.org/10.1186/1755-8794-4-35
- Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays 2006; 28:378-86; PMID:16547952; http://dx.doi.org/10.1002/bies.20390
- Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, Andersen CL, Thorsen K, Ørntoft TF, Mu D, Karni R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol 2013; 229:630-9; PMID:23132731; http://dx.doi.org/10.1002/path.4129
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2ASF is a proto-oncogene. Nat Struct Mol Biol 2007; 14:185-93; PMID:17310252; http://dx.doi.org/10.1038/nsmb1209
- Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakàcs A, Coppola L, Karni R. Splicing factor hnRNP A2B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 2011; 71:4464-72; PMID:21586613; http://dx.doi.org/10.1158/0008-5472.CAN-10-4410
- Lefave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, Pan YX, Cartegni L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J 2011; 30:4084-97; PMID:21915099; http://dx.doi.org/10.1038/emboj.2011.259
- Jia R, Li C, McCoy JP, Deng CX, Zheng ZM. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci 2010; 6:806-26; PMID:21179588; http://dx.doi.org/10.7150/ijbs.6.806
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011; 478:64-9; PMID:21909114; http://dx.doi.org/10.1038/nature10496
- Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013; 368:2059-74; PMID:23634996; http://dx.doi.org/10.1056/NEJMoa1301689
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012; 150:1107-20; PMID:22980975; http://dx.doi.org/10.1016/j.cell.2012.08.029
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 2013; 45:133-5; PMID:23313955; http://dx.doi.org/10.1038/ng.2523
- Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61-70; PMID:23000897; http://dx.doi.org/10.1038/nature11412
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 2011; 365:1384-95; PMID:21995386; http://dx.doi.org/10.1056/NEJMoa1103283
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012; 119:3203-10; PMID:22323480; http://dx.doi.org/10.1182/blood-2011-12-399774
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 2003; 12:525-32; PMID:14536091; http://dx.doi.org/10.1016/j.molcel.2003.08.001
- Munoz MJ, Pérez Santangelo MS, Paronetto MP, de la Mata M, Pelisch F, Boireau S, Glover-Cutter K, Ben-Dov C, Blaustein M, Lozano JJ, et al. DNA damage regulates alternative splicing through inhibition of RNA polymerase II elongation. Cell 2009; 137:708-20; PMID:19450518; http://dx.doi.org/10.1016/j.cell.2009.03.010
- Dujardin G, Lafaille C, de la Mata M, Marasco LE, Muñoz MJ, Le Jossic-Corcos C, Corcos L, Kornblihtt AR. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell 2014; 54:683-90; PMID:24793692; http://dx.doi.org/10.1016/j.molcel.2014.03.044
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science 2010; 327:996-1000; PMID:20133523; http://dx.doi.org/10.1126/science.1184208
- Luco RF, Misteli T. More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr Opin Genet Dev 2011; 21:366-72; PMID:21497503; http://dx.doi.org/10.1016/j.gde.2011.03.004
- Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 2007; 450:893-8; PMID:17982442; http://dx.doi.org/10.1038/nature06358
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061-8; PMID:18772890; http://dx.doi.org/10.1038/nature07385
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455:1069-75; PMID:18948947; http://dx.doi.org/10.1038/nature07423
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314:268-74; PMID:16959974; http://dx.doi.org/10.1126/science.1133427
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 2012; 44:53-7; PMID:22158538; http://dx.doi.org/10.1038/ng.1031
- Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, O'Keefe C, Rogers HJ, Sekeres MA, Maciejewski JP, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 2012; 26:542-5; PMID:21886174; http://dx.doi.org/10.1038/leu.2011.232
- Lasho TL, Jimma T, Finke CM, Patnaik M, Hanson CA, Ketterling RP, Pardanani A, Tefferi A. SRSF2 mutations in primary myelofibrosis: significant clustering with IDH mutations and independent association with inferior overall and leukemia-free survival. Blood 2012; 120:4168-71; PMID:22968464; http://dx.doi.org/10.1182/blood-2012-05-429696
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 2011; 365:2497-506; PMID:22150006; http://dx.doi.org/10.1056/NEJMoa1109016
- Hirabayashi S, Flotho C, Moetter J, Heuser M, Hasle H, Gruhn B, Klingebiel T, Thol F, Schlegelberger B, Baumann I, et al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood 2012; 119:e96-9; PMID:22238327; http://dx.doi.org/10.1182/blood-2011-12-395087
- Wan Y, Wu CJ. SF3B1 mutations in chronic lymphocytic leukemia. Blood 2013; 121:4627-34; PMID:23568491; http://dx.doi.org/10.1182/blood-2013-02-427641
- Scott LM, Rebel VI. Acquired mutations that affect pre-mRNA splicing in hematologic malignancies and solid tumors. J Natl Cancer Inst 2013; 105:1540-9; PMID:24052622; http://dx.doi.org/10.1093/jnci/djt257
- Yoshida K, Ogawa S. Splicing factor mutations and cancer. WIREs RNA, 2014, 5:445-459.
