
Abstract
Adipose tissue serves as both a storage site for excess calories and as an endocrine organ, secreting hormones such as adiponectin that promote metabolic homeostasis. In obesity, adipose tissue expands primarily by hypertrophy (enlargement of existing adipocytes) rather than hyperplasia (generation of new adipocytes via adipogenic differentiation of preadipocytes). Progressive adipocyte hypertrophy leads to inflammation, insulin resistance, dyslipidemia, and ectopic lipid deposition, the hallmark characteristics of metabolic disease. We demonstrate that during chronic high fat feeding in mice, adipogenic differentiation is impaired due to the actions of histone deacetylase 9 (HDAC9), a member of the class II family of HDACs. Mechanistically, upregulated HDAC9 expression blocks the adipogenic differentiation program during chronic high fat feeding, leading to accumulation of improperly differentiated adipocytes with diminished expression of adiponectin. These adipocytes are inefficient at storing lipid, resulting in ectopic lipid deposition in the liver. HDAC9 gene deletion prevents the detrimental effects of chronic high fat feeding on adipogenic differentiation, increases adiponectin expression, and enhances energy expenditure by promoting beige adipogenesis, thus leading to reduced body mass and improved metabolic homeostasis. HDAC9 is therefore emerging as a critical regulator of adipose tissue health and a novel therapeutic target for obesity-related disease.
Introduction
Adipose tissue plays a fundamental metabolic role to store dietary calories as triglyceride lipid within the adipocytes, the major cellular component of the adipose tissue. Adipocytes also function as endocrine cells to produce various hormones (known as “adipokines”) that regulate whole-body insulin sensitivity, systemic glucose tolerance, and other metabolic activities. The number of adipocytes in humans is maintained at a remarkably constant level throughout adult life, despite the fact that adipocytes are constantly turning over.Citation1-6 Since adipocytes are terminally differentiated, they are replenished by adipogenic precursor cells, termed preadipocytes, which are contained within adipose depots and differentiate into mature adipocytes in response to metabolic cues. Conversion of preadipocytes to adipocytes (referred to as “adipogenic differentiation” or “adipogenesis”) is a carefully regulated cellular event, tightly linked to metabolic activities at the whole-body level, and orchestrated at the cellular level by the actions of a number of transcription factors, including C/EBPα and PPARγ.
Adipogenesis is Constrained during Obesity
To accommodate excess dietary calories, adipose tissue can expand by hyperplasia (increasing adipocyte numbers through adipogenic differentiation) and/or hypertrophy (increasing the size of individual adipocytes). During chronic caloric excess, adipogenic differentiation is insufficient to match demand, so adipocyte hypertrophy is the prevailing mechanism of adipose tissue expansion in obesity. Lipid overloading eventually causes these adipocytes to become enlarged beyond their physiological limit, triggering cellular stress. This in turn leads to inflammation, insulin resistance, dyslipidemia, and ectopic lipid deposition in the liver and other organs, hallmark characteristics of metabolic disease ().Citation7,8
Figure 1. Schematic diagram showing adipose tissue dynamics in lean and obese states. Mesenchymal stem cells generate committed preadipocytes that differentiate into unilocular white and multilocular beige (with increased mitochondrial content) adipocytes in response to adipogenic signals to maintain metabolic homeostasis. Chronic high fat diet (HFD)-induced obesity impairs adipogenesis, promotes enlargement of individual adipocytes, decreases beige adipocyte abundance, and increases adipose tissue inflammatory cells, leading to insulin resistance and other consequences of metabolic disease. Decline in histone deacetylase 9 (HDAC9) level precedes activation of the differentiation program, while elevated HDAC9 levels in HFD fed condition impairs activation of this program, promoting adipocyte hypertrophy.
