
Abstract
DNA methylation has been mostly studied in circulating blood cells. Although being readily accessible, metabolically active tissues such as adipose tissue would be more informative for the study of metabolic disorders. However, whether or not the blood DNA methylation profile correlates with that of adipose tissue remains unknown. In this study, DNA methylation patterns of variation at LEP and ADIPOQ gene loci were similar between individual CpGs across the different tissues. We also report that DNA methylation levels at biologically relevant CpGs are correlated between blood, subcutaneous, and visceral adipose tissue, and that these nearby CpGs are associated with LEP and ADIPOQ gene expression in adipose tissues. These results will be highly relevant for future epigenetic studies in metabolic disorders.
Introduction
The LEP and ADIPOQ genes, encoding for leptin and adiponectin respectively, have drawn much attention in candidate gene studies on obesity and its related metabolic disorders. These adipokines are mainly produced and secreted by adipocytes and are important regulators of energy metabolism and insulin sensitivity.Citation1,Citation2 Recent studies suggested that these two adipokines are involved in the fetal metabolic health programing of the newborn through epigenetic adaptations in response to a changing in utero environment. Indeed, we have recently reported correlations between placental DNA methylation levels at the LEP and ADIPOQ gene loci and maternal blood glucose concentrations (2 h post oral glucose tolerance test) at second trimester of pregnancy.Citation3-Citation5 A high maternal glucose concentration has been previously associated with a long-term susceptibility to obesity and related metabolic disorders in the newborn.Citation6-Citation8 Moreover, Tobi et al.Citation9 have observed higher LEP DNA methylation levels in leukocytes of men exposed prenatally to the ending World War 2 Dutch famine in comparison with unexposed siblings, suggesting that epigenetic marks imprinted during fetal life at the LEP locus could last over 60 y after birth.
DNA methylation marks consist in the addition of a methyl group at position 5′ of the pyrimidine ring of the cytosines upstream of a guanine (dinucleotide CpG) catalyzed by DNA methyltransferases.Citation10,Citation11 The establishment of DNA methylation marks during fetal life is crucial for cellular differentiation, X chromosome inactivation, genomic imprinting, and normal embryonic development.Citation12,Citation13 Embryonic DNA methylation marks are mitotically stable thus they can have a long-term functional impact and can be partially transmitted to the next generation.Citation14-Citation18 Although transcriptional regulation through DNA methylation is tissue-specific,Citation19-Citation21 it has been suggested that embryonic epigenetic marks set early during cellular differentiation and division are similar in various tissues.Citation22-Citation24 These similarities could be of importance when access to metabolically active tissues (e.g., adipose tissue) is limited, and especially when studying epigenetically affected genes in the context of in utero or fetal metabolic programming.
The objective of the current study was to compare LEP and ADIPOQ epigenetic profiles in blood, subcutaneous (SAT), and visceral (VAT) adipose tissue samples from the same obese class IIICitation22 subjects in order to characterize tissue-specific DNA methylation at these two adipokine gene loci. The comparison of adipokine DNA methylation levels in blood and adipose tissues will tell us whether or not readily accessible blood cells can reflect the DNA methylation profile of metabolically active tissues and can eventually be used to predict a lifelong susceptibility to obesity and associated metabolic disorders.
Results and Discussion
DNA methylation and mRNA level analyses at the LEP gene locus
Blood, SAT, and VAT were obtained from 73 obese class IIICitation22 subjects with body mass index (BMI) ranging from 40.0 to 60.0 kg/m2 (mean: 49.6 ± 6.0 kg/m2) (sample characteristics in Table S1). DNA methylation levels were assessed in blood, SAT, and VAT at 21 CpGs out of the 31 that weCitation3 and Melzner et al.Citation25 have previously epigenotyped in the proximal promoter CpG island of the LEP gene (Fig. S1).
