
Abstract
The term cancer describes a group of multifaceted diseases characterized by an intricate pathophysiology. Despite significant advances in the fight against cancer, it remains a key public health concern and burden on societies worldwide. Elucidation of key molecular and cellular mechanisms of oncogenic diseases will facilitate the development of better intervention strategies to counter or prevent tumor development. In vivo and in vitro models have long been used to delineate distinct biological processes involved in cancer such as apoptosis, proliferation, angiogenesis, invasion, metastasis, genome instability, and metabolism. In this review, we introduce Caenorhabditis elegans as an emerging animal model for systematic dissection of the molecular basis of tumorigenesis, focusing on the well-established processes of apoptosis and autophagy. Additionally, we propose that C. elegans can be used to advance our understanding of cancer progression, such as deregulation of energy metabolism, stem cell reprogramming, and host–microflora interactions.
Abbreviations
| CSCs | = | cancer stem cells |
| DSBs | = | double-strand breaks |
| DTC | = | distal tip cells |
| EMT | = | epithelial-to-mesenchymal transition |
| GSC | = | germ stem cell |
| ncRNAs | = | non-coding RNAs |
| NER | = | nucleotide excision repair |
| NSCLC | = | non-small cell lung cancer |
| ROS | = | reactive oxygen species |
| RTK | = | receptor tyrosine kinase |
Introducing the Nematode Caenorhabditis elegans: a Compelling Model for the Study of Cancer
Caenorhabditis elegans is a non-parasitic soil nematode that feeds on various bacteria. It can also be easily raised in large numbers in the laboratory on agar plates or in liquid medium using Escherichia coli bacteria. It is one of the simplest multicellular organisms, existing mainly as hermaphrodite although males arise occasionally at a frequency of 0.1%. It has a short generation time of 3.5 d at 20°C and a short lifespan of about 2–3 weeks under favorable conditions. A wild-type hermaphrodite produces approximately 300 progeny by self-fertilization and over 1,000 progeny when fertilized by a male. “Female” and male mature adults contain an invariant number of 959 and 1,031 somatic cells, respectively, with an invariant cell lineage and precise anatomical arrangements. Because of its transparent body at all stages of its life cycle and its small size, C. elegans lends itself to non-invasive optical methodologies that enable manipulation and tracking of normal function and dysfunction at the cellular level during development and aging. Interestingly, although nematodes and humans are separated by almost a billion years of evolution, C. elegans homologs have been identified for 60–80% of human genes and many biological processes, including apoptosis, cell signaling, cell cycle, cell polarity, metabolism, and aging, are conserved between C. elegans and mammals.Citation1 In addition, the ease with which forward and reverse genetics can be applied have led to refined genetic dissection of pathways that regulate development and aging. Collectively, these features make C. elegans an ideal model enabling a systematic approach to the elucidation of genes and pathways involved in diverse pathologies, including neurodegeneration and cancer.
Tumor formation and dissemination are associated with key traits, such as sustained proliferation, immortalization, resisting cell death, genome instability, induction of angiogenesis, invasiveness and metastasis, and deregulated energy metabolism. As emerging findings highlight the importance of the tumor microenvironment, exploring its contribution to tumor growth and metastasis will be crucial for a better understanding of the molecular and physiological requirements of tumorigenesis.Citation2 Taking advantage of the features mentioned above, researchers have successfully used C. elegans to rapidly assess the functional impact of specific gene mutations on tumor development and outcome at the organismal level, and to screen for new anticancer drugs. In this review, we survey new knowledge about cancer development and progression with relevant findings in C. elegans, and discuss the prospects of using the nematode to elucidate the cellular and molecular underpinnings of tumorigenesis. As neoplastic diseases are highly diverse, we focus on worm models that have already fostered a better understanding of the underlying mechanisms relevant to human tumors such as apoptosis and autophagy, and have pioneered the field. Findings from the worm for well-defined processes involved in cancer development including cell cycle progression, invasion, and metastasis will not be extensively covered in this review since ample information is presented in detail elsewhere.Citation3-6 Moreover, we propose that C. elegans could provide valuable insights into cancer development from a less-appreciated perspective, such as cellular metabolism, stem cell reprogramming and dedifferentiation, and host–microflora interactions.
Alterations in Cell Death Pathways and their Implications in Cancer
Different types of cell death have been described based on morphological and biochemical criteria.Citation7 Apoptosis, necrosis, and autophagy are the most frequent modes of cell death. Accumulating findings suggest that, although distinct from each other, cell death mechanisms can effectively crosstalk depending on the cellular context and the initiating stimulus.Citation8 Cell death, which plays pivotal roles in normal development and homeostasis at both the cellular and organismal level, is subjected to tight control and when deregulated contributes to severe pathological conditions.Citation9 Although tumor formation is often linked to the ability of cancer cells to sustain continuous proliferation, it can also result from defects in apoptosis (). It is now clear that evading apoptosis is a core hallmark of cancer cells.Citation2
Figure 1. Apoptosis prevents tumorigenesis. Schematic representation of DNA damage-induced apoptosis that limits tumor growth in mammals and C. elegans. In mammals, activation of the proapoptotic proteins Bax and Bak under stress leads to release of cytochrome c. In turn, cytochrome c interacts with Apaf1, which initiates the activation of a caspase cascade. Defects in apoptosis promote tumorigenesis. In C. elegans, binding of EGL-1 to CED-9/CED-4 complex on the surface of mitochondria releases mitochondrial components allowing CED-4 to activate CED-3. CED-9, C. elegans homolog of antiapoptotic BCL-2; CED-4, C. elegans homolog of apoptotic protease activating factor 1 (APAF-1).
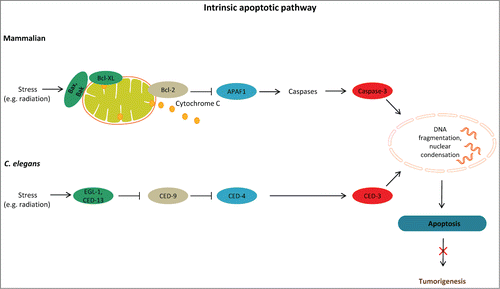
Apoptotic cell death occurs as part of normal development and morphogenesis, but it can also be triggered by a broad range of stimuli, usually mild in nature. Physiological apoptosis in the C. elegans germ line limits the number of oocytes competing for nutrients in the gonad, thereby contributing to tissue homeostasis. Physiological germ cell apoptosis relies on the core apoptotic machinery composed of CED-9, (an antiapoptotic Bcl-2 homolog), CED-4 (a homolog of human APAF-1), and the caspase CED-3, but is independent of CEP-1, a functional homolog of the mammalian p53 tumor suppressor protein. On the other hand, apoptosis induced by genotoxic stress requires both CEP-1 and the proapoptotic BH3-only protein EGL-1.Citation10 Like physiological germ line apoptosis, DNA damage-induced germ cell apoptosis engages the CED-9 (Bcl-2), CED-4 (ApafI), and CED-3 (caspase-3) proteins.Citation10-12 In addition to EGL-1, CED-13 is a conserved BH3-only protein that binds to CED-9 and promotes apoptosis in response to CEP-1 activation.Citation13 Although the BH3-only Bcl-2 family members are key mediators of apoptosis, their mode of action remains elusive.Citation14 Mammalian antiapoptotic Bcl-2–like proteins physically interact with Bax and Bak through the BH3 motifs. It has been shown that Bcl-2 can bind to and suppress the proapoptotic triggering proteins Bax and Bak, thus inhibiting apoptosis and promoting tumor formation. Work over the past decade has produced a considerable body of research establishing that mutations affecting Bcl-2 family members are implicated in cancer.Citation15 Accordingly, overexpression of a prosurvival family member or loss of a proapoptotic Bcl-2 protein can be oncogenic.Citation16,17 Moreover, genes essential for apoptosis confer protection against DNA damage-inducing agents (), including ionizing radiation, or specific anticancer drugs such as cisplatin and camptothecin, both of which block cell proliferation by inducing DNA double-strand breaks (DSBs). An effective response to DSBs is one of the most important mechanisms for maintaining genome integrity and preventing tumorigenesis.Citation18 Finally, apoptosis is activated as part of the nematode innate immunity response against invading pathogens. This pathogenesis-induced germ cell apoptosis also depends on the activity of EGL-1.Citation10
Notably, the cellular and molecular mechanisms that mediate apoptosis are remarkably conserved among metazoans.Citation19 In this regard, C. elegans has proven instrumental in providing critical insights into the mechanisms that link apoptosis to tumor development and anticancer drug resistance. More specifically, several aspects of the C. elegans germ line make it a valuable genetic system for analyzing the cellular and molecular underpinnings of apoptosis and cancer: first, the worm germ line is pluripotent and immortal; second, it is the only tissue in which the pattern of apoptosis is not invariant; and third, it is the only tissue that undergoes apoptosis in adults.Citation10 Furthermore, although cell cycle defects can occur in somatic cells of C. elegans, tumor-like phenotypes have only been observed only in the germ line. When the cell cycle and the apoptotic machinery are compromised, the gonad is filled with mitotic nuclei as a consequence of the expanding stem cell niche, allowing the study of tumor development.