- Cazzola M, Rossi M, Malcovati L. Biologic and clinical significance of somatic mutations of SF3B1 in myeloid and lymphoid neoplasms. Blood 2013; 121:260-9; PMID:23160465; http://dx.doi.org/10.1182/blood-2012-09-399725
- Anczukow O, Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 2012; 19:220-8; PMID:22245967; http://dx.doi.org/10.1038/nsmb.2207
- Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol 2014; 21:189-97; PMID:24440982; http://dx.doi.org/10.1038/nsmb.2756
- Hu J, Ho AL, Yuan L, Hu B, Hua S, Hwang SS, Zhang J, Hu T, Zheng H, Gan B, et al. From the Cover: Neutralization of terminal differentiation in gliomagenesis. Proc Natl Acad Sci U S A 2013; 110:14520-7; PMID:23918370; http://dx.doi.org/10.1073/pnas.1308610110
- Naro C, Sette C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int J Cell Biol 2013; 2013:151839; PMID:24069033; http://dx.doi.org/10.1155/2013/151839
- Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem 2008; 283:1223-7; PMID:18024427; http://dx.doi.org/10.1074/jbc.R700034200
- Shin C, Manley JL. The SR protein SRp38 represses splicing in M phase cells. Cell 2002; 111:407-17; PMID:12419250; http://dx.doi.org/10.1016/S0092-8674(02)01038-3
- Feng Y, Chen M, Manley JL. Phosphorylation switches the general splicing repressor SRp38 to a sequence-specific activator. Nat Struct Mol Biol 2008; 15:1040-8; PMID:18794844; http://dx.doi.org/10.1038/nsmb.1485
- van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, Cáceres JF. The MKK(36)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. J Cell Biol 2000; 149:307-16; PMID:10769024; http://dx.doi.org/10.1083/jcb.149.2.307
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 2012; 47:422-33; PMID:22727668; http://dx.doi.org/10.1016/j.molcel.2012.05.014
- Bielli P, Busa R, Paronetto MP, Sette C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr Relat Cancer 2011; 18:R91-R102; PMID:21565971; http://dx.doi.org/10.1530/ERC-11-0041
- Elliott DJ, Rajan P. The role of the RNA-binding protein Sam68 in mammary tumourigenesis. J Pathol 2010; 222:223-6; PMID:20730808; http://dx.doi.org/10.1002/path.2753
- Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 2013; 52:720-33; PMID:24332178; http://dx.doi.org/10.1016/j.molcel.2013.11.010
- Sutherland LC, Wang K, Robinson AG. RBM5 as a putative tumor suppressor gene for lung cancer. J Thorac Oncol 2010; 5:294-8; PMID:20186023; http://dx.doi.org/10.1097/JTO.0b013e3181c6e330
- Lin JC, Lin CY, Tarn WY, Li FY. Elevated SRPK1 lessens apoptosis in breast cancer cells through RBM4-regulated splicing events. RNA 2014; 20:1621-31; PMID:25140042; http://dx.doi.org/10.1261/rna.045583.114
- Lopez de Silanes I, Fan J, Yang X, Zonderman AB, Potapova O, Pizer ES, Gorospe M. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 2003; 22:7146-54; PMID:14562043; http://dx.doi.org/10.1038/sj.onc.1206862
- Denkert C, Weichert W, Pest S, Koch I, Licht D, Köbel M, Reles A, Sehouli J, Dietel M, Hauptmann S. Overexpression of the embryonic-lethal abnormal vision-like protein HuR in ovarian carcinoma is a prognostic factor and is associated with increased cyclooxygenase 2 expression. Cancer Res 2004; 64:189-95; PMID:14729623; http://dx.doi.org/10.1158/0008-5472.CAN-03-1987
- Heinonen M, Bono P, Narko K, Chang SH, Lundin J, Joensuu H, Furneaux H, Hla T, Haglund C, Ristimäki A. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res 2005; 65:2157-61; PMID:15781626; http://dx.doi.org/10.1158/0008-5472.CAN-04-3765
- Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 2009; 417:15-27; PMID:19061484; http://dx.doi.org/10.1042/BJ20081501
- Zhang Z, Krainer AR. Involvement of SR proteins in mRNA surveillance. Mol Cell 2004; 16:597-607; PMID:15546619; http://dx.doi.org/10.1016/j.molcel.2004.10.031
- Twyffels L, Gueydan C, Kruys V. Shuttling SR proteins: more than splicing factors. FEBS J 2011; 278:3246-55; PMID:21794093; http://dx.doi.org/10.1111/j.1742-4658.2011.08274.x
- Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev 2004; 18:755-68; PMID:15082528; http://dx.doi.org/10.1101/gad.286404
- Li X, Manley JL. Inactivation of the SR protein splicing factor ASFSF2 results in genomic instability. Cell 2005; 122:365-78; PMID:16096057; http://dx.doi.org/10.1016/j.cell.2005.06.008
- Li X, Wang J, Manley JL. Loss of splicing factor ASFSF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev 2005; 19:2705-14; PMID:16260492; http://dx.doi.org/10.1101/gad.1359305
- Wu H, Sun S, Tu K, Gao Y, Xie B, Krainer AR, Zhu J. A splicing-independent function of SF2ASF in microRNA processing. Mol Cell 2010; 38:67-77; PMID:20385090; http://dx.doi.org/10.1016/j.molcel.2010.02.021