DNA methylation levels at these 21 CpG sites analyzed within the LEP gene promoter were found to be highly variable within the region and across the three tissues analyzed. On average, the correlation coefficients between DNA methylation levels at these 21 CpG sites were found to be modest to very high in blood (r = 0.12 to 0.92), SAT (r = 0.01 to 0.82), and VAT (r = 0.01 to 0.86) (Fig. S3). For most of the CpG sites analyzed, methylation values were 1.5 to 2.0 times higher in blood than in SAT and VAT (). These results confirm previous findings from Marchi et al.Citation26 and Stoger et al.Citation27 showing that LEP DNA methylation is higher in tissues known to express lower LEP mRNA levels. The highest DNA methylation differences (>10.0%) between blood and adipose tissues at the LEP gene promoter locus were observed at CpG4, CpG11, CpG24, CpG29, CpG30, and CpG31 with CpG11 being the most significant (Fig. S1). Interestingly, CpG4 is found on the macrophage migration inhibitory factor (MIF-1) transcription factor binding site (TFBS) and CpG11 on or nearby the CCAAT/enhancer binding protein (C/EBP), hepatic leukemia factor (HLF), and c-MYB TFBS suggesting that these CpG sites may be important for tissue-specific LEP expression regulation (Fig. S2). The functional significance of CpG11 is also supported by its location within a highly conserved region across all placental mammals (Fig. S2). This CpG site was previously shown to be strongly differentially methylated between tissues with low (liver) and high (visceral adipocyte) mRNA levels of LEP.Citation26 Moreover, the regulation of LEP gene promoter activity by DNA methylation at C/EBP TFBS has previously been reported.Citation25 No transcriptional regulatory element was identified at CpG24, CpG29, CpG30, and CpG31 of the LEP gene promoter.
Figure 1. DNA methylation levels at LEP gene promoter (A) and ADIPOQ CpG island E (B). CpG sites in blood (white), subcutaneous (black), and visceral (gray) adipose tissues (n = 73). Error bars represent the standard deviation (SD). Mean DNA methylation levels were compared with the paired sample Student t test. Bars with different letters indicate means that are significantly different from each other (P ≤ 0.05) after adjustment for multiple testing.
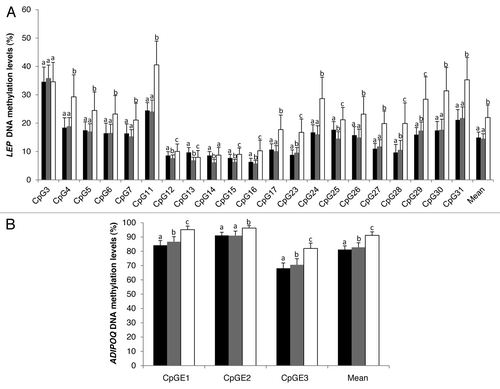
Next, we report that the correlation between DNA methylation levels at each individual CpG site was tissue- and site-specific. After multiple testing corrections, LEP DNA methylation levels at 14 individual CpG sites were found to be significantly correlated between SAT and VAT, whereas 6 and 2 CpGs were significantly correlated between blood and SAT and blood and VAT, respectively (). Interestingly, DNA methylation at CpG11, localized at C/EBP TFBS, was found to be among the most highly correlated between SAT and VAT and between SAT and blood (). For this site, a trend for association was also observed between VAT and blood after multiple testing correction (r = 0.327; P = 0.10). CpG16 was also found to be highly correlated between the three tissues and CpG17 was correlated between SAT and blood (). These two CpG sites are localized within SP1 TFBS and a highly conserved region of the LEP gene promoter locus (Fig. S2). SP1 TFBS was shown to be involved in the regulation of LEP gene expression.Citation25,Citation28,Citation29 High correlation coefficients were observed between DNA methylation levels at LEP CpG28 in the three tissues (). Nevertheless, the cytosine at this CpG site was found to be polymorphic (single nucleotide polymorphism [SNP]; rs791620; C > A), and the genotype is likely driving the correlations observed. Among all the CpG sites analyzed in the current study, CpG11, CpG16, and CpG17 are the most promising as they are located in potentially functional regions and correlated between blood and adipose tissues. Nevertheless, further studies are needed to determine whether these CpGs are associated with the development of obesity in normal weight and obese populations. If such associations are observed, it could eventually raise the possibility of using blood LEP DNA methylation levels at these CpG sites as a surrogate for LEP DNA methylation levels in adipose tissues and as a marker of obesity susceptibility.