Accumulating findings from studies in C. elegans indicate that longevity-influencing genes can modulate tumor susceptibility through induction of germ cell apoptosis, among other mechanisms. In support of this notion, SIR-2.1, the C. elegans homolog of mammalian SIRT1, which has been implicated in modulation of aging, has also been assigned a proapoptotic activity that is confined to DNA damage-induced apoptosis. Specifically, SIR-2.1 has been shown to translocate from the nucleus to the cytoplasm early during apoptosis. Its transient colocalization with CED-4 provides evidence for a functional interaction between these proteins. The proapoptotic function of SIR-2.1 does not require the DAF-16/FOXO transcription factor whereas, in contrast, the longevity effect of sir-2.1 relies on the activity of DAF-16. Further analysis suggests that sir-2.1 acts in parallel to cep-1 during DNA damage-induced apoptosis.Citation20 Interestingly, CEP-1 contains several conserved residues that are frequently mutated in human tumors, making it an attractive model to study p53-mediated pathways with respect to tumor development.Citation21 In addition, regulators of CEP-1 activity include the well-characterized mediators of tumor development Akt/AKT-1, HIF-1, MPK-1/ERK, and LET-60/Ras.Citation22
Further supporting the molecular link between aging and tumor growth, longevity-promoting mutations, such as mutations in the insulin/insulin-like growth factor-1 (IGF-1) receptor abnormal DAuer Formation 2 (DAF-2), also confer protection against germ line tumors caused by mutations in the gld-1 gene, which encodes an RNA binding protein that contains a K homology (KH) domain and primarily functions as a translational repressor.Citation3 The tumor protective effect of daf-2 inhibition during adulthood depends on the activity of DAF-16/FOXO transcription factor, which in turn upregulates diverse target genes that directly or indirectly stimulate germ line apoptosis and inhibit tumor cell proliferation, thus affecting tumor growth. Tumor suppression by daf-2 inhibition also requires CEP-1. Collectively, these findings suggest that the tumor suppressive effects of daf-2 mutations result from decreased cell division and increased DAF-16/CEP-1–dependent apoptosis within the tumors (). Importantly, mutations in the tumor suppressor gene phosphatase and tensin homolog deleted on chromosome 10 (PTEN) induce tumorigenesis in humans through activation of insulin/IGF-1 signaling and consequent inhibition of FOXO3A (the mammalian homolog of DAF-16), leading to increased tumor cell proliferation and decreased apoptosis. Therefore, the role of the insulin/IGF-1 pathway in tumorigenesis appears to be conserved through evolution.Citation25 Further analysis demonstrated that 29 out of the 734 DAF-16 target genes tested influenced germ cell proliferation or cep-1/p53–dependent apoptosis. Specifically, inactivation of many nuclear pore-related genes blocked genotoxic stress-triggered apoptosis.Citation26 The fact that many of the genes identified as modulators of tumor growth are orthologs to known human tumor suppressors or oncogenes validates use of C. elegans as a cancer model. Additionally, recent evidence indicates that daf-2 mutant animals show retardation of dysplastic age-related uterine growths, which is coupled to the transcriptional abundance of cep-1/p53. Uterine growths composed of abnormal nuclei and large chromatin masses resemble the age-related premalignant lesions frequently observed in mammals.Citation28 Other longevity regulating pathways and regimens, such as inhibition of respiration or caloric restriction, delay tumorigenesis by decreasing tumor cell division without affecting apoptosis. Surprisingly, none of these longevity mutations reduce germ cell proliferation in wild-type animals. Taken together, these findings indicate that lifespan-extending mutations may restrict tumorigenesis.Citation26,27
Figure 2. Linking cell death mechanisms to cancer. Deregulation of cell death mechanisms contributes to pathologic conditions, including cancer. Basal autophagy supports metabolism through recycling of cytoplasmic material and serves a quality control function through protein and organelle turnover. Impaired autophagy leads to accumulation of damaged organelles and protein aggregates, thereby promoting cellular damage and increased vulnerability to disease. Stressful conditions, such as low nutrient availability, energy depletion, hypoxia, and oxidative stress, induce autophagy. Excessive autophagy (for example due to activation of AMPK) can also be detrimental. Longevity-influencing genes have a role in tumor suppression; for example, DAF-16/FOXO target genes can induce tumor cell apoptosis and prevent tumor growth. Arrows indicate stimulatory inputs. Bars indicate inhibitory interactions. For clarity, some of the signaling connections are not shown. FOXO/DAF-16, a forkhead box O(FOXO) transcription factor; HIF-1, hypoxia inducible factor-1; LKB1, serine/threonine protein kinase; TOR, target of rapamycin; TSC1/2, tuberosclerosis complexes 1 and 2; ROS, reactive oxygen species.
