Abstract
Fmoc- and Boc-protected modified monomers bearing 5-azidomethyluracil nucleobase were synthesized. Four different solid-phase synthetic strategies were tested in order to evaluate the application of this series of monomers for the solid-phase synthesis of modified PNA. The azide was used as masked amine for the introduction of amide-linked functional groups, allowing the production of a library of compounds starting from a single modified monomer. The azide function was also exploited as reactive group for the modification of PNA in solution via azide-alkyne click cycloaddition.
Introduction
The peptide nucleic acid (PNA) structure,Citation1 on account of its excellent nucleic acid recognition properties and its high chemical and biological stability, has served as a model and a robust scaffold for the synthesis of new compounds aimed at specific applications in diagnosticsCitation2-Citation4 and in the development of gene-targeting drugs,Citation5 and more recently, for the construction of nanostructured materials.Citation6,Citation7 Many modifications of the original scaffold have been proposed in order to address specific problems or to introduce functional groups with specific functions, such as fluorophores, metal binding sites, cellular carriers or reactive sites. Modified PNA have also been used to overcome some of the PNA drawbacksCitation8 such as low DNA vs. RNA specificity, low water solubility, low cellular uptake and tendency to show self-aggregation.
Synthesis of PNA-bioconjugates with additional groups can be obtained by simply coupling the desired compound to the N-terminus or by exploiting the ε-amino group of a lysine (or other amino acid) unit inserted at the C-terminus as a branching point. Although these are the preferred methods for introducing one or two functional groups on a PNA scaffold, using commercially available monomers and simple well-established chemistry, they only allow the placement of groups at the terminal positions, reducing the possibilities for the construction of complex multifunctional molecules based on the PNA structure.
A large variety of PNA backbone modifications have been described in the literatureCitation9-Citation12 and some of them showed improved biological properties in terms of affinity, mismatched recognition, DNA/RNA selectivity or cellular permeation. Some of the backbone-modified PNAs were obtained by introducing substituents at the C2 or C5 carbons of the aminoethylglycine backbone;Citation13-Citation15 these modifications, besides changing the recognition abilities through additional constrains, were also used as branching points for the insertion of lateral functional groups.Citation16 Appella and coworkers reported a C5-modified monomer bearing a Fmoc-protected lysine side chain for the introduction of a series of different groups during solid-phase synthesis.Citation17
Modification of the nucleobases is another complementary approach, and many works have appeared in the literature on this subject.Citation18 The increase of the aromatic portion of nucleobases without alteration of the H-bond donor/acceptor geometry was successfully used to stabilize the PNA:DNA duplex, with good results in terms of complex stability.Citation19-Citation21
Substitution of nucleobases with functional groups has also been used as a tool for inserting specific moieties (mainly fluorophores and intercalating agents) internally in PNA sequences. In this case, the recognition of a specific base must be sacrificed, although other factors such as stacking interactions and size/shape can favor binding to the target sequences containing specific base traits. However, these effects are not always predictable, and selectivity is rather established a posteriori. In some cases the modification of the nucleobase was obtained by replacing the natural with universal bases, which do not present any particular H-bond donor or acceptor groups able to recognize the “complementary” counterpart in the opposite strand,Citation22 but this approach failed in the application to PNA due to their higher sensitivity in mismatch recognition.
A useful approach for the insertion of additional functional groups is also the modification of nucleobases at positions not required for the recognition of the complementary nucleobase (i.e., C8 of adenine and guanine, C5 and C6 in thymine and cytosine). The C5 carbon of uracil is a common site to introduce modifications in nucleic acids, since substituents in this position are protruding into the major groove without affecting the hybridization properties. For oligonucleotides this type of modification is tolerated by polymerase reactions; for this reason there are numerous examples of C5 modified uridines bearing fluorophores, reactive groups, anchoring groups, metal chelators, etc.Citation23 This type of functionalization was, therefore, largely employed for the introduction of modifications in nucleic acids,Citation24 though this type of strategy is still not fully explored in PNA chemistry.
The introduction of modified bases bearing functional moieties along a PNA strand has been achieved by synthesizing modified PNA monomers, as described by different groups.Citation18 However, the development of new solid-phase methods able to allow the production of a large variety of modification can lead to the possibility to a fast development of modified PNA libraries. Introduction of orthogonally protected functional groups or orthogonally reactive functions on the backbone were used to achieve this goal,Citation25 but, to the best of our knowledge, there are no examples of application of these protocols to modified nucleobases. In this paper we present the synthesis of C5-modified uracil-based PNA monomers bearing an azide group, and its application to the solid-phase synthesis of PNAs whith different modifications along the strand.
Results and Discussion
Design and synthesis of the modified monomers
C5 modified PNA monomers were designed in order to introduce a masked amino group for the insertion of functional groups in the major groove. We chose the azide function as a precursor for its ability to unleash an amine function under very mild conditions, such as the Staudinger reaction, which are compatible with the presence of orthogonal protecting groups such as Boc or Fmoc, and for its possible application to Huisgen’s 1,3-dipolar cycloaddition (click reaction). The azide function is stable to both Fmoc- and Boc- chemistries, provided that no sulfur-containing scavengers are used for the deprotection or the cleavage steps.Citation26 Thus, the introduction of this functional group in both Boc- and Fmoc-protected monomers can be used in a general strategy for the rapid construction of libraries of PNA-based compounds by solid-phase or off-resin modifications without the need of an extensive solution-based syntheses of many different modified PNA monomers.
For the preparation of the modified monomers bearing the azide function (), we used as a starting material uracil 1, that can be easily hydroxymethylated with paraformaldehyde following a literature protocol,Citation27 to obtain the hydroxymethyluracil 2. This derivative was preliminarily converted into the chloride derivative 3 by acid catalyzed nucleophilic substitution and the function readily converted into azide of compound 4 using sodium azide. The regioselective alkylation of the N-1 position for the introduction of the carboxymethylene linker was achieved by ethyl or tert-butyl 2-bromoacetate to obtain, respectively, compounds 5a and 5b that, after hydrolysis (basic or acidic, respectively) of the ester functions, gave the acid 6. This derivative was then used as modified nucleobase to be inserted in the PNA scaffold; thus it was linked to the Fmoc- or Boc-protected aminoethylglycine backbone using EDC or DCC/DhBtOH as activating mixtures. Final hydrolysis (TFA in DCM or NaOH in water/MeOH) of the esters gave the final modified monomers 8a and 8b in good yields (86% and 96% respectively).