Table 1. Pearson correlation coefficients between DNA methylation levels at LEP gene promoter CpG sites in blood, subcutaneous (SAT), and visceral adipose (VAT) tissues
LEP gene expression analysis in SAT and VAT confirmed the functional importance of specific CpG sites in both adipose tissues. Considering that LEP expression is influenced by sexCitation30,Citation31 and that associations between LEP DNA methylation and phenotypic variables have been previously reported to be sex-specific,Citation9,Citation32,Citation33 the relationships between LEP DNA methylation and mRNA levels were adjusted for sex. In addition, we observed significant interactions between sex and DNA methylation levels (CpG12 and CpG14) on VAT LEP mRNA levels. In VAT, DNA methylation levels at LEP CpG7, CpG11, and CpG27 to CpG30 were found to be positively associated with LEP mRNA levels (). These results are not supported by previous studies reporting negative associations between mean LEP promoter DNA methylation and expression in the visceral adipocyte fraction,Citation26 placenta,Citation3 and adipocyte cell cultures.Citation25 These contrasting results might be explained by the fact that LEP genetic variations were not taken into account in these studies. Indeed, an exploratory analysis revealed that LEP rs2167270, a variant previously associated with LEP expression,Citation34 tended to be associated with LEP gene expression in VAT (P = 0.06) and was significantly associated with DNA methylation levels at CpG4 in SAT and at CpG3, CpG4, CpG6, and CpG7 in VAT (Table S2). In addition, rs2167270 was found to influence the relationship between LEP DNA methylation and mRNA levels in adipose tissues (). In VAT, positive significant correlation coefficients were found in GG genotypes for CpG11 and nearby CpG sites, while no correlation was observed in GA/AA genotypes (). Interestingly, in SAT a statistical interaction effect was observed between the rs2167270 genotype and LEP DNA methylation levels (CpG5 [pi = 0.03], CpG14 [pi = 0.01], CpG15 [pi = 0.012], and CpG25 [pi = 0.009]) on LEP mRNA levels. We observed that LEP DNA methylation levels (CpG14, CpG17, CpG23, CpG24, and CpG25) were negatively correlated with LEP mRNA levels in carriers of the A allele, and they tended to be positively correlated in the GG genotype at CpG14 and CpG25. These exploratory analysis results highlight, for the first time, the importance of both genetic and epigenetic variations in the regulation of LEP gene expression. Furthermore, they suggest that LEP gene expression regulation in SAT and VAT is independent and restricted to specific CpG sites. In VAT, CpG sites located close to CpG11; nearby the TATA box element and the C/EBP TFBS appear to be the most important regulator of LEP expression whereas in SAT, CpG sites near SP1 TFBS (CpG16 and CpG17) may be central (Fig. S2). Further studies are needed to determine the contribution of the other SNPs (rs791620 and rs34104384) in the regulation of LEP gene expression in both types of adipose tissue. In addition, these preliminary genetic results will need to be confirmed in samples of larger size.
Table 2. Pearson correlation coefficients between DNA methylation and mRNA levels at LEP gene promoter CpG sites in subcutaneous (SAT) and visceral adipose (VAT) tissues according to rs2167270 genotype and adjusted for sex
DNA methylation and mRNA level analyses at the ADIPOQ gene locus
At the ADIPOQ gene locus, DNA methylation levels were assessed in the 73 patients at three polymorphic CpG islands (A, C, and E) as previously described (Fig. S3).Citation4 DNA methylation levels at ADIPOQ CpG islands A and C were found to be hypomethylated (<10.0%) and hypermethylated (>90.0%), respectively in blood and both SAT and VAT (Fig. S3). The CpG sites localized in these CpG islands were considered unmethylated and fully methylated and were thus not further analyzed.
As observed for the LEP gene locus, mean DNA methylation levels at ADIPOQ-CpGE1, CpGE2, and CpGE3 were also found to be higher in blood (91.2 ± 2.4%) in comparison to SAT (81.2 ± 2.7%) and VAT (82.8 ± 3.3%) (). At the ADIPOQ intron 1 locus, DNA methylation levels at CpGE1 and CpGE3 were highly correlated in SAT (r > 0.73; P < 0.001) and VAT (r > 0.73; P < 0.001), whereas in blood, the correlation was modest (r > 0.31; P < 0.01) (Fig. S5). DNA methylation levels at CpGE2 were the only ones significantly correlated between SAT and VAT after corrections for multiple testing (). CpGE2 DNA methylation was found to be negatively correlated with ADIPOQ mRNA levels in VAT, whereas in SAT, DNA methylation at CpGE3 was associated with ADIPOQ mRNA levels (). No transcriptional regulatory elements were identified at or near ADIPOQ CpG island E.