- Das S, Krainer AR. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res 2014; 12:1195-204; PMID:24807918; http://dx.doi.org/10.1158/1541-7786.MCR-14-0131
- Gout S, Brambilla E, Boudria A, Drissi R, Lantuejoul S, Gazzeri S, Eymin B. Abnormal expression of the pre-mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non small cell lung carcinoma. PLoS One 2012; 7:e46539; PMID:23071587; http://dx.doi.org/10.1371/journal.pone.0046539
- Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2ASF activates mTORC1. Proc Natl Acad Sci USA 2008; 105:15323-7; PMID:18832178; http://dx.doi.org/10.1073/pnas.0801376105
- Michlewski G, Sanford JR, Caceres JF. The splicing factor SF2ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell 2008; 30:179-89; PMID:18439897; http://dx.doi.org/10.1016/j.molcel.2008.03.013
- Shimoni-Sebag A, Lebenthal-Loinger I, Zender L, Karni R. RRM1 domain of the splicing oncoprotein SRSF1 is required for MEK1-MAPK-ERK activation and cellular transformation. Carcinogenesis 2013; 34:2498-504; PMID:23843040; http://dx.doi.org/10.1093/carcin/bgt247
- Olshavsky NA, Comstock CE, Schiewer MJ, Augello MA, Hyslop T, Sette C, Zhang J, Parysek LM, Knudsen KE. Identification of ASFSF2 as a critical, allele-specific effector of the cyclin D1b oncogene. Cancer Res 2010; 70:3975-84; PMID:20460515; http://dx.doi.org/10.1158/0008-5472.CAN-09-3468
- Shultz JC, Goehe RW, Murudkar CS, Wijesinghe DS, Mayton EK, Massiello A, Hawkins AJ, Mukerjee P, Pinkerman RL, Park MA, et al. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol Cancer Res 2011; 9:889-900; PMID:21622622; http://dx.doi.org/10.1158/1541-7786.MCR-11-0061
- Maimon A, Mogilevsky M, Shilo A, Golan-Gerstl R, Obiedat A, Ben-Hur V, Lebenthal-Loinger I, Stein I, Reich R, Beenstock J, et al. Mnk2 Alternative Splicing Modulates the p38-MAPK Pathway and Impacts Ras-Induced Transformation. Cell Rep 2014; 7:501-13; PMID:24726367; http://dx.doi.org/10.1016/j.celrep.2014.03.041
- Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R, Delle Fave G, Sette C. Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2eIF4E pathway. Oncogene 2013; 32:2848-57; PMID:22797067; http://dx.doi.org/10.1038/onc.2012.306
- Ben-Hur V, Denichenko P, Siegfried Z, Maimon A, Krainer A, Davidson B, Karni R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep 2013; 3:103-15; PMID:23273915; http://dx.doi.org/10.1016/j.celrep.2012.11.020
- Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, Green MR, Riva S, Biamonti G. Cell motility is controlled by SF2ASF through alternative splicing of the Ron protooncogene. Mol Cell 2005; 20:881-90; PMID:16364913; http://dx.doi.org/10.1016/j.molcel.2005.10.026
- Edmond V, Merdzhanova G, Gout S, Brambilla E, Gazzeri S, Eymin B. A new function of the splicing factor SRSF2 in the control of E2F1-mediated cell cycle progression in neuroendocrine lung tumors. Cell Cycle 2013; 12:1267-78; PMID:23518498; http://dx.doi.org/10.4161/cc.24363
- Shi J, Hu Z, Pabon K, Scotto KW. Caffeine regulates alternative splicing in a subset of cancer-associated genes: a role for SC35. Mol Cell Biol 2008; 28:883-95; PMID:18025108; http://dx.doi.org/10.1128/MCB.01345-07
- Merdzhanova G, Gout S, Keramidas M, Edmond V, Coll JL, Brambilla C, Brambilla E, Gazzeri S, Eymin B. The transcription factor E2F1 and the SR protein SC35 control the ratio of pro-angiogenic versus antiangiogenic isoforms of vascular endothelial growth factor-A to inhibit neovascularization in vivo. Oncogene 2010; 29:5392-403; PMID:20639906; http://dx.doi.org/10.1038/onc.2010.281
- Merdzhanova G, Edmond V, De Seranno S, Van den Broeck A, Corcos L, Brambilla C, Brambilla E, Gazzeri S, Eymin B. E2F1 controls alternative splicing pattern of genes involved in apoptosis through upregulation of the splicing factor SC35. Cell Death Differ 2008; 15:1815-23; PMID:18806759; http://dx.doi.org/10.1038/cdd.2008.135
- Edmond V, Moysan E, Khochbin S, Matthias P, Brambilla C, Brambilla E, Gazzeri S, Eymin B. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J 2011; 30:510-23; PMID:21157427; http://dx.doi.org/10.1038/emboj.2010.333
- Corbo C, Orru S, Salvatore F. SRp20: an overview of its role in human diseases. Biochem Biophys Res Commun 2013; 436:1-5; PMID:23685143; http://dx.doi.org/10.1016/j.bbrc.2013.05.027
- He X, Ee PL, Coon JS, Beck WT. Alternative splicing of the multidrug resistance protein 1ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin Cancer Res 2004; 10:4652-60; PMID:15269137; http://dx.doi.org/10.1158/1078-0432.CCR-03-0439
- He X, Arslan AD, Pool MD, Ho TT, Darcy KM, Coon JS, Beck WT. Knockdown of splicing factor SRp20 causes apoptosis in ovarian cancer cells and its expression is associated with malignancy of epithelial ovarian cancer. Oncogene 2011; 30:356-65; PMID:20856201; http://dx.doi.org/10.1038/onc.2010.426