Figure 1. Synthesis of the modified monomer. (A) CH2O, Et3N in H2O, 81%; (B) HCl 37%, 80%; (C) NaN3 in DMF, 90%; (D) (1) BrCH2COOEt, K2CO3 in DMF, 41% (2) BrCH2COOtBu, K2CO3 in DMF, 49%; (E) (1) NaOH in H2O/MeOH 1:1 then HCl, 96% (2) TFA in DCM, 70%; (F) (1) EDC∙HCl, DhBtOH, DIPEA, tert-butyl 2-[(2-Fmoc-aminoethyl)amino]acetate hydrochloride in DMF, 96% (2) DCC, DhBtOH, DIPEA, ethyl 2-[(2-Fmoc-aminoethyl)amino]acetate in DMF, 77%; (G) (1) TFA in DCM, 86% (2) NaOH in H2O/MeOH 1:1, 96%.
![Figure 1. Synthesis of the modified monomer. (A) CH2O, Et3N in H2O, 81%; (B) HCl 37%, 80%; (C) NaN3 in DMF, 90%; (D) (1) BrCH2COOEt, K2CO3 in DMF, 41% (2) BrCH2COOtBu, K2CO3 in DMF, 49%; (E) (1) NaOH in H2O/MeOH 1:1 then HCl, 96% (2) TFA in DCM, 70%; (F) (1) EDC∙HCl, DhBtOH, DIPEA, tert-butyl 2-[(2-Fmoc-aminoethyl)amino]acetate hydrochloride in DMF, 96% (2) DCC, DhBtOH, DIPEA, ethyl 2-[(2-Fmoc-aminoethyl)amino]acetate in DMF, 77%; (G) (1) TFA in DCM, 86% (2) NaOH in H2O/MeOH 1:1, 96%.](/cms/asset/92336217-1d3e-417a-ab17-b8667faceb9a/kdpx_a_10920158_f0001.gif)
Solid phase modification
Monomer 8 was designed to be applied in both Boc- or Fmoc-based solid phase protocols; thus, both strategies and different modes of insertion of the modified nucleobase were tested, in order to evaluate the suitability of this modification under different conditions ().
Table 1. PNAs sequences synthesized for the evaluation of reactivity of the azide function
As a model sequence, we chose PNA targeting a trait of the cystic fibrosis (CF) gene containing the W1258X mutation, one of the most common point mutations responsible for cystic fibrosis (sequence TCCTTCACT, where the underlined character indicates the position of the mutated base). The azide-modified base was inserted in the position targeting the mutated base. PNA 1 and PNA 2 were synthesized using Fmoc-based chemistry, in order to evaluate the stability of the azide group to both deprotection and cleavage steps needed for this strategy. In the case of PNA 1 the sequence was completed, whereas PNA 2 was obtained by acetylation after the introduction of the modified monomer. Thus, by comparing the post-synthetic modifications of PNA 1 and PNA 2, it was possible to evaluate hindering effects, due to the N-terminal PNA sequence, on the reactivity of the azido group. PNA 2 was synthesized in order to evaluate the stability of the modified monomer under the conditions used in Boc-based solid phase synthesis. PNA 4 was designed in order to evaluate the possibility to use two azide-modified monomers for the simultaneous introduction of multiple functional units after completion of the PNA synthesis.
Preliminary tests for the azide reduction were performed shaking the resin twice for 90 min under a nitrogen atmosphere with a 1 M trimethylphosphine solution in THF containing 10% water according to a literature protocol.Citation28 Due to the long time required for the reduction of the group, improvements were achieved by a double treatment for 10 min (under vigorous shaking) using a water/THF/1M PMe3 3:2:1 mixture, yielding 97% conversion after the first step, with complete reduction after the second cycle, as demonstrated from UPLC-ESI-MS analysis of the crude PNA after cleavage from the resin (data not shown).
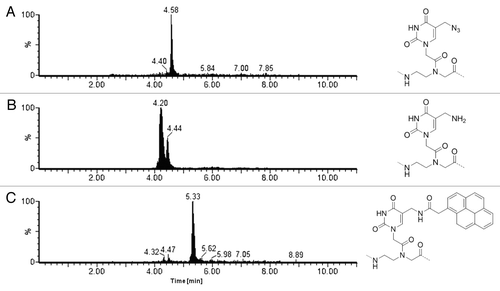
When applying this protocol to PNA 1, an amine function was generated in the middle of the sequence, which was subsequently coupled with 1-pyreneacetic acid using DIC/DhBtOH as activating system, obtaining a complete conversion of the starting material in both steps (). The same protocol was followed for PNA 2, with almost identical results, suggesting that the hindrance induced by the N-terminus of the PNA chain does not affect either the azide reduction or the amine coupling.
Figure 2. UPLC-ESI extracted ion current (XIC) traces of the cleaved resin bearing: (A) the azide function, (B) amine function after reduction with PMe3 (peak at 4.40 min corresponding to the iminophosphorane intermediate of the Staudinger reaction), (C) pyrene moiety after coupling. XIC traces were obtained by extrapolating the multicharge signal of the three different PNAs: 1342.5, 1329.5, 967.4, 895.2, 886.9, 725.7, 671.8, 665.3, 580.9 (for mass spectra see Figs. S1 and S2).
This protocol was also applied to Boc-based solid phase synthesis (), testing the possibility of modifying the nucleobase before the completion of the PNA synthesis with the remaining monomers. For this purpose, we inserted monomer 8b, and then we reduced its azide function to amine, which was then coupled with naphthalene‐2‐carboxylic acid, before the insertion of the remaining unmodified PNA monomers to complete the sequence. Finally, the product was cleaved from the resin using a TFA/TFMSA solution and purified. As shown in , the identity of the PNA was confirmed by HPLC-UV-MS analysis of the pure compound, while in the chromatogram of the crude PNA () only the target sequence is present as PNA product, though the chromatogram shows the peaks of the scavengers used in the cleavage step.
Figure 3. Synthesis of PNA 3. (A) Introduction of monomer 8b during solid-phase synthesis; (B) azide reduction by trimethyl phosphine; (C) coupling of naphthalene‐2‐carboxylic acid activated by DIC/DhBtOH; (D) elongation of the strand to final sequence.
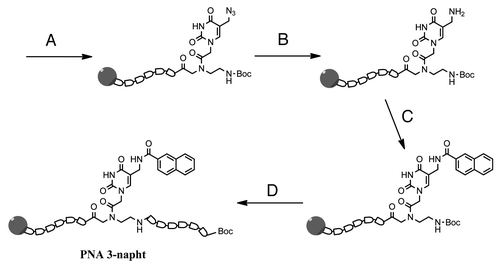
Figure 4. (A) HPLC‐UV traces of crude PNA 3-napht after modification; (B) HPLC‐UV traces of PNA after purification; (C) ESI-MS spectra of modified PNA 3. Peaks marked (*) are multicharge peaks due to source-fragmentation at the C5-methylene position of uracil.