Table 3. Spearman rank correlation coefficients between DNA methylation levels at ADIPOQ CpG island E loci in blood, subcutaneous (SAT), and visceral adipose (VAT) tissues
Table 4. Pearson correlation coefficients between DNA methylation and mRNA levels at ADIPOQ CpG island E in subcutaneous (SAT) and visceral adipose (VAT) tissues
DNA methylation variations between CpG sites at the LEP and ADIPOQ gene loci
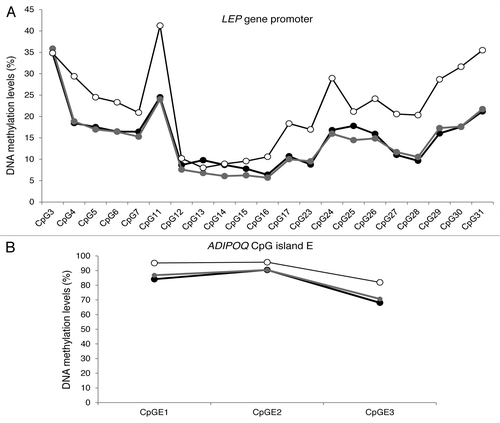
We also report that DNA methylation patterns of variation from one CpG site to the following were highly similar between the three tissues at both the LEP and ADIPOQ gene loci (). These similarities were previously observed by our group in cord blood and placenta samplesCitation5 and by Marchi et al.Citation26 in the liver, visceral adipocyte, and stromal vascular fraction of white adipose tissue at the LEP gene locus. Similarities between inter-individual CpG site variations at specific gene loci have also been reported in several other tissues.Citation24,Citation35 Following these observations, we previously suggested that DNA methylation levels vary concurrently between tissues around predefined (heritable) values, which seem independent from one CpG site to another.Citation15 Recently, Brons et al.Citation36 suggested that a detrimental fetal environment might decrease the acute regulation of the epigenome, which in turn might increase the risk to develop obesity and type 2 diabetes. This likely “predefined” pattern of DNA methylation that we and others have observed might be established early during embryonic development according to the maternal in utero environment and may determine the set point from which stochastic and environmental factors will induce a more or less acute regulation of the epigenome. Since this study was conducted with tissue samples from obese adults, longitudinal and dietary intervention studies will be needed to investigate whether LEP and ADIPOQ tissue-specific epipolymorphisms and associations are present in the newborn and normal weight populations, whether they predict the development of obesity or other metabolic disorders, and whether they respond to dietary and exercise interventions.
Figure 2. Average DNA methylation profile by CpG site for LEP gene promoter locus (A) and ADIPOQ CpG island E gene locus (B) in blood (white circles), subcutaneous (black circles), and visceral (gray circles) adipose tissues (n = 73).
In summary, the current study compared, for the first time, LEP and ADIPOQ DNA methylation levels in blood and adipose tissues. We report that DNA methylation patterns of variation between one CpG to the next at the LEP and ADIPOQ gene loci is highly similar between blood, SAT, and VAT, suggesting that a setting point for DNA methylation at each CpG site may exist. In addition, we showed that DNA methylation levels at potentially functional CpG sites are correlated between blood and adipose tissues. DNA methylation levels at or near these CpG sites were also found to be associated with adipokine gene expression levels. Hence, these specific CpGs might prove useful surrogates for DNA methylation measures, especially with SAT biopsies that require a much less invasive procedure than VAT sampling.
Materials and Methods
Patients
Blood, SAT, and VAT samples were obtained from 33 men and 40 pre-menopausal women (obese class IIICitation22: BMI ≥ 40 kg/m2) undergoing biliopancreatic derivation to treat obesity. The 73 patients were free of treatment for dyslipidemia, diabetes, and hypertension. The surgical procedures have been previously described.Citation37,Citation38 Briefly, fasting blood samples were taken on the morning or the day before the surgery. SAT and VAT samples were obtained at the beginning of the intervention. All samples were stored at −80 °C until nucleic acid extraction. Anthropometric parameters (body weight, height, waist, and hip circumferences), resting blood pressure, and fasting plasma insulin, glucose and lipid levels were also measured before surgery according to standardized procedures.Citation39 All participants provided a written informed consent before their inclusion in the study, and all clinical data were denominalized. This project was performed in collaboration with the Tissue bank for the study of obesity and its complications at the Institut Universitaire de Cardiologie et de Pneumologie de Québec. The project was approved by this institution’s and the Université Laval’s ethics committees and was conducted in accordance with the Declaration of Helsinki.
Nucleic acid extraction
DNA was purified from whole blood samples with the Gentra Puregene Blood Kit (Qiagen). DNA and RNA from SAT and VAT were extracted as previously described.Citation40 RNA quality was assessed with Agilent 2100 Bioanalyzer RNA Nano Chips (Agilent Technologies). Three RNA samples from SAT and VAT had low RNA integrity numbers (RIN < 6.0) and were excluded from the analysis. The other RNA samples in SAT and VAT showed a high quality with mean RIN values of 8.0 ± 0.8 and 8.3 ± 0.6 respectively.