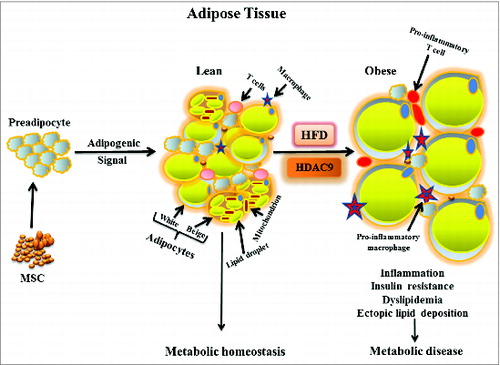
It is not clear why adipogenic differentiation is constrained during chronic caloric excess. Adipose tissue contains a huge reserve of precursor cells (adipose-derived stem cells and committed preadipocytes)—representing approximately 20–40% of the adipose tissue cellularity to support continuous remodeling of this tissue.Citation9 It is estimated that daily turn-over of preadipocytes and adipocytes could be as high as 5% under some conditions,Citation3 and obesity markedly stimulates preadipocyte replication.Citation2 This does not, however, translate into generation of sufficient numbers of mature, functionally competent adipocytes to store the excess calories efficiently. Indeed, adipocytes that accumulate in obese adipose tissues are inadequately differentiated, exhibiting markedly reduced levels of adipogenic differentiation-specific genes.Citation10 Defining the causes and consequences of constrained adipogenic differentiation in obesity is a matter of great interest in obesity research. We recently demonstrated that preadipocytes isolated from high fat diet-induced obese mice exhibit impaired adipogenic differentiation in vitro, suggesting that differentiation is constrained by factors endogenous to the cells themselves rather than the external environmental.Citation10 The fact that impaired differentiation persists in vitro, even after several passages in culture, implies an epigenetic mechanism as the probable cause of impaired adipogenic differentiation in high fat diet-induced obesity.
Histone Deacetylase 9 (HDAC9) is an Epigenetic Regulator of Adipogenesis
Epigenetic processes introduce specific alterations in the chromatin structure which persist through cell divisions, allowing stable changes in gene expression that impart “memory” to the cells.Citation11 To begin to understand how adipogenic differentiation is epigenetically disrupted by diet-induced obesity, we turned our attention to understanding the epigenetic mechanisms that control physiologic adipogenic differentiation in lean mice. Our investigations have focused on the chromatin histone modifying enzyme HDAC9, whose dynamic pattern of expression during adipogenic differentiation stands out among all of the other members of the HDAC/histone acetyltransferase family of genes.Citation12 In lean states, HDAC9 is expressed at high levels in preadipocytes, and its level abruptly falls at the onset of adipogenic differentiation in both humans and mice.Citation10 Elevating HDAC9 levels by transient transfection prevents adipogenic differentiation, while genetic ablation of HDAC9 accelerates this process,Citation12 documenting that HDAC9 negatively regulates adipogenic differentiation. HDAC9 gene deletion, however, does not spontaneously activate adipogenic differentiation, suggesting that the abrupt fall in HDAC9 levels is required to prime responsiveness to adipogenic signals. The mechanisms whereby HDAC9 inhibits adipogenic differentiation are not fully elucidated. Current evidence suggests that HDAC9 blocks the action of key transcription factors to commence the adipogenic differentiation program.Citation12,13 This effect appears to be related to interactions between HDAC9 and transcriptional repressor proteins in the cell nucleus, independent of HDAC9's enzymatic activity. Indeed, as a class II HDAC, HDAC9 possesses very weak histone deacetylase enzymatic activity. These findings are consistent with a growing body of data implicating deacetylase-independent actions of many HDAC family members.Citation14
We subsequently determined that HDAC9 is a nutrient sensitive gene whose expression is upregulated in adipocytes from chronically high fat fed, obese mice.Citation10 Under these conditions, HDAC9 levels fail to decline in preadipocytes during in vitro adipogenic differentiation, suggesting that the regulatory mechanism controlling HDAC9 expression is disrupted in diet-induced obesity.Citation10 Consequently, the adipogenic differentiation program is blocked, leading to generation of improperly differentiated adipocytes with impaired endocrine function and blunted lipid storage capacity.Citation10 Importantly, HDAC9 gene deletion removes this block, allowing appropriate activation of the adipogenic differentiation program in adipocytes during chronic high fat feeding.Citation10 This supports a role for HDAC9 as a molecular mediator of impaired adipogenic differentiation in obesity. How HDAC9 expression is regulated during adipogenic differentiation in lean mice, and how this process is pathologically disrupted in obesity, remain to be elucidated.