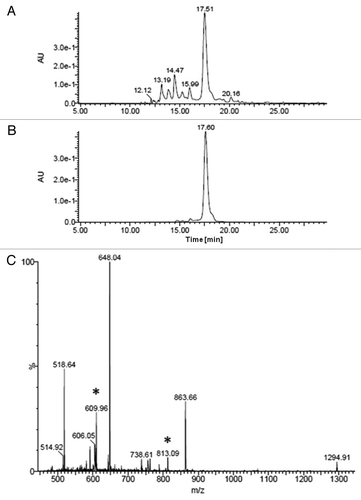
Moving further, we tested the possibility of simultaneously introducing multiple modified monomers, which can be subsequently modified in one step, on a single PNA. Thus PNA 4 was synthesized, and, while still linked to the solid support, both its azide functions were reduced at once and the resulting amines were coupled with a naphthalenediimmide derivative (napht-gly). The introduction of the double modification followed the former protocol obtaining conversion of both amines to the corresponding amides ().
Figure 5. (A) UPLC‐MS traces of PNA 4 after coupling with napht-gly; (B) ESI-MS spectra of the peak at 8.16 min.
Solution click reaction
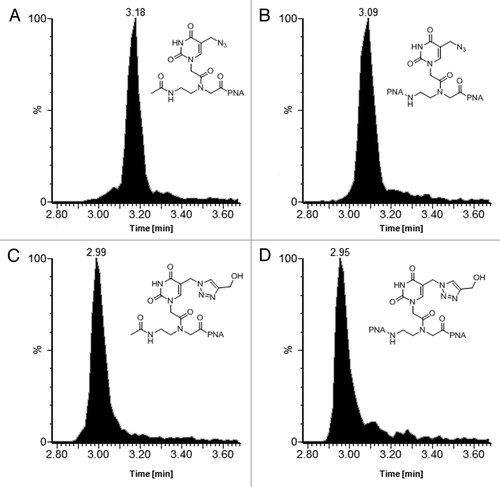
Besides the possibility to use the azide as a masking group for the protection of the amine function and subsequent reaction on the solid phase, the reactivity of this group itself can also be exploited for linking different moieties in solution through Huisgen’s click reaction. As a proof of concept PNA 1 and PNA 2 were cleaved from the resin, and used without purification as substrate for the reaction with propargyl alcohol.
Compared with classical flask click reaction, functionalization of PNA suffers from some problems related to the small (micromolar) scale in which these products are generally synthesized, leading to the necessity to set up reactions in very small amounts, in very small volumes, and this generates difficulties in properly controlling the reaction conditions. For this reason the reactions were performed using a stoichiometric amount of catalyst, generated in situ from copper (II) by the introduction of an excess of reducing agent (sodium ascorbate).
To test the applicability of the reaction, a PNA solution containing two different PNA systems (PNA 1 and PNA 2) was prepared directly from the raw products obtained from cleavage of the resin and testing three different alkyne/catalyst proportions (1, 2 and 5 equivalents). As in the solid phase syntheses, in solution both systems present the same reactivity.
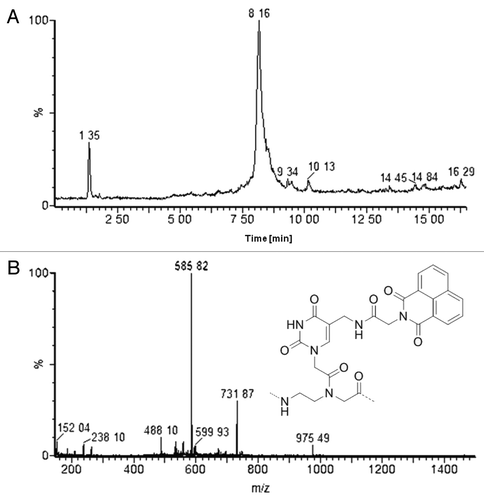
UPLC-ESI-MS analysis showed the complete conversion of the starting material after 45 min in presence of one or two equivalents of the mixture alkyne/catalyst, while with five equivalents the conversion was complete in less than 5 min. In the UPLC-MS traces of PNA 1 and PNA 2 before and after the addition of sodium ascorbate are reported.
Figure 6. UPLC-ESI extracted ion current (XIC) traces of PNA 1 and PNA 2 bearing the unreacted azide (A and B respectively) and after 45 min in presence of 1 equivalent of propargyl alcohol and Cu(I) (C and D respectively). XIC traces were obtained by extrapolating the multicharge signal of the different PNAs: 1468.1, 762.6 and 734.6 for chromatogram (A and C); 1259.5, 1231.6, 840.1, 821.2, 630.3 and 616.2 for chromatogram (B and D) (for mass spectra see Figs. S1 and S2).
Conclusions
In this study we propose the synthesis of modified PNA monomers bearing an azide-containing modified nucleobase for the application to both Boc- or Fmoc-based solid-phase syntheses. We also demonstrated their applicability to PNA synthesis and modification exploiting different conditions such as: Fmoc or Boc chemistries, modification immediately after base insertion or after PNA elongation, the insertion of multiple units and the simultaneous modification of two residues with the same functional group.
Finally, we demonstrated the possibility to use the embedded azide function as a substrate for the mild and chemoselective Huisgen’s click reaction in solution, which can allow the fast synthesis of derivatives not stable under the acid conditions needed for the PNA cleavage from the resin.