DNA methylation analyses and genotyping
DNA methylation levels at CpG sites were assessed using pyrosequencing (Pyromark Q24, QIAGEN-Biotage). Combined with the NaBis DNA treatment, pyrosequencing is a quantitative real-time sequencing technology that allows to measure DNA methylation levels (%) at a single cytosine (CpG) of a given genomic region. NaBis treatment of DNA (EpiTect Bisulfite Kit, Qiagen) specifically converts unmethylated cytosines into uracil, while the methylated cytosines are protected from this transition creating a cytosine/thymine polymorphism. Once treated, NaBis-DNA is amplified (Pyromark PCR kit, Qiagen), and the cytosine and thymine alleles are quantified by pyrosequencing.Citation41 DNA methylation levels at the LEP gene CpG island proximal promoter region were assessed using the PCR and pyrosequencing primers described in Table S3. Specific PCR and pyrosequencing conditions and primer pairs for ADIPOQ DNA methylation analyses were the same as previously described.Citation4
Pyrosequencing technology was also used for genotyping a single nucleotide polymorphism (SNP; rs2167270; G > A) within the LEP proximal promoter region (Fig. S1). The genotype of the 73 patients was determined with the same PCR and sequencing primers as for the methylation analysis of CpG3 to CpG7 (Table S3). The pyrogram signals obtained, representing the amount of G and A nucleotides added to the sequence, allowed the quantification of the two alleles and determination of the genotype.Citation42 The rs2167270 genotype was identical in all three tissues analyzed for the 73 patients. The genotype frequencies (GG: 30 [41.1%]; GA: 35 [47.9%] and AA: 8 [11.0%]) were found to be under Hardy–Weinberg equilibrium (P > 0.05). Carriers of the minor A allele (GA/AA) were grouped together for the analysis since the number of homozygous AA was too low to provide adequate statistical power.
Expression analysis and identification of transcription factor binding sites and conserved regions in LEP and ADIPOQ genes
mRNA levels in SAT and VAT were quantified by quantitative real-time PCR (qRT-PCR). CDNA (cDNA) was generated from total RNA using a random primer hexamer provided with the High Capacity cDNA Archive Kit from Applied Biosystems. Equal amounts of cDNA were run in duplicate and amplified in a 20 µL reaction containing 10 µL of Universal PCR Master Mix (Applied Biosystems). Primers and Taqman probes were obtained from Applied Biosystems (LEP: Hs00174877_m1 and ADIPOQ: Hs00605917_m1). The Glyceraldehyde 3-phosphate dehydrogenase (GAPDH: Hs99999905_m1; Applied Biosystems) housekeeping gene was amplified in parallel. LEP, ADIPOQ, and GAPDH amplifications were performed using the Applied Biosystems 7500 Real Time PCR System, as recommended by the manufacturer (Applied Biosystems). LEP and ADIPOQ mRNA (Ct) levels were quantified relative to change in GAPDH gene expression (Ct). GAPDH/ADIPOQ and GAPDH/LEP Ct ratios (1/x) were used for the analysis.
UCSC genome browserCitation43 and a PubMed literature search were used to identify consensus sequences of TFBS sequence in the LEP gene promoter and ADIPOQ intron 1 DNA sequence. The relationship between LEP and ADIPOQ mRNA regulation and the identified transcription factors was further evaluated with a PubMed literature review.
Statistical analyses
The normal distribution of all variables was assessed using a Kolmogorov–Smirnov test. DNA methylation levels at the ADIPOQ-CpGE1, E2, and E3 loci in blood were not normally distributed. Hence, DNA methylation levels at the ADIPOQ gene locus were compared between blood, SAT, and VAT using the Wilcoxon signed-rank test, whereas paired sample t test was used for LEP gene loci cross-tissue comparisons. In addition, partial Pearson correlation and Spearman correlations were used to determine DNA methylation correlations across the three tissues for the LEP and ADIPOQ gene loci respectively. The associations between adipokines DNA methylation and mRNA levels were also assessed with partial Pearson correlations. Additionally, two-way ANOVA with interaction terms were used to determine the independent variables contributing to LEP mRNA level variance. The Student t test was used to compare DNA methylation and mRNA levels between LEP rs2167270GG genotypes (GG vs GA/AA). Anthropometric characteristics were also compared between men and women using the unpaired Student t test, while categorical variables were compared using the chi-square statistic. Because a large number of tests was performed, Bonferroni multiple testing corrections were applied. Results were therefore considered statistically significant when P values ≤ 4.5 × 10−4 (P ≤ 0.01) and P values ≤ 2.3 × 10−3 (P ≤ 0.05) (two-sided). All statistical analyses were performed with the IBM SPSS Statistics 20 software (release 20.0.0, SPSS).