HDAC9 Gene Deletion Protects Mice against Obesity-Related Metabolic Disease
Based on the observations that upregulated HDAC9 expression constrains adipogenic differentiation in obesity, we postulated that HDAC9 gene deletion would protect against obesity-related metabolic disease by generating more functionally competent adipocytes capable of storing lipid, thus distributing the excess calories into a larger number of adipocytes and reducing the extent of adipocyte hypertrophy leading to cellular stress. Indeed, during chronic high fat diet, HDAC9 knockout mice exhibit smaller adipocytes, increased insulin sensitivity, enhanced glucose tolerance, and reduced hepatic lipid accumulation compared with wild-type mice despite similar food consumption and locomotor activities.Citation10
HDAC9 Knockout Mice Exhibit Increased Energy Expenditure and Enhanced Adaptive Thermogenesis
In addition to their improved metabolic state, HDAC9 knockout mice exhibit lower body weight and smaller adipose tissue mass compared with wild-type mice, both under chow and high fat fed conditions.Citation10 This phenotype was not explained by differences in nutrient absorption between the two strains of mice (data not shown). This suggests an additional mechanism whereby HDAC9 knockout mice are protected against obesity-related disease: enhanced combustion of surplus dietary calories to produce dissipating heat energy. In support of this notion, HDAC9 knockout mice exhibit increased oxygen consumption, elevated energy expenditure, and enhanced adaptive thermogenesis.Citation10
Traditionally, adipocytes are classified as energy storing white adipocytes and energy combusting brown adipocytes, the latter of which turn dietary calories into dissipating heat energy.Citation15 Our investigations, however, revealed that brown adipose tissue in the HDAC9 knockout mice, although phenotypically indistinguishable from wild-type mice, is actually reduced in mass, suggesting that increased calorie burning capacity of HDAC9 knockout mice is not related to differences in their brown adipose tissue content. We therefore sought an alternate explanation for the increased energy expenditure in HDAC9 knockout mice.
HDAC9 Gene Deletion Augments “Beige” Adipocytes during Obesity
In addition to white and brown adipocytes, a third class of adipocytes has recently been identified in subcutaneous adipose tissues of both mice and humans, which, like brown adipocytes, also turn dietary calories to dissipating heat energy.Citation16 The abundance of this new class of adipocytes, referred hereafter as “beige” adipocytes, is inversely related to obesity and insulin resistance in humans,Citation17-19 suggesting a potentially important role for beige adipocytes in modulating obesity and its consequences. Consistent with the human studies, we found that beige adipocyte abundance is reduced in chronically high fat fed obese wild-type mice.Citation10
Interestingly, we observed that HDAC9 gene deletion increases beige adipocyte abundance in subcutaneous adipose tissues of lean mice, as evidenced by increased appearance of multilocular adipocytes (), and expression of beige-specific genes PRDM16, UCP1, CIDEA, and PGC1α. Moreover, HDAC9 gene deletion prevented the reduction in the beige adipocyte gene expression during chronic high fat diet-induced obesity.Citation10 These findings likely help to explain the observations of elevated energy expenditure, increased oxygen consumption, and enhanced adaptive thermogenesis in HDAC9 knockout mice.Citation10 Enhanced beige adipocytes in HDAC9 knockout mice may also cause and/or compensate for the loss of brown adipose tissue mass in these mice. How HDAC9 gene deletion increases beige adipocyte abundance is unknown; nor it is known how beige and brown adipose tissues cross-talk with each other to maintain metabolic energy homeostasis in mice. Brown adipose tissue loss was reported to induce a compensatory increase in beige adipocytes by altering sympathetic innervation of the white adipose tissues.Citation20 Indeed, pharmacological activation of β3 adrenergic receptors augments beige phenotypic changes of white adipose tissue.Citation21 These adipogenic precursor cells also undergo β-adrenergic receptor-independent activation of their beige adipogenic differentiation program,Citation22,23 pointing to involvement of potentially multiple mechanisms. Identification of HDAC9, a nutrient sensitive gene, as a critical regulator of the calorie burning beige adipogenic program provides molecular insight how dietary caloric intake regulates caloric consumption pathways.