Materials and Methods
General
Reagents were purchased from Sigma-Aldrich, Fluka, Merck, Carlo Erba, TCI Europe, Link and ASM and used without further purification. All reactions were performed under a nitrogen atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Anhydrous solvents were obtained by distillation or anhydrification with molecular sieves. Reactions were monitored by TLC performed on 0.25 mm E. Merck silica-gel plates (60F-254) by using UV light as a visualizing agent and ninhydrin solution and heat as developing agents. E. Merck silica gel (60, particle size 0.040–0.063 mm) was used for flash-column chromatography. NMR spectra were recorded on Bruker AC 300 or Bruker Avance 400 instruments and calibrated by using residual undeuterated solvent as an internal reference. The following abbreviations were used to explain the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. IR spectra were measured using a FT-IR Thermo Nicolet 5700 in transmission mode using KBr or NaCl. UPLC-ESI-MS were recorded by using a Waters Acquity Ultra Performance LC with Waters Acquity SQ Detector with ESI interface and equipped with a Waters Acquity UPLC BEH 300 (50 × 2.1 mm, 1.7 µm, C18) (method A, 0.90 min in H2O 0.2% FA, then linear gradient to 50% MeCN 0.2% FA in 5.70 min at a flow rate of 0.25 mL/min; method B, 1.50 min in H2O 0.2% FA, then linear gradient to 80% MeCN 0.2% FA in 5.50 min at a flow rate of 0.25 mL/min) or with Waters Acquity UPLC BEH 300 (150 × 2.1 mm, 1.7 µm, C18) (method C, 1.50 min in H2O 0.2% FA, then linear gradient to 50% MeCN 0.2% FA in 14.30 min at a flow rate of 0.25 mL/min). HPLC-UV-MS were recorded by using a Waters Alliance 2695 HPLC with Micromass Quattro microAPI spectrometer, a Waters 996 PDA and equipped with a Phenomenex Jupiter column (250 × 4.6 mm, 5μ, C18, 300Å) (method D, 5 min in H2O 0.2% FA, then linear gradient to 50% MeCN 0.2% FA in 30 min at a flow rate of 1 mL/min). PNA oligomers were purified with RP-HPLC using a XTerra Prep RP18 column (7.8 × 300 mm, 10 μm) (method E, linear gradient from H2O 0.1% TFA to 50% MeCN 0.1% TFA in 30 min at a flow rate of 4.0 mL/min).
5-hydroxymethyluracil (2)
In a Shlenk tube uracil (10 g, 89.5 mmol) was dispersed together with paraformaldehyde (8.5 g, 283 mmol) in 200 mL H2O then tryethylamine (17.5 mL, 125.9 mmol) was added and the reaction mixture was heated overnight at 60°C. The excess formaldehyde was then purged with nitrogen in a 15% solution of sodium hypochlorite and the reaction solution was concentrated under vacuum to afford an oil that was taken up with 25 mL H2O; the product was precipitated adding 25 mL ethanol, collected through sintered glass funnel and washed with cold ethanol. Subsequent aliquots of product were collected by repeating the concentration-precipitation process to afford 2 as white solid (10.3 g, 81%); TLC (MeOH) Rf: 0.68, MP (°C): decomposes without melt at 215°C; 1H NMR (DMSO-d,Citation6 300 MHz) δ(ppm): 11.05 (1 H, s), 10.71 (1 H, s), 7.24 (1 H, s), 4.85 (1 H, br s), 4.10 (2 H, s); 13C NMR (DMSO-d,Citation6 75 MHz) δ(ppm): 163.8, 151.3, 138.2, 112.7, 55.8; MS (ESI, MeOH): m/z calculated for C5H6N2O3 [M]: 142.0378, found: 165.0 [M + Na]+, 307.2 [2 M + Na]+, 141.0 [M - H]-, 283.2 [2 M - H]-, 305.0 [2 M + Na - H]-; elemental composition: calculated %C 42.26, %H 4.26, %N 19.41, found %C 42.24, %H 4.35, %N 19.33; FT-IR (KBr) ν(cm−1): 3,369 (m), 3,189 (m), 3,038 (m), 2,865 (m), 1,704 (s), 1,672 (s).
5-chloromethyluracil (3)
In a round bottom flask 2 (6.76 g, 47.6 mmol) was dissolved in HCl 37% (25 mL). After 4 h a precipitate was formed and the product was collected by filtration through a sintered glass funnel and dried overnight over P2O5 to afford 3 as white solid (6.09 g, 80%). TLC (MeOH) Rf: 0.60, MP (°C): decomposes without melt at 239°C; 1H NMR (DMSO-d,Citation6 75 MHz) δ(ppm): 11.27 (1H, s), 11.04 (1 H, d, J = 5.0 Hz), 7.73 (1 H, d, J = 6.0 Hz), 4.40 (2 H, s); 13C NMR (DMSO-d,Citation6 300 MHz) δ(ppm): 162.8, 151.0, 142.4, 108.8, 39.8; MS (ESI, MeOH): m/z calculated for C5H5ClN2O2 [M]: 160.0040, found: 198.9 [M + K]+, 159.0 [M - H]-; elemental composition: calculated %C 37.40, %H 3.14, %N 17.45, found %C 37.48, %H 3.20, %N 16.94; FT-IR (KBr) ν(cm−1): 3,305 (s), 3,125 (m), 3,044 (m), 2,829 (m), 1,759 (s), 1,697 (s), 1,656 (s), 1,178 (s), 738 (m).
5-azidomethyluracil (4)
In a round bottom flask sodium azide (2.65 g, 40.78 mmol) was dispersed in 25 mL dry DMF and cooled to 0°C with an ice bath. Compound 3 (5.88 g, 36.63 mmol) dissolved in 80 mL dry DMF was added drop-wise over 30 min. After 2 h the orange solution was quenched by adding 1.0 mL HCl 37% and the resulting white suspension was concentrated under vacuum (t = 40°C). The crude product was sonicated with 25 mL DCM and subsequently with 30 mL cold water. Compound 4 was collected through sintered glass funnel as white solid (5.01 g, 90%). TLC (MeOH) Rf: 0.79, MP (°C): decomposes without melt at 188°C; 1H NMR (DMSO-d,Citation6 300 MHz) δ(ppm): 11.27(1 H, s), 11.17 (1 H, s), 7.65 (1 H, s), 4.02 (2 H, s); 13C NMR (DMSO-d,Citation6 75 MHz) δ(ppm): 163.8, 151.0, 141.9, 106.4, 46.3; MS (ESI, MeOH): m/z calculated for C5H5N5O2 [M]: 167.0443, found: 190.9 [M + Na]+, 166.0 [M - H]-, 333.1 [2 M - H]-; elemental composition: calculated %C 35.93, %H 3.02, %N 41.90, found %C 35.53, %H 2.95, %N 41.51; FT-IR (KBr) ν(cm−1): 3,219 (s), 3,079 (m), 3,026 (m), 2,825 (m), 2,132 (m), 1,757 (s), 1,689 (s), 1,667 (s).