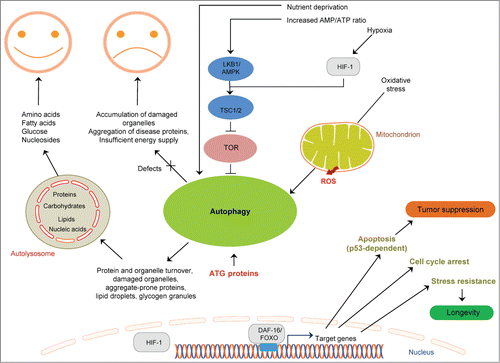
C. elegans has been successfully used to uncover a novel link between hypoxia and apoptosis in tumor progression. A recent study has shown that the worm hypoxia inducible factor-1 (HIF-1) antagonizes the function of CEP-1 in DNA-damage–induced germ cell apoptosis. This inhibitory effect of HIF-1 is mediated to a large part by transcriptional upregulation of the tyrosinase family member TYR-2 in the ASJ neurons. Apoptosis is blocked once TYR-2 enters the gonad. These intriguing findings highlight the important role that 2 single neurons play in the regulation of hypoxic systemic responses. Interestingly, knockdown of the TYR-2 homolog TRP2 in human melanoma cells also increases basal and cisplatin-induced p53 apoptosis, suggesting an evolutionarily conserved link between HIF-1 and apoptosis. This in turn points to TYRP2 as a novel putative target for new treatment strategies against various solid tumors.Citation29
HIF-1 has also been implicated in tumorigenesis induced by loss of the gene encoding the tumor suppressor folliculin (FLCN). Germline mutations in FLCN are associated with the Birt-Hogg-Dubé (BHD) syndrome and an increased cancer risk. FLCN normally binds to and blocks the action of AMP-activated serine/threonine protein kinase (AMPK).Citation30 AMPK is a conserved sensor of cellular energy status that is activated when the cellular AMP/ATP ratio increases under stress conditions such as nutrient deprivation or hypoxia.Citation41 Recent findings indicate that FLCN inhibits tumorigenesis by inhibiting AMPK-dependent HIF-activation. In contrast, loss of FLCN results in activation of the AMPK/PGC-1α/OXPHOS/HIF signaling axis, which has been extensively implicated in the initiation of sporadic tumors in multiple organs.Citation30 It is interesting to note that HIF-1 modulates aging in C. elegans. Specifically, HIF-1 overexpression extends lifespan and enhances resistance to heat and oxidative stress, acting in parallel to DAF-16/FOXO and SKN-1/NRF transcription factors. Surprisingly, loss-of-function mutations in hif-1 also prolong lifespan under laboratory conditions, most likely through different pathways.Citation31
In addition to apoptosis, autophagy has also been implicated in cancer. Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved process through which cytoplasmic components including proteins, lipids, and organelles are engulfed by double-membraned vesicles, the autophagosomes, and delivered to lysosomes for degradation. The resulting breakdown products are exported into the cytoplasm, where they are recycled.Citation32,33 Compelling evidence derived from studies in invertebrate and mammalian models clearly indicates that autophagy plays essential roles under both normal and stress conditions. A basal level of constitutive autophagy is crucial for routine clearance of the cytosol under normal conditions, contributing to protein and organelle homeostasis and thus acting as a quality control mechanism for post-mitotic differentiated cells. Autophagy is also activated in response to various extrinsic and intrinsic stress stimuli, such as low nutrient availability, hypoxia, heat, and reactive oxygen species (ROS), as well as in response to the accumulation of damaged organelles, particularly mitochondria, and proteins, thus acting as a cell survival mechanism.Citation34,35 Accordingly, autophagy inhibition can trigger apoptosis or necrosis in cells that could otherwise survive in a stressful environment.Citation36 Conversely, it is becoming increasingly clear that excessive autophagy can be detrimental.Citation33,37,38 The last decade has witnessed a steady accumulation of findings indicating that autophagy has a key role in the onset and progression of various diseases, including cancer. Several recent studies have revealed that autophagy can either suppress cancer initiation or promote tumor growth in a context-specific manner. Specifically, autophagy can prevent tissue damage, genome instability, and inflammation by maintaining protein and organelle quality control, thereby suppressing tumor initiation. On the other hand, it can promote tumorigenesis by enabling tumor growth and survival through nutrient recycling. Compelling evidence derived from studies in mice has shown that mutant animals with allelic loss of the essential autophagy gene beclin1 show increased occurrence of spontaneous tumors, suggesting a critical role for beclin1 in tumor suppression.Citation39 However, the mechanisms through which autophagy can modulate cancer are only just beginning to be understood. The findings that autophagy-deficient mice accumulate insoluble ubiquitinated proteins, damaged organelles, and lipid droplets and show increased levels of ROS suggest that defective autophagy may cause cellular damage and increased sensitivity to stress, thereby limiting survival (). In fact, these mutant animals show increased vulnerability to disease, including cancer.Citation40 Figuring out how essential components of the autophagy pathway can tip the balance toward either survival or death is critical for the development of autophagy-based anticancer therapeutics.
C. elegans has contributed significant insights into the role of autophagy in cancer. For example, the pathway in which the tumor suppressor FLCN functions was first delineated in nematodes. It has been shown that loss of flcn-1, the C. elegans homolog of FLCN, confers resistance to oxidative stress. This resistance depends on the activity of AAK-2, the worm homolog of AMPK, previously shown to induce autophagy both in mammals and in C. elegans.Citation42 Autophagy induction, in turn, protects against apoptotic cell death and promotes survival under stress. This pathway was shown to be conserved in mammalian cells, suggesting that FCLN prevents tumor formation by negatively regulating the activity of AMPK and consequently preventing AMPK-dependent autophagy activation ().Citation30
A Conserved Signal Transduction Pathway Controlling Vulva Development is Implicated in Tumorigenesis
Receptor tyrosine kinase (RTK)/Ras GTPase/MAP kinase (MAPK) signaling pathways control various biological processes including cellular proliferation and transformation in metazoans. Genetic screens for mutants with vulva defects allowed the identification of several RTK cascade mediators in C. elegans.Citation43-45 LET-23/EGFR and EGL-15/FGFR RTKs stimulate LET-60/Ras and a MAPK cascade consisting of the kinases LIN-45/Raf, MEK-2/MEK, and MPK-1/ERK.Citation46 The let-60 gene, which encodes a C. elegans Ras homolog, plays an important role in inducing vulva formation. Gain-of-function mutations in let-60 are analogous to mutations that constitutively activate mammalian ras leading to dysregulated cell division, thereby contributing to oncogenesis. These mutations result in a multivulva phenotype in the worm whereas partial loss-of-function mutations in let-60 lead to a vulvaless phenotype.Citation47 Ras proteins are small GTP-binding proteins that cycle between a GTP-bound state that is active for signal transduction and an inactive GDP-bound state. One of the 3 human ras alleles is mutated in ∼30% of all tumors, and the incidence of ras mutation in pancreatic cancer is approximately 90%.Citation48,49 Vulval development provides an excellent model system for studying cell proliferation and differentiation, processes that are tightly controlled in normal tissues. Deregulation of cell growth and division and inhibition of cell differentiation favor tumor development.
There are also clear examples of how C. elegans has proved to be a powerful system for investigating the causative relationship between gain-of-function mutations in c-Met receptor tyrosine kinase and lung cancer in a whole organism context. Transgenic worms expressing the most frequently observed mutant proteins c-MetR988C and c-MetT1010I display locomotion defects, significantly reduced fecundity, and abnormal vulva development resulting in hyperplasia. These mutant phenotypes are intensified following nicotine treatment. Taken together, these findings imply that C. elegans is a rewarding model organism for studying the impact of mutations in known human cancer genes and also rapidly assessing gene–environment interactions in the pathogenesis of the disease, given that smoking is the major risk factor for lung cancer.Citation50,51 Overexpression, activation, or unique mutations and sequence variants of c-Met receptor have been linked with non-small cell lung cancer (NSCLC), a disease that is difficult to cure. Inhibition of c-Met in NSCLC cell lines and tumor tissues has been reported to reduce cell viability.Citation52 In light of these encouraging results, new nematode models expressing c-Met alterations in the semaphorin domain and the juxtamembrane domain, which are also implicated in NSCLC, could be generated with the aim of validating c-Met as a therapeutic target in a whole animal setting.