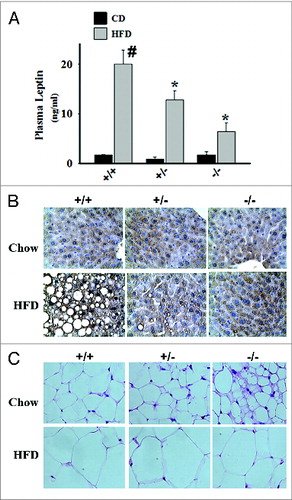
Figure 2. Effects of HDAC9 gene deletion on plasma leptin levels, hepatic lipid accumulation, and multilocular beige adipocyte abundance during chronic high fat diet. (A) Compared with chow diet, feeding 8-wk old male C57BL/6J mice with a high fat diet (60% calories from fat) for 12 wk dramatically increases plasma leptin levels. Deletion of one or both alleles of the HDAC9 gene causes a gene-dose dependent reduction in plasma leptin levels in these mice. (B) Chronic high fat diet causes significant hepatic lipid accumulation in wild type mice, which is prevented by the deletion of one or both alleles of HDAC9 gene. (C) HDAC9 gene deletion (–/–) noticeably increased the abundance of multilocular beige adipocytes in subcutaneous adipose tissue, especially in chow fed mice. Data are mean ± SEM of 4 experiments; #P < 0.05 compared with chow fed wild-type (+/+) mice, *P < 0.05 compared with high fat fed wild-type (+/+) mice.
HDAC9 Gene Deletion Prevents Fibroblast Growth Factor 21 (FGF21) Downregulation in Adipocytes of Obese Mice
FGF21, a member of the FGF family of proteins, functions as an endocrine hormone and exhibits therapeutic potential as an anti-obesity and anti-diabetic agent in preclinical studies.Citation24-26 In addition to liver,Citation27,28 FGF21 is secreted by the adipocytes,Citation29 where it acts as a local autocrine/paracrine factor to enhance beige adipogenesis, presumably through a PGC1α-dependent mechanism.Citation30 FGF21 stimulates thermogenic gene expression in white adipose tissue, supporting adaptive thermogenesis, fatty acid oxidation, and lipid metabolism.Citation31-33 We demonstrated significant FGF21 expression in subcutaneous, but not in visceral, adipose tissues of lean mice, which was markedly blunted in the setting of chronic high fat diet-induced obesity,Citation10 indicating that FGF21 is a nutrient sensitive gene. While HDAC9 gene deletion did not affect FGF21 expression in lean mice, it completely prevented the reduction in FGF21 expression in obese mice.Citation10 How chronic high fat diet-induced obesity reduces FGF21 levels in subcutaneous adipose issue is not known, nor is it known how HDAC9 gene deletion reverses this process. FGF21 is expressed in beige adipocytes,Citation29 so it is possible that the reduction in FGF21 levels during obesity could be consequent to reduced beige adipocyte abundance. In such case, HDAC9 could indirectly regulate FGF21 levels in subcutaneous adipose tissue through its influence on beige adipogenesis. Future investigations with sorted cells from subcutaneous adipose depots are required to demonstrate whether FGF21 is expressed exclusively by the beige adipocytes to act in autocrine fashion, or whether it is expressed by other cells in the adipose depot.
HDAC9 as a Potential Therapeutic Target in Obesity: Present Understanding and Future Perspectives
HDAC9 is emerging as an important regulatory molecule capable of profoundly influencing adipose tissue biology and metabolic homeostasis. The marked upregulation of HDAC9 expression in adipose tissues during chronic high fat diet-induced obesity clearly contributes to impaired adipogenic differentiation, as evidenced by our findings in HDAC9 knockout mice.Citation10 It appears that HDAC9 gene deletion improves metabolic state not only through its favorable effects on adipogenic differentiation, but also by upregulating beige adipogenesis.Citation10 Thus, inhibiting the actions of HDAC9 may favorably regulate both the balance between adipocyte hypertrophy and hyperplasia and the balance between energy storage and consumption in obesity. This dual effect potentially sets HDAC9 apart from many other potential therapeutic targets for obesity-related metabolic disease.