Ethyl 2-(5-azidomethyluracil-1-yl)acetate (5a)
In a round bottom flask 4 (1.02 g, 6.07 mmol) and K2CO3 (0.84 g, 6.07 mmol) were dispersed in 10 mL dry DMF and cooled to 0°C with an ice bath. Ethyl 2-bromoacetate (0.674 mL, 6.07 mmol) was diluted with 1 mL dry DMF and added drop-wise over 1 h. The reaction was then left to warm to room temperature and left to stir overnight. The solvent was evaporated under vacuum and the resulting oil was partitioned between AcOEt (40 mL) and water (40 mL) and transferred in a separatory funnel, the aqueous phase was then extracted with AcOEt (2 × 40 mL). The combined organic fractions were dried together with Na2SO4 and concentrated under vacuum. Flash chromatography (EtOAc:hexane 8:2) yielded 5a as a white solid (0.63 g, 41%). TLC (AcOEt) Rf: 0.46, MP (°C): 128.5–129.4; 1H NMR (CDCl3, 300 MHz) δ(ppm): 8.83 (1 H, s), 7.17 (1 H, s), 4.48 (2 H, s), 4.26 (2 H, q, J = 7.1 Hz), 4.18 (2 H, s), 1.31 (3 H, t, J = 7.1 Hz); 13C NMR (CDCl3, 75 MHz) δ(ppm): 167.1, 162.4, 150.2, 142.4, 110.0, 62.4, 48.8, 46.9, 14.0; MS (ESI, MeOH): m/z calculated for C9H11N5O4 [M]: 253.0811, found: 276.2 [M + Na]+, 292.1 [M + K]+, 252.2 [M - H]-; elemental composition: calculated %C 42.69, %H 4.38, %N 27.66, found %C 42.82, %H 4.28, %N 27.37; FT-IR (KBr) ν(cm−1): 3,160 (m), 3,036 (s), 2,841 (m), 2,109 (s), 2,082 (s), 1,739 (s), 1,701(s), 1,647 (s).
Tert-butyl 2-(5‐azidomethyluracil-1-yl)acetate (5b)
In a round bottom flask 4 (510 mg, 3.1 mmol) and K2CO3 (422 mg, 3.1 mmol) were dispersed in 5 mL dry DMF and cooled to 0°C. Then t-butyl 2-bromoacetate (0.493 mL, 3.1 mmol) was diluted with 1 mL dry DMF and added drop-wise over 1 h. The reaction was then left to warm to room temperature and stirred overnight. The solvent was evaporated under vacuum and the resulting oil was partitioned between AcOEt (40 mL) and water (40 mL) and transferred in a separatory funnel, the aqueous phase was then extracted with AcOEt (2 × 40 mL). The combined organic fractions were dried together with Na2SO4 and concentrated under vacuum. Flash chromatography (AcOEt:hexane 5:3) yielded 5b as a white solid (0.42 g, 49%). TLC (AcOEt/hexane 5:3) Rf: 0.27, MP (°C): 141.0–142.2; 1H NMR (CDCl3, 300 MHz) δ(ppm): 9.07 (1 H, s), 7.17 (1 H, s), 4.38 (2 H, s), 4.17 (2 H, s), 1.49 (9 H, s); 13C NMR (CDCl3, 75 MHz) δ(ppm): 166.2, 162.4, 150.2, 142.7, 109.7, 83.8, 49.4, 46.9, 28.0; MS (ESI, MeOH): m/z calculated for C11H15N5O4 [M]: 281.1124, found: 280.2 [M - H]-; elemental composition: calculated %C 46.97, %H 5.38, %N 24.90, found %C 47.05, %H 5.33, %N 24.56; FT-IR (KBr) ν(cm−1): 3,178 (m), 3,054 (m), 2,998 (m), 2,824 (m), 2,117 (s), 1,736 (s), 1,705 (s).
2-(5-azidomethyluracil-1-yl)acetic acid (6)
Method A: in a round bottom flask 5a (1.15 g, 4.54 mmol) was dispersed in 10 mL MeOH, 10 mL NaOH 1 M were added and the mixture was left to react. After 1 h the solution became clear; the organic solvent was then evaporated under vacuum, the pH was lowered to 3 with HCl 37% and 6 was collected over Buchner as a white solid (0.98 g, 96%). Method B: in a round bottom flask 5b (386 mg, 1.36 mmol) was dissolved in 5 mL DCM, 3 mL TFA were added at 0°C and the mixture and left to react. After 2 h the solution was evaporated to give a yellow oil that was partitioned between water (40 mL) and AcOEt (40 mL). The organic layer was dried over Na2SO4 and concentrated under vacuum to obtain 6 as a white solid (216 mg, 70%). TLC (AcOEt/MeOH) Rf: 0.31, MP (°C): decomposes without melt at 175°C; 1H NMR (DMSO-d,Citation6 300 MHz) δ(ppm): 13.21 (1 H, br s), 11.65 (1 H, s), 7.83 (1 H, s), 4.43 (2 H, s), 4.06 (2 H, s); 13C NMR (DMSO-d,Citation6 75 MHz) δ(ppm): 169.3, 163.3, 150.6, 145.3, 107.3, 48.6, 46.5; MS (ESI, MeOH): m/z calculated for C7H7N5O4 [M]: 225.04980, found: 226.4 [M + H]+, 248.3 [M + Na]+, 264.2 [M + K]+, 473.2 [2 M + Na]+, 224.3 [M - H]-, 449.3 [2 M - H]-; elemental composition: calculated %C 37.34, %H 3.13, %N 31.10, found %C 37.45, %H 3.32, %N 30.24; FT-IR (KBr) ν(cm−1): 3,412(w), 3,002(br), 2,825(m), 2,108(s), 1,751(s), 1,700(s), 1,691(s), 1,482(s).
Tert-butyl 2-[N-(2-Fmoc-aminoethyl)-2-(5-azidomethyluracil-1-yl)acetamido]acetate (7a)
In a round bottom flask 6 (251 mg, 1.12 mmol) was dissolved in 3 mL dry DMF at 0°C together with EDC ∙ HCl (213 mg, 1.12 mmol), DhBtOH (181 mg, 1.12 mmol) and DIPEA (290 μl, 1.68 mmol) and left to react for 5 min. Tert-butyl 2-[(2-Fmoc-aminoethyl)amino]acetate hydrochloride (240 mg, 0.56 mmol) was added and the reaction mixture was stirred for 15 min at 0°C and further 2.5 h at room temperature. The reaction was then diluted with 200 mL AcOEt and washed with saturated KHSO4 (2 × 200 mL), saturated NaHCO3 (2 × 200 mL) and brine (200 mL). The organic fraction was dried over Na2SO4 and concentrated under vacuum. Flash chromatography (from AcOEt/hexane 1:1 to AcOEt/hezane 3:1) yielded 7b as a white solid (323 mg, 96%). TLC (AcOEt) Rf: 0.52; MP (°C): 83.1–86.0°C; 1H NMR (CDCl3, 400 MHz, major rotamer) δ(ppm): 9.65 (1 H, s), 7.77 (2 H, d, J = 7.5 Hz), 7.63 (2 H, d, J = 7.4 Hz), 7.41 (2 H, t, J = 7.4 Hz), 7.32 (2 H, t, J = 7.4 Hz), 7.11, 6.08 (1 H, br t), 4.50 (2 H, s), 4.46 (2 H, t, J = 7.6 Hz), 4.23 (1 H, t, J = 5.9 Hz, 3 H), 4.09 (2 H, s), 4.07 (2 H, s), 3.63–3.48 (2 H, m), 3.45–3.33 (2 H, m), 1.48 (9 H, s); 13C NMR (CDCl3, 100 MHz, major rotamer) δ(ppm): 168.7, 167.0, 163.2, 156.8, 150.8, 144.0, 143.8, 141.3, 127.8, 127.1, 125.1, 120.0, 109.5, 82.8, 66.8, 51.1, 49.8, 48.8, 48.0, 47.0, 39.2, 28.0; MS (ESI, MeOH): m/z calculated for C30H33N7O7 [M]: 603.24415, found: 626.3 [M + Na]+, 642.5 [M + K]+; HRMS (LTQ-Orbitrap, MeOH) m/z found: 604.2510 [C30H33O7N7]+.