- Chen W, Itoyama T, Chaganti RS. Splicing factor SRP20 is a novel partner of BCL6 in a t(3;6)(q27;p21) translocation in transformed follicular lymphoma. Genes Chromosomes Cancer 2001; 32:281-4; PMID:11579468; http://dx.doi.org/10.1002/gcc.1191
- Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AM, Deeley RG. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992; 258:1650-4; PMID:1360704; http://dx.doi.org/10.1126/science.1360704
- Fu Y, Huang B, Shi Z, Han J, Wang Y, Huangfu J, Wu W. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing beta-catenin biosynthesis. EMBO Mol Med 2013; 5:737-50; PMID:23592547; http://dx.doi.org/10.1002/emmm.201202218
- Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem 1993; 62:289-321; PMID:8352591; http://dx.doi.org/10.1146/annurev.bi.62.070193.001445
- Carpenter B, MacKay C, Alnabulsi A, MacKay M, Telfer C, Melvin WT, Murray GI. The roles of heterogeneous nuclear ribonucleoproteins in tumour development and progression. Biochim Biophys Acta 2006; 1765:85-100; PMID:16378690
- Ushigome M, Ubagai T, Fukuda H, Tsuchiya N, Sugimura T, Takatsuka J, Nakagama H. Up-regulation of hnRNP A1 gene in sporadic human colorectal cancers. Int J Oncol 2005; 26:635-40; PMID:15703818
- Boukakis G, Patrinou-Georgoula M, Lekarakou M, Valavanis C, Guialis A. Deregulated expression of hnRNP AB proteins in human non-small cell lung cancer: parallel assessment of protein and mRNA levels in paired tumournon-tumour tissues. BMC Cancer 2010; 10:434; PMID:20716340; http://dx.doi.org/10.1186/1471-2407-10-434
- Zhou ZJ, Dai Z, Zhou SL, Fu XT, Zhao YM, Shi YH, Zhou J, Fan J. Overexpression of HnRNP A1 promotes tumor invasion through regulating CD44v6 and indicates poor prognosis for hepatocellular carcinoma. Int J Cancer 2013; 132:1080-9; PMID:22821376; http://dx.doi.org/10.1002/ijc.27742
- Biamonti G, Bassi MT, Cartegni L, Mechta F, Buvoli M, Cobianchi F, Riva S. Human hnRNP protein A1 gene expression. Structural and functional characterization of the promoter. J Mol Biol 1993; 230:77-89; PMID:8383772; http://dx.doi.org/10.1006/jmbi.1993.1127
- Zerbe LK, Pino I, Pio R, Cosper PF, Dwyer-Nield LD, Meyer AM, Port JD, Montuenga LM, Malkinson AM. Relative amounts of antagonistic splicing factors, hnRNP A1 and ASFSF2, change during neoplastic lung growth: implications for pre-mRNA processing. Mol Carcinog 2004; 41:187-96; PMID:15390079; http://dx.doi.org/10.1002/mc.20053
- Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. The alternative splicing repressors hnRNP A1A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci USA 2010; 107:1894-9; PMID:20133837; http://dx.doi.org/10.1073/pnas.0914845107
- David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 2010; 24:2343-64; PMID:21041405; http://dx.doi.org/10.1101/gad.1973010
- Shilo A, Ben Hur V, Denichenko P, Stein I, Pikarsky E, Rauch J, Kolch W, Zender L, Karni R. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development. RNA 2014; 20:505-15; PMID:24572810; http://dx.doi.org/10.1261/rna.042259.113
- Bonomi S, di Matteo A, Buratti E, Cabianca DS, Baralle FE, Ghigna C, Biamonti G. HnRNP A1 controls a splicing regulatory circuit promoting mesenchymal-to-epithelial transition. Nucleic Acids Res 2013; 41:8665-79; PMID:23863836; http://dx.doi.org/10.1093/nar/gkt579
- Zhou J, Allred DC, Avis I, Martínez A, Vos MD, Smith L, Treston AM, Mulshine JL. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2B1 (hnRNP-A2B1) in normal breast and neoplastic breast cancer. Breast Cancer Res Treat 2001; 66:217-24; PMID:11510693; http://dx.doi.org/10.1023/A:1010631915831
- Lee CH, Lum JH, Cheung BP, Wong MS, Butt YK, Tam MF, Chan WY, Chow C, Hui PK, Kwok FS, et al. Identification of the heterogeneous nuclear ribonucleoprotein A2B1 as the antigen for the gastrointestinal cancer specific monoclonal antibody MG7. Proteomics 2005; 5:1160-6; PMID:15759317; http://dx.doi.org/10.1002/pmic.200401159
- Fielding P, Turnbull L, Prime W, Walshaw M, Field JK. Heterogeneous nuclear ribonucleoprotein A2B1 up-regulation in bronchial lavage specimens: a clinical marker of early lung cancer detection. Clin Cancer Res 1999; 5:4048-52; PMID:10632338
- Zhou J, Nong L, Wloch M, Cantor A, Mulshine JL, Tockman MS. Expression of early lung cancer detection marker: hnRNP-A2B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 2001; 34:341-50; PMID:11714531; http://dx.doi.org/10.1016/S0169-5002(01)00254-9