How could one potentially therapeutically target HDAC9 in obesity? First, it is important to point out that non-selective or selective HDAC enzymatic inhibitors would most likely not mimic the effects of HDAC9 gene deletion in obesity, since HDAC9 appears to regulate adipogenesis independent of its deacetylase enzymatic activity.Citation12 Indeed, overexpression of a truncated C-terminal mutant of HDAC9 that lacks the enzymatic domain produced equivalent impairment of adipogenic differentiation as compared with the full-length molecule, while overexpression of an N-terminal mutant that lacks the nuclear localization sequence but possesses full enzymatic activity had no effect.Citation12 Consistent with this notion, a recent study reported that a class II HDAC enzymatic inhibitor, MC1568, did not improve metabolic disease in obese mice.Citation34 Class II HDACs (HDAC 4–7, 9, 11) exhibit weak or no histone deacetylase enzymatic activity and are considered as pseudoenzymes;Citation35 their deacetylase activity relies primarily on recruitment of catalytically potent class I HDACs (HDAC 1–3, 8, 10). Interestingly, deacetylase enzymatic activity likewise does not underlie all of the functional effects of class I HDACs. For example, HDAC3, a class I HDAC, regulates gene expression and metabolic activities in the liver, but its histone deacetylase activity is completely dispensable for these functions.Citation14 Thus, it is perhaps not surprising that HDAC enzymatic inhibitors do not always phenocopy the effects of HDAC knockdown or gene deletion, and in some cases, may yield opposite results.
Evidence is emerging that HDAC9, like other class II and class I HDACs, modulates gene expression by interacting with nuclear repressor complexes.Citation36-38 HDAC9 likely acts as a scaffold protein in the cell nucleus by physically interacting with transcriptional repressors and transcription factors (USF1, PPARγ, etc.) at discreet transcription sites to influence gene expression.Citation12,13,39 HDAC9 also regulates epigenetic histone modifications at selective gene targets to influence gene expression.Citation40-44 More research is required to define HDAC9's nuclear binding partners in order to understand how it impairs adipogenesis; this information can then be used to develop a therapeutic strategy that is likely to be effective. It is important to point out that there is a clear gene-dose effect of HDAC9 to influence metabolic disease in chronic high fat fed mice. HDAC9 heterozygote mice display an “intermediate phenotype” with regard to many aspects of diet-induced obesity: they gain less weight (not shown) and exhibit lower plasma leptin levels as compared with wild-type mice (), and the extent of ectopic lipid accumulation in the liver is dramatically reduced in the heterozygotes, approaching that seen in HDAC9 homozygous knockout mice (). This remarkable gene-dose effect of HDAC9 expression on metabolic parameters suggests that a moderate degree of inhibition of HDAC9, such as is commonly achieved with pharmacotherapy, could favorably impact obesity-related metabolic disease.
How HDAC9's molecular actions are functionally linked to the global metabolic state in obesity remains to be ascertained. It should be pointed out that, in addition to adipose tissue, HDAC9 is expressed in other metabolically relevant tissues, including liver, skeletal muscle, and pancreas,Citation45-47 as well as in immune cells.Citation48 This expression pattern suggests that HDAC9 is globally positioned to influence metabolic disease by influencing multiple target tissues and cells. Our laboratory is currently examining the tissue-specific role of HDAC9 in metabolic disease during chronic high fat diet-induced obesity and in genetic models of obesity. Hopefully, these investigations will reveal which tissues are critical in transducing the actions of