Altered Cellular Energetics and Fat Metabolism in Cancer: Lessons Learned from C. elegans
Cancer development is manifested by alterations in metabolic regulation. The initial observation was made several decades ago by Warburg, who demonstrated that cancer cells exhibit abnormal energy production and utilization toward a favorable carcinogenic outcome. Adaptation of energy metabolism during cancer development is considered a key event and efforts to delineate the underlying mechanisms have attracted much attention over past years. Remarkably, but perhaps not surprisingly, various cancer types are attributed to obesity; however, the complex mechanisms involved remain widely elusive. In recent years, C. elegans has increasingly been used to explore molecular mechanisms related to energy homeostasis, since the most common metabolic pathways and the network of genes that are involved in food sensation, endocrine signaling, nutrient uptake, and transport and storage of fat are conserved. The use of C. elegans can enable a better understanding of both intrinsic and extrinsic mechanisms at an organismal level and ultimately aid the design of better strategies against cancer.
During tumor progression the rapidly proliferating carcinogenic cells require high amounts of energy in order to cope with increased metabolic needs. At the same time, high availability of macromolecules is vital. Apart from the mechanisms described by the Warburg effect, alternative pathways and processes are proposed to endorse cancer. Mitochondrial metabolism is highly implicated in cell division by providing energy and molecules for de novo cell membrane synthesis. Consequently, mitochondria are considered attractive targets against cancer and it is suggested that induction of mitochondrial dysfunction could serve as a way to fight cancer cells with metabolic dysregulation.Citation53 However, the underlying mechanisms remain enigmatic. Pathways that are implicated in mitochondrial repair during mitochondrial dysfunction have been identified in C. elegans. Specifically, a genome-wide RNAi screen revealed 45 genes that are required for mitochondrial repair, detoxification, and pathogen response during compromised mitochondrial function.Citation54 These genes were involved in ceramide and mevalonate metabolism. Deterioration in ceramide generation or inhibition of the mevalonate pathway resulted in compromised mitochondrial surveillance and promoted mitochondrial dysfunction. Interestingly, the mevalonate pathway has been proposed as a target for anticancer therapy, whereas defects in ceramide biosynthesis and metabolism contribute to tumor cell survival and resistance to chemotherapy.Citation55,56 These findings further complicate the already complex mechanisms by which defects in ceramide generation that sustain mitochondrial dysfunction also contribute to tumor survival. Therefore, it is imperative to better understand the exact mechanisms governing mitochondrial function in order to more efficiently treat tumors. Furthermore, in the tumor microenvironment where harsh conditions prevail, mitochondrial mass is altered by mechanisms controlling mitochondrial biogenesis and mitophagy, a selective type of autophagy responsible for mitochondria elimination.Citation53 C. elegans has proved to be a powerful model to further understand such mechanisms from diverse aspects. For example, the mitochondrial prohibitin complex, which has been associated with various cancer types, has been shown to be involved in mitochondrial biogenesis and function, exerting opposing effects in respect to energy metabolism, fat utilization, and aging.Citation57 Finally, during mitochondrial metabolism, ROS are produced as a by-product of oxygen metabolism and their levels determine cell fate in mechanisms best described by mitohormesis.Citation58 ROS are thought to be a double-edged sword for cancer cells since they are required for cancer development, but at the same time can be exploited in order to specifically eliminate cancer cells.Citation59 Studies performed on C. elegans have shed light on how stressful conditions may be regulated or affect whole organisms.Citation60
The tumor microenvironment is characterized by extreme conditions in which hypoxia and nutrient deprivation stress the cells to a great extent. The autophagic machinery supports the breakdown and recycling of macromolecules and cellular organelles under such stressful conditions in order to maintain energy homeostasis and provide new resources that are required for cell division and growth. Lipophagy, another type of selective autophagy for lipid breakdown, links lipid metabolism and autophagy; however, how lipophagy is regulated remains unclear. C. elegans has been proposed as an attractive animal model to investigate lipophagy.Citation61,62 Moreover, endocrine and paracrine effects of adipose tissue are greatly implicated in cancer development.Citation63 Obesity causes adipocytes to malfunction, which in turn may facilitate the development of obesity-related metabolic disorders and cancer, mainly through paracrine and endocrine effects mediated by adipokines. The systemic effects exerted by adipose tissue are hard to elucidate. Thus, there is an imperative need to investigate the mechanisms involved and studies in C. elegans may contribute to a better understanding of such endocrine effects at a systemic level. Indeed, the nematode has long been used to elucidate such effects and the fact that several metabolic pathways are conserved suggests that the worm is well suited to investigate paracrine and endocrine effects exerted by the adipose tissue. Overall, C. elegans could serve as a compelling animal model to study the basics of cellular metabolism in a systemic fashion, with important implications for various disorders such as metabolic and oncogenic diseases.
Pluripotent Cells and Cancer Stem Cells: Gaining Insight into Stem Cell Reprogramming and Dedifferentiation from an Invertebrate Perspective
Over the past years an intriguing but controversial concept has emerged—that solid tumors may contain cancer stem cells (CSCs). Despite limited data, the prevailing hypothesis surmises that CSCs, like their normal counterparts, are more resistant to anticancer treatment and are responsible for tumor relapse once treatment has been halted.Citation64 Whether, and how, CSCs are involved in the intricate mechanisms that confer resistance to cancer therapy and enable tumors to regenerate, needs to be defined in order to increase the chance of successful treatment. Accumulating evidence suggests that epithelial-to-mesenchymal transition (EMT) and the tumor microenvironment play a major role.Citation64 However, the molecular and cellular components of the stem cell niches and the pathways involved in EMT transition remain generally unknown. Because of the lack of appropriate experimental systems, studies in C. elegans could rapidly provide valuable information about the nature of CSCs and the mechanisms by which they support tumors.
Although C. elegans consists mainly of post-mitotic cells, it is widely accepted as a simple model system for elucidating key aspects of stem cell biology.Citation65 The C. elegans hermaphrodite germ line is considered the main pool of pluripotent cells and seam cells are acknowledged as an epidermal stem cell lineage. Moreover, the germ line is the only tissue in C. elegans that can lead to bona fide tumors caused by germline hyperproliferation and, as already mentioned above, the only tissue capable of undergoing apoptosis throughout adulthood. The flexibility and advantages of such a powerful system for studying biological phenomena pertinent to cancer and stem cells are increasingly becoming acknowledged.