| Abbreviations: | ||
| ADIPOQ | = | adiponectin |
| BMI | = | body mass index |
| C/EBP | = | CCAAT/enhancer binding protein |
| HLF | = | hepatic leukemia factor |
| IGT | = | impaired glucose tolerance |
| LEP | = | leptin |
| MIF-1 | = | macrophage migration inhibitory factor |
| RIN | = | RNA integrity number |
| SAT | = | subcutaneous adipose tissue |
| TFBS | = | transcription factor binding site |
| VAT | = | visceral adipose tissue |
Additional material
Download Zip (1.1 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors acknowledge the contribution of bariatric surgeons (S. Biron, S. Marceau, O. Lescelleur, L. Biertho) for tissue collection. The authors also express their gratitude to Céline Bélanger, Chicoutimi Hospital, for her thoughtful language revision of the manuscript. A.T. is the director of a Research Chair in Bariatric and Metabolic Surgery. M.C.V. is the recipient of the Canada Research Chair in Genomics Applied to Nutrition and Health. During this research, A.A.H. was a recipient of a FRQS doctoral training award. M.F.H. is supported by a Canadian Diabetes Association clinical scientist award. L.B. and M.F.H. are junior research scholars from the Fonds de recherche du Québec – Santé (FRQS) and members of the FRQS-funded Centre de recherche clinique Étienne-Le Bel (affiliated to Centre Hospitalier de l’Université de Sherbrooke). This project was supported by the Canadian Institutes of Health Research and the FRQS.
References
- Rabe K, Lehrke M, Parhofer KG, Broedl UC. Adipokines and insulin resistance. Mol Med 2008; 14:741 - 51; http://dx.doi.org/10.2119/2008-00058.Rabe; PMID: 19009016
- Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab 2008; 93:Suppl 1 S64 - 73; http://dx.doi.org/10.1210/jc.2008-1613; PMID: 18987272
- Bouchard L, Thibault S, Guay SP, Santure M, Monpetit A, St-Pierre J, Perron P, Brisson D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010; 33:2436 - 41; http://dx.doi.org/10.2337/dc10-1024; PMID: 20724651
- Bouchard L, Hivert MF, Guay SP, St-Pierre J, Perron P, Brisson D. Placental adiponectin gene DNA methylation levels are associated with mothers’ blood glucose concentration. Diabetes 2012; 61:1272 - 80; http://dx.doi.org/10.2337/db11-1160; PMID: 22396200
- Houde AA, Hivert MF, Bouchard L. Fetal epigenetic programming of adipokines. Adipocyte 2013; 2:41 - 6; http://dx.doi.org/10.4161/adip.22055; PMID: 23700551
- Clausen TD, Mathiesen ER, Hansen T, Pedersen O, Jensen DM, Lauenborg J, Schmidt L, Damm P. Overweight and the metabolic syndrome in adult offspring of women with diet-treated gestational diabetes mellitus or type 1 diabetes. J Clin Endocrinol Metab 2009; 94:2464 - 70; http://dx.doi.org/10.1210/jc.2009-0305; PMID: 19417040
- Hillier TA, Pedula KL, Schmidt MM, Mullen JA, Charles MA, Pettitt DJ. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care 2007; 30:2287 - 92; http://dx.doi.org/10.2337/dc06-2361; PMID: 17519427
- Deierlein AL, Siega-Riz AM, Chantala K, Herring AH. The association between maternal glucose concentration and child BMI at age 3 years. Diabetes Care 2011; 34:480 - 4; http://dx.doi.org/10.2337/dc10-1766; PMID: 21216858
- Tobi EW, Lumey LH, Talens RP, Kremer D, Putter H, Stein AD, Slagboom PE, Heijmans BT. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum Mol Genet 2009; 18:4046 - 53; http://dx.doi.org/10.1093/hmg/ddp353; PMID: 19656776
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev 2002; 16:6 - 21; http://dx.doi.org/10.1101/gad.947102; PMID: 11782440
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet 2000; 9:2395 - 402; http://dx.doi.org/10.1093/hmg/9.16.2395; PMID: 11005794
- Hackett JA, Surani MA. DNA methylation dynamics during the mammalian life cycle. Philos Trans R Soc Lond B Biol Sci 2013; 368:20110328; http://dx.doi.org/10.1098/rstb.2011.0328; PMID: 23166392
- Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet 2013; 14:204 - 20; http://dx.doi.org/10.1038/nrg3354; PMID: 23400093
- Kangaspeska S, Stride B, Métivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, Benes V, Gannon F, Reid G. Transient cyclical methylation of promoter DNA. Nature 2008; 452:112 - 5; http://dx.doi.org/10.1038/nature06640; PMID: 18322535
- Houde AA, Guay SP, Desgagné V, Hivert MF, Baillargeon JP, St-Pierre J, Perron P, Gaudet D, Brisson D, Bouchard L. Adaptations of placental and cord blood ABCA1 DNA methylation profile to maternal metabolic status. Epigenetics 2013; 8:1289 - 302; http://dx.doi.org/10.4161/epi.26554; PMID: 24113149
- Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, et al, MuTHER Consortium. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet 2012; 8:e1002629; http://dx.doi.org/10.1371/journal.pgen.1002629; PMID: 22532803
- Lesseur C, Armstrong DA, Paquette AG, Koestler DC, Padbury JF, Marsit CJ. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol Cell Endocrinol 2013; 381:160 - 7; http://dx.doi.org/10.1016/j.mce.2013.07.024; PMID: 23911897
- Zhang Y, Kent JW 2nd, Lee A, Cerjak D, Ali O, Diasio R, Olivier M, Blangero J, Carless MA, Kissebah AH. Fa-tty acid binding protein 3 (fabp3) is associated with insulin, lipids and cardiovascular phenotypes of the metabolic syndrome through epigenetic modifications in a Northern European family population. BMC Med Genomics 2013; 6:9; http://dx.doi.org/10.1186/1755-8794-6-9; PMID: 23510163
- Liang P, Song F, Ghosh S, Morien E, Qin M, Mahmood S, Fujiwara K, Igarashi J, Nagase H, Held WA. Genome-wide survey reveals dynamic widespread tissue-specific changes in DNA methylation during development. BMC Genomics 2011; 12:231; http://dx.doi.org/10.1186/1471-2164-12-231; PMID: 21569359
- Wan J, Oliver VF, Zhu H, Zack DJ, Qian J, Merbs SL. Integrative analysis of tissue-specific methylation and alternative splicing identifies conserved transcription factor binding motifs. Nucleic Acids Res 2013; 41:8503 - 14; http://dx.doi.org/10.1093/nar/gkt652; PMID: 23887936
- Yagi S, Hirabayashi K, Sato S, Li W, Takahashi Y, Hirakawa T, Wu G, Hattori N, Hattori N, Ohgane J, et al. DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression. Genome Res 2008; 18:1969 - 78; http://dx.doi.org/10.1101/gr.074070.107; PMID: 18971312
- Obesity WHO. preventing and managing the global epidemic. Report of a WHO Consultation. WHO Technical Report Series 894. Geneva: World Health Organization, 2000.
- Heijmans BT, Tobi EW, Lumey LH, Slagboom PE. The epigenome: archive of the prenatal environment. Epigenetics 2009; 4:526 - 31; http://dx.doi.org/10.4161/epi.4.8.10265; PMID: 19923908
- Slieker RC, Bos SD, Goeman JJ, Bovée JV, Talens RP, van der Breggen R, Suchiman HE, Lameijer EW, Putter H, van den Akker EB, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 2013; 6:26; http://dx.doi.org/10.1186/1756-8935-6-26; PMID: 23919675
- Melzner I, Scott V, Dorsch K, Fischer P, Wabitsch M, Brüderlein S, Hasel C, Möller P. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J Biol Chem 2002; 277:45420 - 7; http://dx.doi.org/10.1074/jbc.M208511200; PMID: 12213831
- Marchi M, Lisi S, Curcio M, Barbuti S, Piaggi P, Ceccarini G, Nannipieri M, Anselmino M, Di Salvo C, Vitti P, et al. Human leptin tissue distribution, but not weight loss-dependent change in expression, is associated with methylation of its promoter. Epigenetics 2011; 6:1198 - 206; http://dx.doi.org/10.4161/epi.6.10.16600; PMID: 21931275
- Stöger R. In vivo methylation patterns of the leptin promoter in human and mouse. Epigenetics 2006; 1:155 - 62; http://dx.doi.org/10.4161/epi.1.4.3400; PMID: 17965621