- Yan-Sanders Y, Hammons GJ, Lyn-Cook BD. Increased expression of heterogeneous nuclear ribonucleoprotein A2B1 (hnRNP) in pancreatic tissue from smokers and pancreatic tumor cells. Cancer Lett 2002; 183:215-20; PMID:12065097; http://dx.doi.org/10.1016/S0304-3835(02)00168-4
- Kozu T, Henrich B, Schafer KP. Structure and expression of the gene (HNRPA2B1) encoding the human hnRNP protein A2B1. Genomics 1995; 25:365-71; PMID:7789969; http://dx.doi.org/10.1016/0888-7543(95)80035-K
- Matsuyama S, Goto Y, Sueoka N, Ohkura Y, Tanaka Y, Nakachi K, Sueoka E. Heterogeneous nuclear ribonucleoprotein B1 expressed in esophageal squamous cell carcinomas as a new biomarker for diagnosis. Jpn J Cancer Res 2000; 91:658-63; PMID:10874220; http://dx.doi.org/10.1111/j.1349-7006.2000.tb00996.x
- Wu S, Sato M, Endo C, Sakurada A, Dong B, Aikawa H, Chen Y, Okada Y, Matsumura Y, Sueoka E, et al. hnRNP B1 protein may be a possible prognostic factor in squamous cell carcinoma of the lung. Lung Cancer 2003; 41:179-86; PMID:12871781; http://dx.doi.org/10.1016/S0169-5002(03)00226-5
- Snead DR, Perunovic B, Cullen N, Needham M, Dhillon DP, Satoh H, Kamma H. hnRNP B1 expression in benign and malignant lung disease. J Pathol 2003; 200:88-94; PMID:12692846; http://dx.doi.org/10.1002/path.1292
- Sueoka E, Sueoka N, Goto Y, Matsuyama S, Nishimura H, Sato M, Fujimura S, Chiba H, Fujiki H. Heterogeneous nuclear ribonucleoprotein B1 as early cancer biomarker for occult cancer of human lungs and bronchial dysplasia. Cancer Res 2001; 61:1896-902; PMID:11280744
- Tominaga M, Sueoka N, Irie K, Iwanaga K, Tokunaga O, Hayashi S, Nakachi K, Sueoka E. Detection and discrimination of preneoplastic and early stages of lung adenocarcinoma using hnRNP B1 combined with the cell cycle-related markers p16, cyclin D1, and Ki-67. Lung Cancer 2003; 40:45-53; PMID:12660006; http://dx.doi.org/10.1016/S0169-5002(02)00529-9
- Patry C, Bouchard L, Labrecque P, Gendron D, Lemieux B, Toutant J, Lapointe E, Wellinger R, Chabot B. Small interfering RNA-mediated reduction in heterogeneous nuclear ribonucleoparticule A1A2 proteins induces apoptosis in human cancer cells but not in normal mortal cell lines. Cancer Res 2003; 63:7679-88; PMID:14633690
- He Y, Brown MA, Rothnagel JA, Saunders NA, Smith R. Roles of heterogeneous nuclear ribonucleoproteins A and B in cell proliferation. J Cell Sci 2005; 118:3173-83; PMID:16014382; http://dx.doi.org/10.1242/jcs.02448
- David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010; 463:364-8; PMID:20010808; http://dx.doi.org/10.1038/nature08697
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008; 452:230-3; PMID:18337823; http://dx.doi.org/10.1038/nature06734
- Kim JH, Paek KY, Choi K, Kim TD, Hahm B, Kim KT, Jang SK. Heterogeneous nuclear ribonucleoprotein C modulates translation of c-myc mRNA in a cell cycle phase-dependent manner. Mol Cell Biol 2003; 23:708-20; PMID:12509468; http://dx.doi.org/10.1128/MCB.23.2.708-720.2003
- Rauch J, O'Neill E, Mack B, Matthias C, Munz M, Kolch W, Gires O. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res 2010; 70:1679-88; PMID:20145135; http://dx.doi.org/10.1158/0008-5472.CAN-09-2740
- Rauch J, Moran-Jones K, Albrecht V, Schwarzl T, Hunter K, Gires O, Kolch W. c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res 2011; 71:4664-74; PMID:21512137; http://dx.doi.org/10.1158/0008-5472.CAN-10-4447
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M Jr, Black DL. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 2007; 21:1636-52; PMID:17606642; http://dx.doi.org/10.1101/gad.1558107
- Keppetipola N, Sharma S, Li Q, Black DL. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol 2012; 47:360-78; PMID:22655688; http://dx.doi.org/10.3109/10409238.2012.691456
- He X, Pool M, Darcy KM, Lim SB, Auersperg N, Coon JS, Beck WT. Knockdown of polypyrimidine tract-binding protein suppresses ovarian tumor cell growth and invasiveness in vitro. Oncogene 2007; 26:4961-8; PMID:17310993; http://dx.doi.org/10.1038/sj.onc.1210307
- Jin W, McCutcheon IE, Fuller GN, Huang ES, Cote GJ. Fibroblast growth factor receptor-1 alpha-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res 2000; 60:1221-4; PMID:10728679
- He X, Arslan AD2, Ho TT2, Yuan C3, Stampfer MR4, Beck WT5. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis 2014; 3:e84; PMID:24418892; http://dx.doi.org/10.1038/oncsis.2013.47
- Chen S, Zhang J, Duan L, Zhang Y, Li C, Liu D, Ouyang C, Lu F, Liu X. Identification of HnRNP M as a novel biomarker for colorectal carcinoma by quantitative proteomics. Am J Physiol Gastrointest Liver Physiol 2014; 306:G394-403; PMID:24381081; http://dx.doi.org/10.1152/ajpgi.00328.2013
- Xu Y, Gao XD, Lee JH, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev 2014; 28:1191-203; PMID:24840202; http://dx.doi.org/10.1101/gad.241968.114