Conserved mechanisms for stem cell maintenance and differentiation have been derived from studies in C. elegans. The proper balance of proliferation versus differentiation has been suggested to be mediated by cell cycle regulation through a mechanism involving repression of the Cyclin E/Cdk2 inhibitor CKI-2Cip/Kip and the RNA binding proteins FBF/Pumilio and GLD-1/Quaking.Citation66,67 PUF-8, another RNA binding protein, regulates the balance between cell proliferation and differentiation by preventing cells re-entering the mitotic phase.Citation68 In addition to the discussed RNA-binding proteins, non-coding RNAs (ncRNAs) are also implicated in pluripotency regulation and the processes of somatic cell reprogramming.Citation69 These studies highlight the importance of RNA regulation in pluripotency maintenance. Furthermore, the germline stem cell niche is a key determinant of stem cell fate. C. elegans offers a powerful in vivo model to study such mechanisms in a physiological cellular microenvironment, allowing investigation of niche–stem cells interactions at a single-cell resolution. Several extrinsic mechanisms have been identified in C. elegans for the maintenance of the progenitor pool, revealing the importance of the micro- and macro-environment (). The best-examined mechanisms are those mediated by the 2 distal tip cells (DTCs), which exert their effects from the distal terminus of each gonad. The DTC of each gonad arm promotes the mitotically proliferating population of cells through a conserved GLP-1/Notch signaling pathway. DTCs express Delta/Serrate-like ligands, which bind on the GLP-1/Notch receptor of distal germ cells resulting in cleavage of the GLP-1/Notch intracellular domain, which then translocates into the nucleus to inhibit meiosis and promote mitosis.Citation70 Whereas glp-1 knockout causes germ cell differentiation and its knockdown is responsible for the balance between cell proliferation and differentiation, hyperactivation of the GLP-1/Notch signaling cascade causes germ cells to persist in the mitotic cycle, leading to tumors.Citation71-73 These phenotypes are at least partially independent of the insulin/IGF-1 and TOR/S6K pathways that are also involved in germline proliferation and maintaining the balance between meiosis and mitosis.Citation73,74 In addition, S6K/RSKS-1 was shown to link cell cycle, cell fate, and nutrient response in progenitor cells in the hyperactive GLP-1/Notch germ line model.Citation74 In fact, in the absence of S6K/RSKS-1 germ cells were resistant to adverse effects mediated by dietary restriction, suggesting the importance of the TOR/S6K pathway in stem cell and germ cell metabolism, and eventually fertility. Furthermore, lst-1 (lateral signaling target) and sygl-1 (synthetic Glp) are targets of GLP-1/Notch signaling within the niche and function redundantly to maintain germ line stem cells (GSCs).Citation75 Neuroendocrine mechanisms have also been shown to influence stem cell fate through the stem cell niche. DAF-7/TGF-β expressed from ASI neurons has been implicated in mechanisms that influence the balance of proliferation versus differentiation in the C. elegans germ line by acting on its receptor located on DTCs.Citation76 Furthermore, adhesion molecules (such as cadherins and integrins) also play a key role in germ cell renewal orchestrated by the niche.Citation77
Figure 3. Mechanisms for stem cell maintenance, differentiation and reprogramming derived from studies in C. elegans. (A) A wild-type gonad of a hermaphrodite worm. GLP-1/Notch signaling from the DTC cell is required for normal germ cell renewal. Neuroendocrine effects mediated by ASI-expressed DAF-7/TGF-β are also implicated. (B) Examples of how epigenetic factors may influence reprogramming. i) Reduction of a histone chaperone (LIN-53) and concomitant ectopic expression of a specific neurogenic transcription factor leads to differentiation of germ cells into specific neuronal ones (depending on the neurogenic factor expressed). ii) Germ cells are reprogrammed into specific neurons in SRP-5 (a histone demethylase) and LET-418 (a chromatin remodeler) double mutants. (C) Example of germ cell reprogramming mediated by RNA. GLD-1 and MEX-3 are RNA binding proteins that regulate germ cell differentiation.
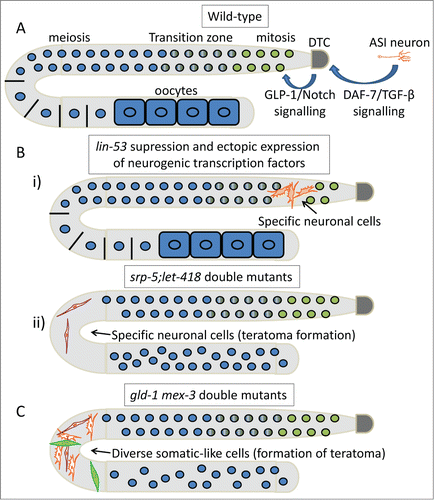
Finally, a better understanding of the sophisticated mechanisms implicated in the process by which fully differentiated cells can be reprogrammed into other cell types will have a huge impact in regenerative medicine, stem cell biology, and cancer biology. Such information could prove crucial for designing alternative strategies to control tumor progression. Interestingly, natural cell reprogramming events have also been observed, allowing a better understanding of mechanisms involved in cell dedifferentiation. In vivo observations of natural reprogramming in C. elegans during development suggest that cell division is not a prerequisite for cell reprogramming. In C. elegans, an epithelial cell transdifferentiates into a neuron via a conserved pathway that requires the NODE complex and SOX-2, suggesting that cell plasticity phenomena might share mechanistic similarities across phyla.Citation78,79 Furthermore, various manipulations have been used to stimulate reprogramming of cells into a desired phenotype.Citation80 Notably, a few years ago it was demonstrated that depletion of GLD-1 and MEX-3, or GLD-1 alone, causes germ cells to ectopically transdifferentiate into various somatic cell types, forming human germ cell tumor-like teratomas.Citation67,81 However, epigenetic factors may confer resistance to efficient reprogramming to specific cell types.Citation82 It was recently demonstrated that ectopic expression of a specific neurogenic transcription factor in worms with suppression of LIN-53 (a histone chaperone) leads to the conversion of mitotic germ cells into specific neuronal ones, whereas the histone H3K4 demethylase SPR-5/LSD1 and the chromatin remodeler LET-418/Mi2 cooperatively function to impede somatic dedifferentiation of germ cells.Citation83,84 The information that has been acquired from C. elegans indicates that it could be a powerful in vivo model for the investigation of key aspects of pluripotency with respect to cancer development, relapse, and treatment.
Interactions between Microbiota and C. elegans: an Emerging Model system for Investigating the Effects of Symbiosis and Dysbiosis in Cancer Development
Gastrointestinal microflora is believed to be linked to the genesis and development of cancer. Emerging evidence suggests that gut microbiota, which account for the largest percentage of the human microbiome, are directly associated with colorectal cancer, although the mode of action remains largely enigmatic.Citation85,86 Paradoxically, both tumor promoting and antitumor effects have been attributed to microbiota.Citation86–90 The tumor promoting effects are believed to be exerted by specific bacterial pathogens (i.e., Helicobacter pylori) or by the metabolic action of microbiota.Citation87,89,90 Xenobiotics, as well as prescribed drugs and anticancer agents or their metabolites that are further metabolized by gut microbiota, may lead to cell toxicity and the induction of intestinal tumors.Citation90-92 On the other hand, a “healthy” microbiome may act protectively by facilitating maturation of the immune system.Citation93
The direct interplay between bacteria and eukaryotes could be investigated in a simpler in vivo model that lacks an adaptive immune system. C. elegans grown on a bacteria lawn in common laboratory settings is considered a fundamental model organism to study host–microbe–interactions at the organismal, cellular, and molecular level.Citation42 Just recently, it was demonstrated that bacteria-derived nitric oxide (NO) increases C. elegans stress resistance and enhances longevity through a mechanism regulated by HSF-1 and DAF-16/FoxO transcription factors.Citation94 This study shed light on an mechanism that is shared among species and underlies the effect of a signaling molecule with multifaceted and controversial outcomes. The role of NO in cancer development remains ambiguous. It has been suggested to promote different cancer-related events such as angiogenesis, apoptosis, and metastasis; however, its role as a potential antioncogenic agent is currently being evaluated. The increased expression of iNOS and eNOS in human colorectal cancers might suggest a hormetic mode of action of NO and additive effects of microbiota-derived NO should be also taken into account.Citation95 Further understanding of the role of NO in tumor biology will reduce the controversy and confusion and aid the development of novel NO-based therapies to prevent and treat various human cancers. In another example, the microbiome-dependent effect of metformin on C. elegans revealed the importance of microbiota metabolism on drug efficacy.Citation96 Metformin is a widely prescribed antihyperglycemic drug used for the treatment of type-2 diabetes. Anticancer properties of metformin have also been identified; however, the mechanisms mediating these beneficial effects are still unknown. A possible explanation is that drug efficacy is indirectly induced through gut microbiota metabolism; thus the microbiota might determine the diverse responses of drugs observed in individuals. Studies on C. elegans may significantly contribute toward a better understanding of complex interspecies interactions that impact diverse diseases such as cancer, thus enabling specific individualized anticancer treatments based on microflora composition.