Ethyl 2-[N-(2-Boc-aminoethyl)-2-(5-azidomethyluracil-1-yl)acetamido]acetate (7b)
In a round bottom flask 6 (0.70 g, 3.1 mmol) was solubilized in 10 mL dry DMF at 0°C together with DCC (0.63 g, 3.05 mmol), DhBtOH (0.40 g, 2.5 mmol) and left to react for 30 min. Ethyl 2-[(2-Boc-aminoethyl)amino]acetate (0.93 g, 0.33 mmol) and DIPEA (0.8 mL, 4.63 mmol) were added and the reaction mixture was left to stir overnight at room temperature. The reaction mixture was then filtered to remove the DCU, diluted with 100 mL AcOEt, and washed with saturated KHSO4 (3 × 100 mL), saturated NaHCO3 (3 × 100 mL) and brine (100 mL). The organic fraction was dried over Na2SO4 and concentrated under vacuum. Flash chromatography (AcOEt) yielded 7b as a white solid (1.06 g, 77%). TLC (AcOEt) Rf: 0.33; MP (°C): 62.5–69.8; 1H NMR (DMSO-d,Citation6 300 MHz, major rotamer) δ(ppm): 11.59 (1 H, s, 1 H), 7.64 (1 H, s), 6.94 (1 H, t, 1 H, J = 5.5 Hz), 4.72 (2 H, s), 4.25–3.95 (6 H, m), 3.41 (2 H, t, J = 6.5 Hz), 3.17 (2 H, q, J = 6.0 Hz), 1.38 (9 H, s), 1.24 (3 H, t, J = 7.1 Hz); 13C NMR (DMSO-d,Citation6 75 MHz, major rotamer) δ(ppm): 168.8, 167.0, 163.3, 155.6, 150.5, 145.4, 107.1, 77.9, 60.4, 47.7, 47.6, 46.7, 46.5, 46.4, 38.0, 28.0, 13.9; MS (ESI, MeOH): m/z calculated for C18H27N7O7 [M]: 453.1972, found: 452.6 [M - H]-; elemental composition: calculated %C 47.78, %H 6.00, %N 21.62, found %C 48.22, %H 6.29, %N 20.20; FT-IR (KBr) ν(cm−1): 3,329 (m), 3,210 (m), 3,065 (m), 2,981 (m), 2,934 (m), 2,851 (m), 2,108 (s), 1,750–1,600 (s), 1,466 (m).
2-[N-(2-Fmoc-aminoethyl)-2-(5-azidomethyluracil-1-yl)acetamido]acetic acid (8a)
In a round bottom flask 7a (311 mg, 0.51 mmol) was solubilized in 6 mL DCM at 0°C, then TFA (4 mL) was added and the reaction mixture was left to react. After 30 min the reaction mixture was left to warm to room temperature and to react for another 1 h. The solvent was then co-evaporated with MeOH and CHCl3 under reduced pressure. The resulting oil was dispersed in 20 mL H2O and filtered through Buchner to yield 8a as a white solid (241 mg, 86%). TLC (AcOEt/MeOH 1:1) Rf: 0.41; MP (°C): decomposes without melt at 110°C; 1H NMR (DMSO-d,Citation6 400 MHz, major rotamer) δ(ppm): 12.81 (1 H, br s), 11.60 (1 H, s), 7.90 (2 H, d, J = 7.5 Hz), 7.77–7.62 (3 H, m), 7.42 (2 H, t, J = 7.3 Hz), 7.33 (2 H, t, J = 7.4 Hz), 4.73 (2 H, s), 4.32 (2 H, dd, J = 18.3, 6.8 Hz), 4.23 (1 H, t, J = 7.0 Hz), 4.05 (2 H, s), 4.00 (2 H, s), 3.48–3.17 (4 H, m); 13C NMR (DMSO-d,Citation6 100 MHz, major rotamer) δ(ppm): 170.8, 167.5, 163.9, 156.8, 151.2, 146.3, 144.3, 141.2, 128.1, 127.5, 125.6, 120.6, 107.7, 66.0, 49.7, 48.6, 48.4, 48.2, 47.2, 38.4; MS (ESI, MeOH): m/z calculated for C26H25N7O7 [M]: 547.18155, found: 570.5 [M + Na]+, 586.5 [M + K]+, 546.4 [M - H]-; HRMS (LTQ-Orbitrap, MeOH) m/z found: 546.17335 [C26H24N7O7]-.
2-[N-(2-Boc-aminoethyl)-2-(5-azidomethyluracil-1-yl)acetamido]acetic acid (8b)
In a round bottom flask 7b (1.09 g, 2.40 mmol) was dissolved in 10 mL MeOH, 10 mL NaOH 1M were added and the mixture and left to react for 1 h 30 min. The reaction was then quenched with HCl 37%, and, after MeOH evaporation, the solution was diluted with saturated KHSO4 (50 mL) and extracted with AcOEt (2 × 50 mL). The combined organic phases were dried over Na2SO4 and evaporated under pressure to afford 8b as a white solid (980 mg, 96%). TLC (AcOEt/MeOH 6:4) Rf: 0.27; MP (°C): decomposes without melt at 156°C; 1H NMR (DMSO-d,Citation6 300 MHz, major rotamer) δ(ppm): 12.80 (1 H, s), 11.59 (1 H, s), 7.67 (1 H s, 6.94 (1 H, t, J = 5.4 Hz), 4.71 (2 H, s), 4.06 (2 H, s), 3.97 (2 H, s), 3.39 (2 H, t, J = 6.5 Hz), 3.16 (2 H, q, J = 5.7 Hz), 1.38 (9 H, s); 13C NMR (DMSO-d,Citation6 75 MHz, major rotamer) δ(ppm): 170.2, 166.7, 163.3, 155.6, 150.5, 145.5, 77.9, 47.7, 47.4, 47.0–46.0, 37.9, 28.0; MS (ESI, MeOH): m/z calculated for C16H23N7O7 [M]: 425.1659, found: 426.3 [M + H]+; FT-IR (KBr) ν(cm−1): 3,360 (m), 3,178 (m), 3,061 (m), 2,980 (m), 2,935 (m), 2,853 (m), 2,109 (s), 1,800–1,600 (s), 1,472 (m).