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 2009; 33:591-601; PMID:19285943; http://dx.doi.org/10.1016/j.molcel.2009.01.025
- Pino I, Pío R, Toledo G, Zabalegui N, Vicent S, Rey N, Lozano MD, Torre W, García-Foncillas J, Montuenga LM. Altered patterns of expression of members of the heterogeneous nuclear ribonucleoprotein (hnRNP) family in lung cancer. Lung Cancer 2003; 41:131-43; PMID:12871776; http://dx.doi.org/10.1016/S0169-5002(03)00193-4
- Ostrowski J, Bomsztyk K. Nuclear shift of hnRNP K protein in neoplasms and other states of enhanced cell proliferation. Br J Cancer 2003; 89:1493-501; PMID:14562022; http://dx.doi.org/10.1038/sj.bjc.6601250
- Matta A, Tripathi SC, DeSouza LV, Grigull J, Kaur J, Chauhan SS, Srivastava A, Thakar A, Shukla NK, Duggal R, et al. Heterogeneous ribonucleoprotein K is a marker of oral leukoplakia and correlates with poor prognosis of squamous cell carcinoma. Int J Cancer 2009; 125:1398-406; PMID:19548310; http://dx.doi.org/10.1002/ijc.24517
- Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 2008; 14:818-29; PMID:18539112; http://dx.doi.org/10.1016/j.devcel.2008.05.009
- Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A 2007; 104:10069-74; PMID:17537911; http://dx.doi.org/10.1073/pnas.0703900104
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117:927-39; PMID:15210113; http://dx.doi.org/10.1016/j.cell.2004.06.006
- Biamonti G, Bonomi S, Gallo S, Ghigna C. Making alternative splicing decisions during epithelial-to-mesenchymal transition (EMT). Cell Mol 2012; Life Sci 69:2515-26; PMID:22349259; http://dx.doi.org/10.1007/s00018-012-0931-7
- Pino MS, Balsamo M, Di Modugno F, Mottolese M, Alessio M, Melucci E, Milella M, McConkey DJ, Philippar U, Gertler FB, et al. Human Mena+11a isoform serves as a marker of epithelial phenotype and sensitivity to epidermal growth factor receptor inhibition in human pancreatic cancer cell lines. Clin Cancer Res 2008; 14:4943-50; PMID:18676769; http://dx.doi.org/10.1158/1078-0432.CCR-08-0436
- Keirsebilck A, Bonné S, Staes K, van Hengel J, Nollet F, Reynolds A, van Roy F. Molecular cloning of the human p120ctn catenin gene (CTNND1): expression of multiple alternatively spliced isoforms. Genomics 1998; 50:129-46; PMID:9653641; http://dx.doi.org/10.1006/geno.1998.5325
- Jordan P, Brazao R, Boavida MG, Gespach C, Chastre E. Cloning of a novel human Rac1b splice variant with increased expression in colorectal tumors. Oncogene 1999; 18:6835-9; PMID:10597294; http://dx.doi.org/10.1038/sj.onc.1203233
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 2011; 7:e1002218; PMID:21876675; http://dx.doi.org/10.1371/journal.pgen.1002218
- Braeutigam C, Rago L, Rolke A, Waldmeier L, Christofori G, Winter J. The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 2014; 33:1082-92; PMID:23435423; http://dx.doi.org/10.1038/onc.2013.50
- Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, Guo W, Xing Y, Carstens RP. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. EMBO J 2010; 29:3286-300; PMID:20711167; http://dx.doi.org/10.1038/emboj.2010.195
- Warzecha CC, Carstens RP. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin Cancer Biol 2012; 22:417-27; PMID:22548723; http://dx.doi.org/10.1016/j.semcancer.2012.04.003
- Di Modugno F, Iapicca P, Boudreau A, Mottolese M, Terrenato I, Perracchio L, Carstens RP, Santoni A, Bissell MJ, Nisticò P. Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc Natl Acad Sci U S A 2012; 109:19280-5; PMID:23129656; http://dx.doi.org/10.1073/pnas.1214394109
- Solit DB, Rosen N. Towards a unified model of RAF inhibitor resistance. Cancer Discov 2014; 4:27-30; PMID:24402945; http://dx.doi.org/10.1158/2159-8290.CD-13-0961
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 2014; 4:94-109; PMID:24265153; http://dx.doi.org/10.1158/2159-8290.CD-13-0617
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 2014; 4:80-93; PMID:24265155; http://dx.doi.org/10.1158/2159-8290.CD-13-0642
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011; 480:387-90; PMID:22113612[http://dx.doi.org/10.1038/nature10662
- Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocr Relat Cancer 2011; 18:R183-96; PMID:21778211; http://dx.doi.org/10.1530/ERC-11-0141
- Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res 2013; 73:483-9; PMID:23117885; http://dx.doi.org/10.1158/0008-5472.CAN-12-3630
- Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 2012; 11:125-40; PMID:22262036
- Rymond B. Targeting the spliceosome. Nat Chem Biol 2007; 3:533-5; PMID:17710096; http://dx.doi.org/10.1038/nchembio0907-533