Concluding Remarks and Outlook
Studies in C. elegans have provided valuable information that extends beyond the customary insights and have contributed to current understanding of the complex molecular and cellular mechanisms governing cancer initiation and development. In this review, we survey well-conserved processes that have been characterized as hallmarks of cancer.Citation2 In addition to the knowledge gained from such an approach, the nematode C. elegans could also be used as an in vivo model for high-throughput anticancer drug screening in a whole animal context without the limitations of ethical boundaries. Drug efficacy and adverse effects could be rapidly and readily assessed in a simple but well-characterized animal, as recently demonstrated for anticancer agents.Citation97,98 In this regard, the nematode could be even used in drug screening against tumor-induced angiogenesis. Despite the lack of a circulatory system, proteins vital for angiogenesis, such as the PDGF/VEGF-like factor PVF-1, are expressed in C. elegans and, more importantly, can induce angiogenesis in vertebrate assay systems.Citation99 In summary, C. elegans offers a powerful platform for the deconvolution of carcinogenesis and thereby the identification of new drug targets, thus contributing to the development of new therapeutic interventions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We apologize to those colleagues whose work could not be referenced directly owing to space limitations.
Funding
E.K. is supported by the General Secretariat for Research and Technology of the Greek Ministry of Education. Work in the authors’ laboratory is also funded by grants from the European Research Council (ERC) and the European Commission Framework Programmes.
References
- Harris TW, Chen N, Cunningham F, Tello-Ruiz M, Antoshechkin I, Bastiani C, Bieri T, Blasiar D, Bradnam K, Chan J. et al. WormBase: a multi-species resource for nematode biology and genomics. Nucleic Acids Res 2004; 32:D411-7; PMID:14681445; http://dx.doi.org/10.1093/nar/gkh066
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID:21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Kirienko NV, Mani K, Fay DS. Cancer models in Caenorhabditis elegans. Dev Dyn 2010; 239:1413-48; PMID:20175192; http://dx.doi.org/10.1002/dvdy.22247
- Hyenne V, Chartier NT, Labbe JC. Understanding the role of asymmetric cell division in cancer using C. elegans. DevDyn 2010; 239:1378-87; PMID:20140912; http://dx.doi.org/10.1002/dvdy.22237
- McCarthy N. Pushing on through. Nat Rev Cancer 2010; 10:452; PMID:20589968; http://dx.doi.org/10.1038/nrc2878
- Kipreos ET. C. elegans cell cycles: invariance and stem cell divisions. Nat Rev Mol Cell Biol 2005; 6:766-76; PMID:16314866; http://dx.doi.org/10.1038/nrm1738
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16:3-11; PMID:18846107; http://dx.doi.org/10.1038/cdd.2008.150
- Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 2013; 1833:3448-59; PMID:23770045; http://dx.doi.org/10.1016/j.bbamcr.2013.06.001
- Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ 2007; 14:1237-43; PMID:17431418; http://dx.doi.org/10.1038/sj.cdd.4402148
- Gartner A, Boag PR, Blackwell TK. Germline survival and apoptosis. WormBook 2008:1-20; PMID:18781708; http://dx.doi.org/10.1895/wormbook.1.145.1
- Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO. A conserved checkpoint pathway mediates DNA damage–induced apoptosis and cell cycle arrest in C. elegans. Mol Cell 2000; 5:435-43; PMID:10882129; http://dx.doi.org/10.1016/S1097-2765(00)80438-4
- Lettre G, Hengartner MO. Developmental apoptosis in C. elegans: a complex CEDnario. Nat Rev Mol Cell Biol 2006; 7:97-108; PMID:16493416; http://dx.doi.org/10.1038/nrm1836
- Schumacher B, Schertel C, Wittenburg N, Tuck S, Mitani S, Gartner A, Conradt B, Shaham S. C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ 2005; 12:153-61; PMID:15605074; http://dx.doi.org/10.1038/sj.cdd.4401539
- Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene 2008; 27 Suppl 1:S2-19; PMID:19641503; http://dx.doi.org/10.1038/onc.2009.39
- Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science 1998; 281:1322-6; PMID:9735050; http://dx.doi.org/10.1126/science.281.5381.1322
- Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007; 26:1324-37; PMID:17322918; http://dx.doi.org/10.1038/sj.onc.1210220
- Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 1994; 76:665-76; PMID:7907274; http://dx.doi.org/10.1016/0092-8674(94)90506-1
- van Haaften G, Romeijn R, Pothof J, Koole W, Mullenders LH, Pastink A, Plasterk RH, Tijsterman M. Identification of conserved pathways of DNA-damage response and radiation protection by genome-wide RNAi. Current biology : CB 2006; 16:1344-50; PMID:16824923; http://dx.doi.org/10.1016/j.cub.2006.05.047
- Kaufmann SH, Vaux DL. Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene 2003; 22:7414-30; PMID:14576849; http://dx.doi.org/10.1038/sj.onc.1206945
- Greiss S, Hall J, Ahmed S, Gartner A. C. elegans SIR-2.1 translocation is linked to a proapoptotic pathway parallel to cep-1/p53 during DNA damage-induced apoptosis. Genes Dev 2008; 22:2831-42; PMID:18923081; http://dx.doi.org/10.1101/gad.482608
- Schumacher B, Hofmann K, Boulton S, Gartner A. The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 2001; 11:1722-7; PMID:11696333; http://dx.doi.org/10.1016/S0960-9822(01)00534-6
- Jolliffe AK, Derry WB. The TP53 signaling network in mammals and worms. Brief Funct Genomics 2013; 12:129-41; PMID:23165352; http://dx.doi.org/10.1093/bfgp/els047
- Derry WB, Putzke AP, Rothman JH. Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 2001; 294:591-5; PMID:11557844; http://dx.doi.org/10.1126/science.1065486
- Stergiou L, Doukoumetzidis K, Sendoel A, Hengartner MO. The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differentiation 2007; 14:1129-38; PMID:17347667; http://dx.doi.org/10.1038/sj.cdd.4402115
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005; 37:19-24; PMID:15624019; http://dx.doi.org/10.1038/ng1494
- Pinkston-Gosse J, Kenyon C. DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nat Genet 2007; 39:1403-9; PMID:17934462; http://dx.doi.org/10.1038/ng.2007.1
- Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science 2006; 313:971-5; PMID:16917064; http://dx.doi.org/10.1126/science.1121908
- McGee MD, Day N, Graham J, Melov S. cep-1/p53-dependent dysplastic pathology of the aging C. elegans gonad. Aging 2012; 4:256-69; PMID:22562940