HDAC9 in obesity, which could be important from a therapeutic perspective. In addition, HDAC9 is expressed in vascular cells and tissues.Citation49 Elevated vascular HDAC9 expression was reported to be associated with aortic and femoral atherosclerosis as well as carotid artery disease and large vessel ischemic stroke in humans.Citation49 Moreover, a recent genome-wide association study found two single nucleotide polymorphic (SNP) variants of HDAC9 to be strongly linked to large blood vessel disease in humans.Citation50 These findings raise the intriguing possibility that therapeutically targeting HDAC9 in obesity could also favorably modulate the development of cardiovascular complications, a major cause of morbidity and mortality in this population.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Berry R, Jeffery E, Rodeheffer MS. Weighing in on Adipocyte Precursors. Cell Metab 2014; 19:8-20; PMID:24239569; http://dx.doi.org/10.1016/j.cmet.2013.10.003
- Joe AW, Yi L, Even Y, Vogl AW, Rossi FM. Depot-specific differences in adipogenic progenitor abundance and proliferative response to high-fat diet. Stem Cells 2009; 27:2563-70; PMID:19658193; http://dx.doi.org/10.1002/stem.190
- Rigamonti A, Brennand K, Lau F, Cowan CA. Rapid cellular turnover in adipose tissue. PLoS One 2011; 6:e17637; PMID:21407813; http://dx.doi.org/10.1371/journal.pone.0017637
- Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Näslund E, Britton T, et al. Dynamics of fat cell turnover in humans. Nature 2008; 453:783-7; PMID:18454136; http://dx.doi.org/10.1038/nature06902
- Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab 2013; 18:355-67; PMID:24011071; http://dx.doi.org/10.1016/j.cmet.2013.08.003
- Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med 2013; 19:1338-44; PMID:23995282; http://dx.doi.org/10.1038/nm.3324
- Blüher M. Adipose tissue dysfunction contributes to obesity related metabolic diseases. Best Pract Res Clin Endocrinol Metab 2013; 27:163-77; PMID:23731879; http://dx.doi.org/10.1016/j.beem.2013.02.005
- Hauner H. Adipose tissue inflammation: are small or large fat cells to blame? Diabetologia 2010; 53:223-5; PMID:19937224; http://dx.doi.org/10.1007/s00125-009-1605-3
- Hauner H. Secretory factors from human adipose tissue and their functional role. Proc Nutr Soc 2005; 64:163-9; PMID:15960861; http://dx.doi.org/10.1079/PNS2005428
- Chatterjee TK, Basford JE, Knoll E, Tong WS, Blanco V, Blomkalns AL, Rudich S, Lentsch AB, Hui DY, Weintraub NL. HDAC9 knockout mice are protected from adipose tissue dysfunction and systemic metabolic disease during high-fat feeding. Diabetes 2014; 63:176-87; PMID:24101673; http://dx.doi.org/10.2337/db13-1148
- Cantone I, Fisher AG. Epigenetic programming and reprogramming during development. Nat Struct Mol Biol 2013; 20:282-9; PMID:23463313; http://dx.doi.org/10.1038/nsmb.2489
- Chatterjee TK, Idelman G, Blanco V, Blomkalns AL, Piegore MG Jr., Weintraub DS, Kumar S, Rajsheker S, Manka D, Rudich SM, et al. Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J Biol Chem 2011; 286:27836-47; PMID:21680747; http://dx.doi.org/10.1074/jbc.M111.262964
- Chen YH, Yeh FL, Yeh SP, Ma HT, Hung SC, Hung MC, Li LY. Myocyte enhancer factor-2 interacting transcriptional repressor (MITR) is a switch that promotes osteogenesis and inhibits adipogenesis of mesenchymal stem cells by inactivating peroxisome proliferator-activated receptor gamma-2. J Biol Chem 2011; 286:10671-80; PMID:21247904; http://dx.doi.org/10.1074/jbc.M110.199612
- Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, Everett LJ, Nabel CS, Li Y, Selvakumaran V, et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell 2013; 52:769-82; PMID:24268577; http://dx.doi.org/10.1016/j.molcel.2013.10.022
- Herzig S, Wolfrum C. Brown and white fat: from signaling to disease. Biochim Biophys Acta 2013; 1831:895; PMID:23561705; http://dx.doi.org/10.1016/j.bbalip.2013.03.009
- Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev 2013; 27:234-50; PMID:23388824; http://dx.doi.org/10.1101/gad.211649.112
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med 2009; 360:1500-8; PMID:19357405; http://dx.doi.org/10.1056/NEJMoa0808718
- Vijgen GH, Bouvy ND, Teule GJ, Brans B, Schrauwen P, van Marken Lichtenbelt WD. Brown adipose tissue in morbidly obese subjects. PLoS One 2011; 6:e17247; PMID:21390318; http://dx.doi.org/10.1371/journal.pone.0017247
- Vijgen GH, Bouvy ND, Teule GJ, Brans B, Hoeks J, Schrauwen P, van Marken Lichtenbelt WD. Increase in brown adipose tissue activity after weight loss in morbidly obese subjects. J Clin Endocrinol Metab 2012; 97:E1229-33; PMID:22535970; http://dx.doi.org/10.1210/jc.2012-1289
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, Tseng YH. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 2013; 495:379-83; PMID:23485971; http://dx.doi.org/10.1038/nature11943
- Lee YH, Petkova AP, Mottillo EP, Granneman JG. In vivo identification of bipotential adipocyte progenitors recruited by β3-adrenoceptor activation and high-fat feeding. Cell Metab 2012; 15:480-91; PMID:22482730; http://dx.doi.org/10.1016/j.cmet.2012.03.009
- Ye L, Wu J, Cohen P, Kazak L, Khandekar MJ, Jedrychowski MP, Zeng X, Gygi SP, Spiegelman BM. Fat cells directly sense temperature to activate thermogenesis. Proc Natl Acad Sci U S A 2013; 110:12480-5; PMID:23818608; http://dx.doi.org/10.1073/pnas.1310261110
- Chen KY, Brychta RJ, Linderman JD, Smith S, Courville A, Dieckmann W, Herscovitch P, Millo CM, Remaley A, Lee P, et al. Brown fat activation mediates cold-induced thermogenesis in adult humans in response to a mild decrease in ambient temperature. J Clin Endocrinol Metab 2013; 98:E1218-23; PMID:23780370; http://dx.doi.org/10.1210/jc.2012-4213
- Foltz IN, Hu S, King C, Wu X, Yang C, Wang W, Weiszmann J, Stevens J, Chen JS, Nuanmanee N, et al. Treating diabetes and obesity with an FGF21-mimetic antibody activating the βKlotho/FGFR1c receptor complex. Sci Transl Med 2012; 4:ra153; PMID:23197570; http://dx.doi.org/10.1126/scitranslmed.3004690
- Alisi A, Panera N, Nobili V. Commentary: FGF21 holds promises for treating obesity-related insulin resistance and hepatosteatosis. Endocrinology 2014; 155:343-6; PMID:24248463; http://dx.doi.org/10.1210/en.2013-1828
- Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab 2013; 18:333-40; PMID:24011069; http://dx.doi.org/10.1016/j.cmet.2013.08.005
- Yilmaz Y, Eren F, Yonal O, Kurt R, Aktas B, Celikel CA, Ozdogan O, Imeryuz N, Kalayci C, Avsar E. Increased serum FGF21 levels in patients with nonalcoholic fatty liver disease. Eur J Clin Invest 2010; 40:887-92; PMID:20624171; http://dx.doi.org/10.1111/j.1365-2362.2010.02338.x
- Hondares E, Rosell M, Gonzalez FJ, Giralt M, Iglesias R, Villarroya F. Hepatic FGF21 expression is induced at birth via PPARalpha in response to milk intake and contributes to thermogenic activation of neonatal brown fat. Cell Metab 2010; 11:206-12; PMID:20197053; http://dx.doi.org/10.1016/j.cmet.2010.02.001
- Lee P, Werner CD, Kebebew E, Celi FS. Functional thermogenic beige adipogenesis is inducible in human neck fat. Int J Obes 2014; 38:170-6; PMID:23736373; http://dx.doi.org/10.1038/ijo.2013.82
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 2012; 26:271-81; PMID:22302939; http://dx.doi.org/10.1101/gad.177857.111
- De Sousa-Coelho AL, Relat J, Hondares E, Pérez-Martí A, Ribas F, Villarroya F, Marrero PF, Haro D. FGF21 mediates the lipid metabolism response to amino acid starvation. J Lipid Res 2013; 54:1786-97; PMID:23661803; http://dx.doi.org/10.1194/jlr.M033415
- Cuevas-Ramos D, Almeda-Valdes P, Aguilar-Salinas CA, Cuevas-Ramos G, Cuevas-Sosa AA, Gomez-Perez FJ. The role of fibroblast growth factor 21 (FGF21) on energy balance, glucose and lipid metabolism. Curr Diabetes Rev 2009; 5:216-20; PMID:19531026; http://dx.doi.org/10.2174/157339909789804396