- Mason MM, He Y, Chen H, Quon MJ, Reitman M. Regulation of leptin promoter function by Sp1, C/EBP, and a novel factor. Endocrinology 1998; 139:1013 - 22; PMID: 9492033
- Wrann CD, Rosen ED. New insights into adipocyte-specific leptin gene expression. Adipocyte 2012; 1:168 - 72; http://dx.doi.org/10.4161/adip.20574; PMID: 23700528
- Montague CT, Prins JB, Sanders L, Digby JE, O’Rahilly S. Depot- and sex-specific differences in human leptin mRNA expression: implications for the control of regional fat distribution. Diabetes 1997; 46:342 - 7; http://dx.doi.org/10.2337/diab.46.3.342; PMID: 9032087
- Kristensen K, Pedersen SB, Fisker S, Nørrelund H, Rosenfalck AM, Jørgensen JO, Richelsen B. Serum leptin levels and leptin expression in growth hormone (GH)-deficient and healthy adults: influence of GH treatment, gender, and fasting. Metabolism 1998; 47:1514 - 9; http://dx.doi.org/10.1016/S0026-0495(98)90079-8; PMID: 9867083
- Lesseur C, Armstrong DA, Murphy MA, Appleton AA, Koestler DC, Paquette AG, Lester BM, Marsit CJ. Sex-specific associations between placental leptin promoter DNA methylation and infant neurobehavior. Psychoneuroendocrinology 2014; 40:1 - 9; http://dx.doi.org/10.1016/j.psyneuen.2013.10.012; PMID: 24485470
- Lesseur C, Armstrong DA, Paquette AG, Koestler DC, Padbury JF, Marsit CJ. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol Cell Endocrinol 2013; 381:160 - 7; http://dx.doi.org/10.1016/j.mce.2013.07.024; PMID: 23911897
- Hoffstedt J, Eriksson P, Mottagui-Tabar S, Arner P. A polymorphism in the leptin promoter region (-2548 G/A) influences gene expression and adipose tissue secretion of leptin. Horm Metab Res 2002; 34:355 - 9; http://dx.doi.org/10.1055/s-2002-33466; PMID: 12189581
- Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 2012; 13:R43; http://dx.doi.org/10.1186/gb-2012-13-6-r43; PMID: 22703893
- Brøns C, Jacobsen S, Nilsson E, Rönn T, Jensen CB, Storgaard H, Poulsen P, Groop L, Ling C, Astrup A, et al. Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J Clin Endocrinol Metab 2010; 95:3048 - 56; http://dx.doi.org/10.1210/jc.2009-2413; PMID: 20410232
- Marceau P, Hould FS, Simard S, Lebel S, Bourque RA, Potvin M, Biron S. Biliopancreatic diversion with duodenal switch. World J Surg 1998; 22:947 - 54; http://dx.doi.org/10.1007/s002689900498; PMID: 9717420
- Vohl MC, Sladek R, Robitaille J, Gurd S, Marceau P, Richard D, Hudson TJ, Tchernof A. A survey of genes differentially expressed in subcutaneous and visceral adipose tissue in men. Obes Res 2004; 12:1217 - 22; http://dx.doi.org/10.1038/oby.2004.153; PMID: 15340102
- Robitaille J, Després JP, Pérusse L, Vohl MC. The PPAR-gamma P12A polymorphism modulates the relationship between dietary fat intake and components of the metabolic syndrome: results from the Québec Family Study. Clin Genet 2003; 63:109 - 16; http://dx.doi.org/10.1034/j.1399-0004.2003.00026.x; PMID: 12630956
- Bouchard L, Faucher G, Tchernof A, Deshaies Y, Lebel S, Hould FS, Marceau P, Vohl MC. Comprehensive genetic analysis of the dipeptidyl peptidase-4 gene and cardiovascular disease risk factors in obese individuals. Acta Diabetol 2009; 46:13 - 21; http://dx.doi.org/10.1007/s00592-008-0049-4; PMID: 18682883
- Dejeux E, El abdalaoui H, Gut IG, Tost J. Identification and quantification of differentially methylated loci by the pyrosequencing technology. Methods Mol Biol 2009; 507:189 - 205; http://dx.doi.org/10.1007/978-1-59745-522-0_15; PMID: 18987816
- Royo JL, Galán JJ. Pyrosequencing for SNP genotyping. Methods Mol Biol 2009; 578:123 - 33; http://dx.doi.org/10.1007/978-1-60327-411-1_7; PMID: 19768590
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996 - 1006; http://dx.doi.org/10.1101/gr.229102; PMID: 12045153