- Sendoel A, Kohler I, Fellmann C, Lowe SW, Hengartner MO. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 2010; 465:577-83; PMID:20520707; http://dx.doi.org/10.1038/nature09141
- Possik E, Jalali Z, Nouet Y, Yan M, Gingras MC, Schmeisser K, Panaite L, Dupuy F, Kharitidi D, Chotard L, et al. Folliculin regulates ampk-dependent autophagy and metabolic stress survival. PLoS genetics 2014; 10:e1004273; PMID:24763318; http://dx.doi.org/10.1371/journal.pgen.1004273
- Zhang Y, Shao Z, Zhai Z, Shen C, Powell-Coffman JA. The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PloS one 2009; 4:e6348; PMID:19633713; http://dx.doi.org/10.1371/journal.pone.0006348
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/10.1126/science.1193497
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463-77; PMID:15068787; http://dx.doi.org/10.1016/S1534-5807(04)00099-1
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/10.1016/j.cell.2007.10.035
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol 2010; 12:842-6; PMID:20811357; http://dx.doi.org/10.1038/ncb0910-842
- Kourtis N, Tavernarakis N. Autophagy and cell death in model organisms. Cell Death Differ 2009; 16:21-30; PMID:19079286; http://dx.doi.org/10.1038/cdd.2008.120
- Samara C, Syntichaki P, Tavernarakis N. Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ 2008; 15:105-12; PMID:17901876; http://dx.doi.org/10.1038/sj.cdd.4402231
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100:15077-82; PMID:14657337; http://dx.doi.org/10.1073/pnas.2436255100
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401-10; PMID:22534666; http://dx.doi.org/10.1038/nrc3262
- Akman HO, Sampayo JN, Ross FA, Scott JW, Wilson G, Benson L, Bruno C, Shanske S, Hardie DG, Dimauro S. Fatal infantile cardiac glycogenosis with phosphorylase kinase deficiency and a mutation in the gamma2-subunit of AMP-activated protein kinase. Pediatr Res 2007; 62:499-504; PMID:17667862; http://dx.doi.org/10.1203/PDR.0b013e3181462b86
- Cabreiro F, Gems D. Worms need microbes too: microbiota, health and aging in Caenorhabditis elegans. EMBO Mol Med 2013; 5:1300-10; PMID:23913848; http://dx.doi.org/10.1002/emmm.201100972
- Horvitz HR, Sulston JE. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics 1980; 96:435-54; PMID:7262539
- Ferguson EL, Horvitz HR. Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics 1985; 110:17-72; PMID:3996896
- Seydoux G, Savage C, Greenwald I. Isolation and characterization of mutations causing abnormal eversion of the vulva in Caenorhabditis elegans. Dev Biol 1993; 157:423-36; PMID:8500652; http://dx.doi.org/10.1006/dbio.1993.1146
- Sundaram MV. RTK/Ras/MAPK signaling. WormBook 2006:1-19; http://dx.doi.org/10.1895/wormbook.1.80.1
- Eisenmann DM, Kim SK. Mechanism of activation of the Caenorhabditis elegans ras homologue let-60 by a novel, temperature-sensitive, gain-of-function mutation. Genetics 1997; 146:553-65; PMID:9178006
- Bos JL. The ras gene family and human carcinogenesis. Mutat Res 1988; 195:255-71; PMID:3283542; http://dx.doi.org/10.1016/0165-1110(88)90004-8
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res 1989; 49:4682-9; PMID:2547513
- Siddiqui SS, Loganathan S, Krishnaswamy S, Faoro L, Jagadeeswaran R, Salgia R. C. elegans as a model organism for in vivo screening in cancer: effects of human c-Met in lung cancer affect C. elegans vulva phenotypes. Cancer Biol Ther 2008; 7:856-63; PMID:18340114; http://dx.doi.org/10.4161/cbt.7.6.5842
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55:74-108; PMID:15761078
- Ma PC, Jagadeeswaran R, Jagadeesh S, Tretiakova MS, Nallasura V, Fox EA, Hansen M, Schaefer E, Naoki K, Lader A, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res 2005; 65:1479-88; PMID:15735036
- Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Front Oncol 2013; 3:292; PMID:24350057; http://dx.doi.org/10.3389/fonc.2013.00292
- Liu Y, Samuel BS, Breen PC, Ruvkun G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 2014; 508:406-10; PMID:24695221; http://dx.doi.org/10.1038/nature13204
- Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer 2013; 13:51-65; PMID:23235911; http://dx.doi.org/10.1038/nrc3398
- Fritz G. Targeting the mevalonate pathway for improved anticancer therapy. Curr Cancer Drug Targets 2009; 9:626-38; PMID:19508172
- Artal-Sanz M, Tavernarakis N. Prohibitin couples diapause signalling to mitochondrial metabolism during ageing in C. elegans. Nature 2009; 461:793-7; PMID:19812672; http://dx.doi.org/10.1038/nature08466
- Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radic Biol Med 2011; 51:327-36; PMID:21619928; http://dx.doi.org/10.1016/j.freeradbiomed.2011.05.010
- Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 2006; 10:175-6; PMID:16959608; http://dx.doi.org/10.1016/j.ccr.2006.08.015
- Rodriguez M, Snoek LB, De Bono M, Kammenga JE. Worms under stress: C. elegans stress response and its relevance to complex human disease and aging. Trends Genet 2013; 29:367-74; PMID:23428113; http://dx.doi.org/10.1016/j.tig.2013.01.010
- Lapierre LR, Melendez A, Hansen M. Autophagy links lipid metabolism to longevity in C. elegans. Autophagy 2012; 8:144-6; PMID:22186228; http://dx.doi.org/10.4161/auto.8.1.18722
- Yang P, Zhang H. You are what you eat: multifaceted functions of autophagy during C. elegans development. Cell Res 2014; 24:80-91; PMID:24296782; http://dx.doi.org/10.1038/cr.2013.154
- Paz-Filho G, Lim EL, Wong ML, Licinio J. Associations between adipokines and obesity-related cancer. Front Biosci (Landmark Ed) 2011; 16:1634-50
- Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov 2014; 13:497-512; PMID:24981363; http://dx.doi.org/10.1038/nrd4253
- Joshi PM, Riddle MR, Djabrayan NJ, Rothman JH. Caenorhabditis elegans as a model for stem cell biology. Dev Dyn 2010; 239:1539-54; PMID:20419785; http://dx.doi.org/10.1002/dvdy.22296
- Kalchhauser I, Farley BM, Pauli S, Ryder SP, Ciosk R. FBF represses the Cip/Kip cell-cycle inhibitor CKI-2 to promote self-renewal of germline stem cells in C. elegans. Embo J 2011; 30:3823-9; PMID:21822213; http://dx.doi.org/10.1038/emboj.2011.263
- Biedermann B, Wright J, Senften M, Kalchhauser I, Sarathy G, Lee MH, Ciosk R. Translational repression of cyclin E prevents precocious mitosis and embryonic gene activation during C. elegans meiosis. Dev Cell 2009; 17:355-64; PMID:19758560; http://dx.doi.org/10.1016/j.devcel.2009.08.003