2-[1,3-dioxo-1H-benzo(de)isoquinoline-2(3H)-yl]acetic acid (napht-gly)
In a Shlenk tube 1,8-naphthalic anhydride (598 mg, 1.00 mmol) was dispersed in 7 mL DMF together with glycine (199 mg, 2.00 mmol). The reaction mixture was then warmed to 130°C for 5 h, after cooling down to room temperature the reaction was quenched with 250 mL AcOEt, transferred in a separatory funnel and washed with a saturated solution of KHSO4 (2 × 250 mL) and brine (250 mL). The organic phase was dried over Na2SO4 and the solvent removed under reduced pressure to afford the desired compound as a gray pounder (253 mg, 99%). TLC (AcOEt) Rf: 0.04; MP (°C): decomposes without melt at 247°C; 1H NMR (C6D6, 400 MHz) δ(ppm): 13.30 (1 H, s), 8.44 (2 H, d, J = 7.2 Hz), 7.78 (2 H, d, J = 8.3 Hz), 7.28 (2 H, d, J = 7.7 Hz), 4.95 (2 H, s); 13C NMR (C6D6, 100 MHz) δ(ppm): 169.9, 163.4, 134.0, 131.5, 130.9, 126.7, 122.4, 41.4; MS (ESI, MeOH): m/z calculated for C16H9NO4 [M]: 255.05316, found: 278.1 [M + Na]+, 254.1 [M - H]-; HRMS (LTQ-Orbitrap, MeOH) m/z found: 254.04615 [C16H9NO4]-; FT-IR (KBr) ν(cm−1): 3,456 (s), 2,359 (m), 1,702 (s), 1,651 (s), 1,392 (m).
Solid phase synthesis
PNAs synthesis was performed on MBHA resin (for Boc-protocol, PNA 3) or AM Champion (for Fmoc-protocol, PNA 1, PNA 2 and PNA 4) pre-loaded with 0.2 mmol/g Fmoc-Gly-OH. In all cases manual synthesis using HBTU/DIPEA as a coupling mixture was used and standard commercial monomer were employed together with 8a or 8b. For the insertion of monomer 8b, 10 equivalents and DIC/DhBtOH as activating mixture were used, in combination with 15 min pre-activation and overnight coupling. All coupling reactions on the deprotected amine were conducted using 10 equivalents of acid and DIC/DhBtOH as activating mixture, with no preactivating time and 1 h 30 min coupling. Where specified, PNA were purified by RP-HPLC. The identity of the PNAs were confirmed by UPLC-ESI-MS or HPLC-UV-MS for purified PNA. PNA 1: tR = 2.70 min (method A); ESI-MS: m/z calculated 2,458.9758 [M]: 1,231.2 [MH2]2+, 821.1 [MH3]3+, 616.2 [MH4]4+, 493.1 [MH4]4+; PNA 2: tR = 2.82 min (method A); ESI-MS: m/z calculated 1,466.5797 [M]: 1,468.1 [MH]+, 734.5 [MH2]2+, 490.2 [MH3]3+; PNA 3-napht: 12% yield, tR = 17.6 min (method E); ESI-MS: m/z calculated 2,587.1827 [M]: 1,294.9 [MH2]2+, 863.5 [MH3]3+, 647.8 [MH4]4+, 518.6 [MH5]5+; PNA 4-(naphy-gly)2: tR = 8.16 min (Method C); ESI-MS: m/z calculated 2,922.3990 [M]: 1,462.6 [MH2]2+, 975.9 [MH3]3+, 731.9 [MH4]4+, 585.2 [MH5]5+, 488.1 [MH6]6+.
Solution click reaction
Different water solutions were prepared: 2.5 mM solution of PNA, 200 mM solution of propargyl alcohol, 200 mM solution of copper sulfate, 200 mM solution of sodium ascorbate. Reactions were performed with a final PNA concentration of 1 mM using different proportion of propargyl alcohol maintaining a molar ratio alkyne/ascorbate/Cu(II) of 1:2:1. All solutions were analyzed by UPLC-MS using method B.
| Abbreviations: | ||
| AcOEt | = | ethyl acetate |
| Boc | = | t-butoxycarbonyl |
| DCC | = | N,N′-dicyclohexylcarbodiimide |
| DCM | = | dichloromethane |
| DhBtOH | = | 3-hydroxy-1,2,3-benzotriazin-4(3H)-one |
| DIC | = | N,N′-diisopropylcarbodiimide |
| DIPEA | = | diisopropylethylamine |
| DMF | = | N,N-dimethylformamide |
| DMSO | = | dimethylsulfocide |
| EDC | = | N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide |
| FA | = | formic acid |
| Fmoc | = | 9-fluorenylmethoxycarbonyl |
| MeOH | = | methanol |
| TFA | = | trifluoroacetic acid |
| TFMSA | = | trifluoromethanesulfonic acid |
| THF | = | tetrahydrofuran |
| XIC | = | extracted ion current |
Additional material
Download Zip (365.6 KB)Acknowledgments
This work was partially supported by a grant from MIUR [PRIN09 20093N774P “Molecular recognition of micro-RNA (miR) by modified PNA: from structure to activity”].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991; 254:1497 - 500; http://dx.doi.org/10.1126/science.1962210; PMID: 1962210
- Nielsen PE, Egholm M. Peptide Nucleic Acids: Protocols and Applications. 2. Wymondham: Horizon Press, 2004.