- Murata Y, Konishi M, Itoh N. FGF21 as an Endocrine Regulator in Lipid Metabolism: From Molecular Evolution to Physiology and Pathophysiology. J Nutr Metab 2011; 2011:981315; PMID:21331285; http://dx.doi.org/10.1155/2011/981315
- Galmozzi A, Mitro N, Ferrari A, Gers E, Gilardi F, Godio C, Cermenati G, Gualerzi A, Donetti E, Rotili D, et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes 2013; 62:732-42; PMID:23069623; http://dx.doi.org/10.2337/db12-0548
- Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, Sambucini S, Bottomley MJ, Lo Surdo P, Carfí A, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A 2007; 104:17335-40; PMID:17956988; http://dx.doi.org/10.1073/pnas.0706487104
- Zhang CL, McKinsey TA, Olson EN. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc Natl Acad Sci U S A 2001; 98:7354-9; PMID:11390982; http://dx.doi.org/10.1073/pnas.131198498
- Zhang CL, McKinsey TA, Lu JR, Olson EN. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem 2001; 276:35-9; PMID:11022042; http://dx.doi.org/10.1074/jbc.M007364200
- Sparrow DB, Miska EA, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, Spohr G, Kouzarides T, Mohun TJ. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J 1999; 18:5085-98; PMID:10487760; http://dx.doi.org/10.1093/emboj/18.18.5085
- Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 2009; 136:1056-72; PMID:19303849; http://dx.doi.org/10.1016/j.cell.2008.12.040
- Méjat A, Ramond F, Bassel-Duby R, Khochbin S, Olson EN, Schaeffer L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat Neurosci 2005; 8:313-21; PMID:15711539; http://dx.doi.org/10.1038/nn1408
- Muralidhar SA, Ramakrishnan V, Kalra IS, Li W, Pace BS. Histone deacetylase 9 activates gamma-globin gene expression in primary erythroid cells. J Biol Chem 2011; 286:2343-53; PMID:21078662; http://dx.doi.org/10.1074/jbc.M110.115725
- Sugo N, Oshiro H, Takemura M, Kobayashi T, Kohno Y, Uesaka N, Song WJ, Yamamoto N. Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur J Neurosci 2010; 31:1521-32; PMID:20525066
- Yan K, Cao Q, Reilly CM, Young NL, Garcia BA, Mishra N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J Biol Chem 2011; 286:28833-43; PMID:21708950; http://dx.doi.org/10.1074/jbc.M111.233932
- Yuan Z, Peng L, Radhakrishnan R, Seto E. Histone deacetylase 9 (HDAC9) regulates the functions of the ATDC (TRIM29) protein. J Biol Chem 2010; 285:39329-38; PMID:20947501; http://dx.doi.org/10.1074/jbc.M110.179333
- Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Genet Genomics 2010; 283:341-9; PMID:20165882; http://dx.doi.org/10.1007/s00438-010-0522-y
- Lenoir O, Flosseau K, Ma FX, Blondeau B, Mai A, Bassel-Duby R, Ravassard P, Olson EN, Haumaitre C, Scharfmann R. Specific control of pancreatic endocrine β- and δ-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011; 60:2861-71; PMID:21953612; http://dx.doi.org/10.2337/db11-0440
- Zhang CL, McKinsey TA, Olson EN. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol 2002; 22:7302-12; PMID:12242305; http://dx.doi.org/10.1128/MCB.22.20.7302-7312.2002
- Beier UH, Akimova T, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr Opin Immunol 2011; 23:670-8; PMID:21798734; http://dx.doi.org/10.1016/j.coi.2011.07.002
- Markus HS, Mäkelä KM, Bevan S, Raitoharju E, Oksala N, Bis JC, O’Donnell C, Hainsworth A, Lehtimäki T. Evidence HDAC9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. Stroke 2013; 44:1220-5; PMID:23449258; http://dx.doi.org/10.1161/STROKEAHA.111.000217
- Bellenguez C, Bevan S, Gschwendtner A, Spencer CC, Burgess AI, Pirinen M, Jackson CA, Traylor M, Strange A, Su Z, et al.; International Stroke Genetics Consortium (ISGC); Wellcome Trust Case Control Consortium 2 (WTCCC2). Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat Genet 2012; 44:328-33; PMID:22306652; http://dx.doi.org/10.1038/ng.1081