- Subramaniam K, Seydoux G. Dedifferentiation of primary spermatocytes into germ cell tumors in C. elegans lacking the pumilio-like protein PUF-8. Curr Biol 2003; 13:134-9; PMID:12546787; http://dx.doi.org/10.1016/S0960-9822(03)00005-8
- Wright JE, Ciosk R. RNA-based regulation of pluripotency. Trends Genet 2013; 29:99-107; PMID:23146412; http://dx.doi.org/10.1016/j.tig.2012.10.007
- Byrd DT, Kimble J. Scratching the niche that controls Caenorhabditis elegans germline stem cells. Semin Cell Dev Biol 2009; 20:1107-13; PMID:19765664; http://dx.doi.org/10.1016/j.semcdb.2009.09.005
- Berry LW, Westlund B, Schedl T. Germ-line tumor formation caused by activation of glp-1, a Caenorhabditis elegans member of the Notch family of receptors. Development 1997; 124:925-36; PMID:9043073
- Austin J, Kimble J. glp-1 is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell 1987; 51:589-99; PMID:3677168; http://dx.doi.org/10.1016/0092-8674(87)90128-0
- Michaelson D, Korta DZ, Capua Y, Hubbard EJ. Insulin signaling promotes germline proliferation in C. elegans. Development 2010; 137:671-80; PMID:20110332; http://dx.doi.org/10.1242/dev.042523
- Korta DZ, Tuck S, Hubbard EJ. S6K links cell fate, cell cycle and nutrient response in C. elegans germline stem/progenitor cells. Development 2012; 139:859-70; PMID:22278922; http://dx.doi.org/10.1242/dev.074047
- Kershner AM, Shin H, Hansen TJ, Kimble J. Discovery of two GLP-1/Notch target genes that account for the role of GLP-1/Notch signaling in stem cell maintenance. Proc Natl Acad Sci U S A 2014; 111:3739-44; PMID:24567412; http://dx.doi.org/10.1073/pnas.1401861111
- Dalfo D, Michaelson D, Hubbard EJ. Sensory regulation of the C. elegans germline through TGF-beta-dependent signaling in the niche. Curr Biol 2012; 22:712-9; PMID:22483938; http://dx.doi.org/10.1016/j.cub.2012.02.064
- Chen S, Lewallen M, Xie T. Adhesion in the stem cell niche: biological roles and regulation. Development 2013; 140:255-65; PMID:23250203; http://dx.doi.org/10.1242/dev.083139
- Jarriault S, Schwab Y, Greenwald I. A Caenorhabditis elegans model for epithelial-neuronal transdifferentiation. Proc Natil Acad Sci U S A 2008; 105:3790-5; PMID:18308937; http://dx.doi.org/10.1073/pnas.0712159105
- Kagias K, Ahier A, Fischer N, Jarriault S. Members of the NODE (Nanog and Oct4-associated deacetylase) complex and SOX-2 promote the initiation of a natural cellular reprogramming event in vivo. Proc Natl Acad Sci U S A 2012; 109:6596-601; PMID:22493276; http://dx.doi.org/10.1073/pnas.1117031109
- Hajduskova M, Ahier A, Daniele T, Jarriault S. Cell plasticity in Caenorhabditis elegans: from induced to natural cell reprogramming. Genesis 2012; 50:1-17; PMID:21932439; http://dx.doi.org/10.1002/dvg.20806
- Ciosk R, DePalma M, Priess JR. Translational regulators maintain totipotency in the Caenorhabditis elegans germline. Science 2006; 311:851-3; PMID:16469927; http://dx.doi.org/10.1126/science.1122491
- Pasque V, Jullien J, Miyamoto K, Halley-Stott RP, Gurdon JB. Epigenetic factors influencing resistance to nuclear reprogramming. Trends Genet 2011; 27:516-25; PMID:21940062; http://dx.doi.org/10.1016/j.tig.2011.08.002
- Tursun B, Patel T, Kratsios P, Hobert O. Direct conversion of C. elegans germ cells into specific neuron types. Science 2011; 331:304-8; PMID:21148348; http://dx.doi.org/10.1126/science.1199082
- Kaser-Pebernard S, Muller F, Wicky C. LET-418/Mi2 and SPR-5/LSD1 Cooperatively Prevent Somatic Reprogramming of C. elegans Germline Stem Cells. Stem Cell Reports 2014; 2:547-59; PMID:24749077; http://dx.doi.org/10.1016/j.stemcr.2014.02.007
- Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014; 15:317-28; PMID:24629338; http://dx.doi.org/10.1016/j.chom.2014.02.007
- Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer 2013; 13:800-12; PMID:24132111; http://dx.doi.org/10.1038/nrc3610
- Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology 2011; 140:210-20; PMID:20950613; http://dx.doi.org/10.1053/j.gastro.2010.09.048
- Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC, Karre K, Pettersson S, Greicius G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis 2012; 33:1231-8; PMID:22461519; http://dx.doi.org/10.1093/carcin/bgs137
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M. et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013; 499:97-101; PMID:23803760; http://dx.doi.org/10.1038/nature12347
- Reddy BS, Narisawa T, Wright P, Vukusich D, Weisburger JH, Wynder EL. Colon carcinogenesis with azoxymethane and dimethylhydrazine in germ-free rats. Cancer Res 1975; 35:287-90; PMID:162868
- Saitta KS, Zhang C, Lee KK, Fujimoto K, Redinbo MR, Boelsterli UA. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 2014; 44:28-35; PMID:23829165; http://dx.doi.org/10.3109/00498254.2013.811314
- Takada H, Hirooka T, Hiramatsu Y, Yamamoto M. Effect of beta-glucuronidase inhibitor on azoxymethane-induced colonic carcinogenesis in rats. Cancer Res 1982; 42:331-34; PMID:7053860
- Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell 2012; 148:1258-70; PMID:22424233; http://dx.doi.org/10.1016/j.cell.2012.01.035
- Gusarov I, Gautier L, Smolentseva O, Shamovsky I, Eremina S, Mironov A, Nudler E. Bacterial nitric oxide extends the lifespan of C. elegans. Cell 2013; 152:818-30; PMID:23415229; http://dx.doi.org/10.1016/j.cell.2012.12.043
- Yagihashi N, Kasajima H, Sugai S, Matsumoto K, Ebina Y, Morita T, Murakami T, Yagihashi S. Increased in situ expression of nitric oxide synthase in human colorectal cancer. Virchows Arch 2000; 436:109-14; PMID:10755599; http://dx.doi.org/10.1007/PL00008208
- Cabreiro F, Au C, Leung KY, Vergara-Irigaray N, Cocheme HM, Noori T, Weinkove D, Schuster E, Greene ND, Gems D. Metformin retards aging in C. elegans C. elegans by altering microbial folate and methionine metabolism. Cell 2013; 153:228-39; PMID:23540700; http://dx.doi.org/10.1016/j.cell.2013.02.035
- Artal-Sanz M, de Jong L, Tavernarakis N. Caenorhabditis elegans: a versatile platform for drug discovery. Biotechnol J 2006; 1:1405-18; PMID:17109493; http://dx.doi.org/10.1002/biot.200600176
- Bae YK, Sung JY, Kim YN, Kim S, Hong KM, Kim HT, Choi MS, Kwon JY, Shim J. An in vivo C. elegans model system for screening EGFR-inhibiting anti-cancer drugs. PloS one 2012; 7:e42441; PMID:22957020; http://dx.doi.org/10.1371/journal.pone.0042441
- Tarsitano M, De Falco S, Colonna V, McGhee JD, Persico MG. The C. elegans pvf-1 gene encodes a PDGF/VEGF-like factor able to bind mammalian VEGF receptors and to induce angiogenesis. FASEB J 2006; 20:227-33; PMID:16449794; http://dx.doi.org/10.1096/fj.05-4147com