- Demidov VV. PNA and LNA throw light on DNA. Trends Biotechnol 2003; 21:4 - 7; http://dx.doi.org/10.1016/S0167-7799(02)00008-2; PMID: 12480343
- Zhang N, Appella DH. Advantages of peptide nucleic acids as diagnostic platforms for detection of nucleic acids in resource-limited settings. J Infect Dis 2010; 201:Suppl 1 S42 - 5; http://dx.doi.org/10.1086/650389; PMID: 20225945
- Nielsen PE. Gene targeting using peptide nucleic acid. Methods Mol Biol 2005; 288:343 - 58; PMID: 15333914
- Gourishankar A, Shukla S, Sastry M, Ganesh KN. DNA and PNA as templates for building nanoassemblies via electrostatic complexation with gold nanoparticles. Curr Appl Phys 2005; 5:102 - 7; http://dx.doi.org/10.1016/j.cap.2004.06.020
- Corradini R, Tedeschi T, Sforza S, Green MM, Marchelli R. Control of Helical Handedness in DNA and PNA Nanostructures. In: Zuccheri G, Samorì B, eds. DNA Nanotechnology: Methods in Molecular Biology. 749. Springer Science+Business Media, 2001, 79-92. doi:10.1007/978-1-61779-142-0_6.
- Howarth NM. Recent advances in the field of peptide nucleic acids. Lett Org Chem 2006; 3:495 - 503; http://dx.doi.org/10.2174/157017806778341852
- Falkiewicz B. Peptide nucleic acids and their structural modifications. Acta Biochim Pol 1999; 46:509 - 29; PMID: 10698263
- Ganesh KN, Nielsen PE. Peptide Nucleic Acids: analogs and derivatives. Curr Org Chem 2000; 4:931 - 43; http://dx.doi.org/10.2174/1385272003375969
- Corradini R, Sforza S, Tedeschi T, Totsingan F, Manicardi A, Marchelli R. Peptide nucleic acids with a structurally biased backbone. Updated review and emerging challenges. Curr Top Med Chem 2011; 11:1535 - 54; http://dx.doi.org/10.2174/156802611795860979; PMID: 21510833
- Kumar VA, Ganesh KN. Conformationally constrained PNA analogues: structural evolution toward DNA/RNA binding selectivity. Acc Chem Res 2005; 38:404 - 12; http://dx.doi.org/10.1021/ar030277e; PMID: 15895978
- Menchise V, De Simone G, Tedeschi T, Corradini R, Sforza S, Marchelli R, et al. Insights into peptide nucleic acid (PNA) structural features: the crystal structure of a D-lysine-based chiral PNA-DNA duplex. Proc Natl Acad Sci U S A 2003; 100:12021 - 6; http://dx.doi.org/10.1073/pnas.2034746100; PMID: 14512516
- Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH. A simple γ-backbone modification preorganizes peptide nucleic acid into a helical structure. J Am Chem Soc 2006; 128:10258 - 67; http://dx.doi.org/10.1021/ja0625576; PMID: 16881656
- Sforza S, Tedeschi T, Corradini R, Marchelli R. Induction of Helical Handedness and DNA Binding Properties of Peptide Nucleic Acids (PNAs) with Two Stereogenic Centres. Eur J Org Chem 2007; 35:5879 - 85; http://dx.doi.org/10.1002/ejoc.200700644
- de Koning MC, Petersen L, Weterings J, Overhand JM, van der Marel GA, Filippov DV. Synthesis of thiol-modified peptide nucleic acids designed for post-assembly conjugation reactions. Tetrahedron 2006; 62:3248 - 58; http://dx.doi.org/10.1016/j.tet.2006.01.065
- Englung EA, Appella DH. Gamma-substituted peptide nucleic acids constructed from L-lysine are a versatile scaffold for multifunctional display. Angew Chem Int Ed 2007; 46:1414 - 8; http://dx.doi.org/10.1002/anie.200603483
- Wojciechowski F, Hudson RHE. Nucleobase modifications in peptide nucleic acids. Curr Top Med Chem 2007; 7:667 - 79; http://dx.doi.org/10.2174/156802607780487795; PMID: 17430208
- Eldrup AB, Christensen C, Haaima G, Nielsen PE. Substituted 1,8-naphthyridin-2(1H)-ones are superior to thymine in the recognition of adenine in duplex as well as triplex structures. J Am Chem Soc 2002; 124:3254 - 62; http://dx.doi.org/10.1021/ja0117027; PMID: 11916408
- Wilhelmsson LM, Holmén A, Lincoln P, Nielsen PE, Nordén B. A highly fluorescent DNA base analogue that forms Watson-Crick base pairs with guanine. J Am Chem Soc 2001; 123:2434 - 5; http://dx.doi.org/10.1021/ja0025797; PMID: 11456897
- Rajeev KG, Maier MA, Lesnik EA, Manoharan M. High-affinity peptide nucleic acid oligomers containing tricyclic cytosine analogues. Org Lett 2002; 4:4395 - 8; http://dx.doi.org/10.1021/ol027026a; PMID: 12465896
- Loakes D. Survey and summary: The applications of universal DNA base analogues. Nucleic Acids Res 2001; 29:2437 - 47; http://dx.doi.org/10.1093/nar/29.12.2437; PMID: 11410649
- Luyten L, Herdewijn P. Hybridization properties of base-modified oligonucleotides within the double and triple helix motif. Eur J Med Chem 1998; 33:515 - 76; http://dx.doi.org/10.1016/S0223-5234(98)80016-0
- Weisbrod SH, Marx A. Novel strategies for the site-specific covalent labelling of nucleic acids. Chem Commun (Camb) 2008; 5675 - 85; http://dx.doi.org/10.1039/b809528k; PMID: 19009049
- Englund EA, Appella DH. γ-substituted peptide nucleic acids constructed from L-lysine are a versatile scaffold for multifunctional display. Angew Chem Int Ed Engl 2007; 46:1414 - 8; http://dx.doi.org/10.1002/anie.200603483; PMID: 17133633
- Schneggenburger PE, Worbs B, Diederichsen U. Azide reduction during peptide cleavage from solid support-the choice of thioscavenger?. J Pept Sci 2010; 16:10 - 4; http://dx.doi.org/10.1002/psc.1202; PMID: 19950105
- Hudson RHE, Liu X, Wojciechowski F. Hydrophilic Nucleobase Modifications in Peptide Nucleic Acid: Synthesis and Properties of PNA Containing 5-Hydroxymethyl- Uracil and Cytosine. Can J Chem 2007; 85:302 - 12; http://dx.doi.org/10.1139/v07-030
- Debaene F, Winssinger N. Azidopeptide nucleic acid. An alternative strategy for solid-phase peptide nucleic acid (PNA) synthesis. Org Lett 2003; 5:4445 - 7; http://dx.doi.org/10.1021/ol0358408; PMID: 14602021