- Chang JG, Yang DM, Chang WH, Chow LP, Chan WL, Lin HH, Huang HD, Chang YS, Hung CH, Yang WK. Small molecule amiloride modulates oncogenic RNA alternative splicing to devitalize human cancer cells. PLoS One 2011; 6:e18643; PMID:21694768; http://dx.doi.org/10.1371/journal.pone.0018643
- Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo) 1996; 49:1204-11; PMID:9031665; http://dx.doi.org/10.7164/antibiotics.49.1204
- Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 2007; 3:576-83; PMID:17643111; http://dx.doi.org/10.1038/nchembio.2007.18
- Roybal GA, Jurica MS. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res 2010; 38:6664-72; PMID:20529876; http://dx.doi.org/10.1093/nar/gkq494
- Corrionero A, Minana B, Valcarcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 2011; 25:445-59; PMID:21363963; http://dx.doi.org/10.1101/gad.2014311
- Convertini P, Shen M, Potter PM, Palacios G, Lagisetti C, de la Grange P, Horbinski C, Fondufe-Mittendorf YN, Webb TR, Stamm S. Sudemycin E influences alternative splicing and changes chromatin modifications. Nucleic Acids Res 2014; 42:4947-61; PMID:24623796; http://dx.doi.org/10.1093/nar/gku151
- Xiao SH, Manley JL. Phosphorylation of the ASFSF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev 1997; 11:334-44; PMID:9030686; http://dx.doi.org/10.1101/gad.11.3.334
- Cao W, Jamison SF, Garcia-Blanco MA. Both phosphorylation and dephosphorylation of ASFSF2 are required for pre-mRNA splicing in vitro. RNA 1997; 3:1456-67; PMID:9404896
- Gui JF, Tronchere H, Chandler SD, Fu XD. Purification and characterization of a kinase specific for the serine- and arginine-rich pre-mRNA splicing factors. Proc Natl Acad Sci USA 1994; 91:10824-8; PMID:7526381; http://dx.doi.org/10.1073/pnas.91.23.10824
- Kuroyanagi N, Onogi H, Wakabayashi T, Hagiwara M. Novel SR-protein-specific kinase, SRPK2, disassembles nuclear speckles. Biochem Biophys Res Commun 1998; 242:357-64; PMID:9446799; http://dx.doi.org/10.1006/bbrc.1997.7913
- Kojima T, Zama T, Wada K, Onogi H, Hagiwara M. Cloning of human PRP4 reveals interaction with Clk1. J Biol Chem 2001; 276:32247-56; PMID:11418604; http://dx.doi.org/10.1074/jbc.M103790200
- Rossi F, Labourier E, Forné T, Divita G, Derancourt J, Riou JF, Antoine E, Cathala G, Brunel C, Tazi J. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature 1996; 381:80-2; PMID:8609994; http://dx.doi.org/10.1038/381080a0
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The ClkSty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J 1996; 15:265-75; PMID:8617202
- Pilch B, Allemand E, Facompré M, Bailly C, Riou JF, Soret J, Tazi J. Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res 2001; 61:6876-84; PMID:11559564
- Soret J, Bakkour N, Maire S, Durand S, Zekri L, Gabut M, Fic W, Divita G, Rivalle C, Dauzonne D, et al. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors. Proc Natl Acad Sci U S A 2005; 102:8764-9; PMID:15939885; http://dx.doi.org/10.1073/pnas.0409829102
- Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, Sumi K, Yomoda J, Murray MV, Kimura H, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem 2004; 279:24246-54; PMID:15010457; http://dx.doi.org/10.1074/jbc.M314298200
- Fukuhara T, Hosoya T, Shimizu S, Sumi K, Oshiro T, Yoshinaka Y, Suzuki M, Yamamoto N, Herzenberg LA, Herzenberg LA, et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc Natl Acad Sci U S A 2006; 103:11329-33; PMID:16840555; http://dx.doi.org/10.1073/pnas.0604616103
- Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc Natl Acad Sci U S A 1993; 90:8673-7; PMID:8378346; http://dx.doi.org/10.1073/pnas.90.18.8673
- Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 2010; 50:259-93; PMID:20055705; http://dx.doi.org/10.1146/annurev.pharmtox.010909.105654
- Arechavala-Gomeza V, Anthony K, Morgan J, Muntoni F. Antisense oligonucleotide-mediated exon skipping for Duchenne muscular dystrophy: progress and challenges. Curr Gene Ther 2012; 12:152-60; PMID:22533380; http://dx.doi.org/10.2174/156652312800840621
- Porensky PN, Burghes AH. Antisense oligonucleotides for the treatment of spinal muscular atrophy. Hum Gene Ther 2013; 24:489-98; PMID:23544870; http://dx.doi.org/10.1089/hum.2012.225
- Rigo F, Hua Y, Krainer AR, Bennett CF. Antisense-based therapy for the treatment of spinal muscular atrophy. J Cell Biol 2012; 199:21-5; PMID:23027901; http://dx.doi.org/10.1083/jcb.201207087
- Siva K, Covello G, Denti MA. Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid Ther 2014; 24:69-86; PMID:24506781; http://dx.doi.org/10.1089/nat.2013.0461
- Ghigna C, De Toledo M, Bonomi S, Valacca C, Gallo S, Apicella M, Eperon I, Tazi J, Biamonti G. Pro-metastatic splicing of Ron proto-oncogene mRNA can be reversed: therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol 2010; 7:495-503; PMID:20864806; http://dx.doi.org/10.4161/rna.7.4.12744
- Wang Z, Jeon HY, Rigo F, Bennett CF, Krainer AR. Manipulation of PK-M mutually exclusive alternative splicing by antisense oligonucleotides. Open Biol 2012; 2:120133; PMID:23155487; http://dx.doi.org/10.1098/rsob.120133
- Shiraishi T, Eysturskarth J, Nielsen PE. Modulation of mdm2 pre-mRNA splicing by 9-aminoacridine-PNA (peptide nucleic acid) conjugates targeting intron-exon junctions. BMC Cancer 2010; 10:342; PMID:20591158; http://dx.doi.org/10.1186/1471-2407-10-342
- Shieh JJ, Liu KT, Huang SW, Chen YJ, Hsieh TY. Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J Invest Dermatol 2009; 129:2497-506; PMID:19369967; http://dx.doi.org/10.1038/jid.2009.83
- Bruno IG, Jin W, Cote GJ. Correction of aberrant FGFR1 alternative RNA splicing through targeting of intronic regulatory elements. Hum Mol Genet 2004; 13:2409-20; PMID:15333583; http://dx.doi.org/10.1093/hmg/ddh272
- Mercatante DR, Mohler JL, Kole R. Cellular response to an antisense-mediated shift of Bcl-x pre-mRNA splicing and antineoplastic agents. J Biol Chem 2002; 277:49374-82; PMID:12381725; http://dx.doi.org/10.1074/jbc.M209236200
- Hanamura A, Caceres JF, Mayeda A, Franza BR Jr, Krainer AR. Regulated tissue-specific expression of antagonistic pre-mRNA splicing factors. RNA 1998; 4:430-44; PMID:9630249
- Zahler AM, Neugebauer KM, Lane WS, Roth MB. Distinct functions of SR proteins in alternative pre-mRNA splicing. Science 1993; 260:219-22; PMID:8385799; http://dx.doi.org/10.1126/science.8385799
- Chaudhury A, Chander P, Howe PH. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1's multifunctional regulatory roles. RNA 2010; 16:1449-62; PMID:20584894; http://dx.doi.org/10.1261/rna.2254110
- Han SP, Tang YH, Smith R. Functional diversity of the hnRNPs: past, present and perspectives. Biochem J 2010; 430:379-92; PMID:20795951; http://dx.doi.org/10.1042/BJ20100396
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR Jr, Ye Z, Liu F, et al. ASFSF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 2005; 120:59-72; PMID:15652482; http://dx.doi.org/10.1016/j.cell.2004.11.036
- Wang HY, Xu X, Ding JH, Bermingham JR Jr, Fu XD. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell 2001; 7:331-42; PMID:11239462; http://dx.doi.org/10.1016/S1097-2765(01)00181-2
- Jumaa H, Wei G, Nielsen PJ. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr Biol 1999; 9:899-902; PMID:10469594; http://dx.doi.org/10.1016/S0960-9822(99)80394-7
- Sen S, Jumaa H, Webster NJ. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun 2013; 4:1336; PMID:23299886; http://dx.doi.org/10.1038/ncomms2342
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 2010; 17:909-15; PMID:20601959; http://dx.doi.org/10.1038/nsmb.1838
- Ule J, Stefani G, Mele A, Ruggiu M, Wang X, Taneri B, Gaasterland T, Blencowe BJ, Darnell RB. An RNA map predicting Nova-dependent splicing regulation. Nature 2006; 444:580-6; PMID:17065982; http://dx.doi.org/10.1038/nature05304
- Jangi M, Boutz PL, Paul P, Sharp PA. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev 2014; 28:637-51; PMID:24637117; http://dx.doi.org/10.1101/gad.235770.113
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 2009; 16:130-7; PMID:19136955; http://dx.doi.org/10.1038/nsmb.1545
- Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 2009; 36:996-1006; PMID:20064465; http://dx.doi.org/10.1016/j.molcel.2009.12.003
- Sanford JR, Coutinho P, Hackett JA, Wang X, Ranahan W, Caceres JF. Identification of nuclear and cytoplasmic mRNA targets for the shuttling protein SF2ASF. PLoS One 2008; 3:e3369; PMID:18841201; http://dx.doi.org/10.1371/journal.pone.0003369
- Anko ML, Müller-McNicoll M, Brandl H, Curk T, Gorup C, Henry I, Ule J, Neugebauer KM. The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol 2012; 13:R17; PMID:22436691; http://dx.doi.org/10.1186/gb-2012-13-3-r17
- Rossbach O, Hung LH, Khrameeva E, Schreiner S, König J, Curk T, Zupan B, Ule J, Gelfand MS, Bindereif A. Crosslinking-immunoprecipitation (iCLIP) analysis reveals global regulatory roles of hnRNP L. RNA Biol 2014; 11:146-55; PMID:24526010; http://dx.doi.org/10.4161/rna.27991
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 2012; 1:167-78; PMID:22574288; http://dx.doi.org/10.1016/j.celrep.2012.02.001
- Rauch J, Ahlemann M, Schaffrik M, Mack B, Ertongur S, Andratschke M, Zeidler R, Lang S, Gires O. Allogenic antibody-mediated identification of head and neck cancer antigens. Biochem Biophys Res Commun 2004; 323:156-62; PMID:15351715; http://dx.doi.org/10.1016/j.bbrc.2004.08.071